
- Department of Gastroenterology and Hepatology, Kansai Medical University, Moriguchi, Osaka, Japan
Current evidence suggests that regulation of extracellular matrix (ECM) accumulation by fibrogenic transforming growth factor (TGF)-β and platelet-derived growth factor (PDGF) signals involves different mechanisms in acute and chronic liver injuries, even though hepatic stellate cells (HSC) are the principal effecter in both cases. As a result of chronic liver damage, HSC undergo progressive activation to become myofibroblasts (MFB)-like cells. Our current review will discuss the differential regulation of TGF-β signaling between HSC and MFB in vitro and in vivo. Smad proteins, which convey signals from TGF-β receptors to the nucleus, have intermediate linker regions between conserved Mad-homology (MH) 1 and MH2 domains. TGF-β type I receptor and Ras-associated kinases differentially phosphorylate Smad2 and Smad3 to create COOH-terminally (C), linker (L), or dually (L/C) phosphorylated (p) isoforms. After acute liver injury, TGF-β and PDGF synergistically promote collagen synthesis in the activated HSC via pSmad2L/C and pSmad3L/C pathways. To avoid unlimited ECM deposition, Smad7 induced by TGF-β negatively regulates the fibrogenic TGF-β signaling. In contrast, TGF-β and PDGF can transmit the fibrogenic pSmad2L/C and mitogenic pSmad3L signals in MFB throughout chronic liver injury, because Smad7 cannot be induced by the pSmad3L pathway. This lack of Smad7 induction might lead to constitutive fibrogenesis in MFB, which eventually develop into accelerated liver fibrosis.
Introduction
Current evidence suggests that regulation of extracellular matrix (ECM) accumulation in acute and chronic liver injuries involves different mechanisms, even though hepatic stellate cells (HSC) are the principal effecter in both cases (Friedman, 2010). HSC characterized by retinoid droplets in the cytoplasm (Figure 1), are present in the space of Disse in close contact with hepatocytes and sinusoidal endothelial cells. When HSC are activated, they lose retinoid, and show increased proliferation, motility, and ECM production (Brenner, 2009). As a result of chronic liver damage, HSC undergo progressive activation to become myofibroblasts (MFB)-like cells (Inagaki and Okazaki, 2007).
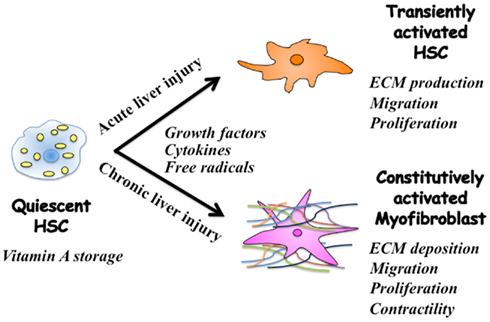
Figure 1. Phenotypic alternation of HSC during acute and chronic liver injuries. Quiescent hepatic stellate cells (HSC) are characterized by retinoid droplets in the cytoplasm. Following single liver injury, HSC transiently acquire an activated phenotype, which includes extracellular matrix (ECM) synthesis, proliferation, and migration. HSC move from the space of Disse to sites of damage where the activated HSC contribute to tissue repair by producing large amounts of collagens. Prolonged exposure to chronic injury, HSC undergo constitutive activation to become myofibroblasts (MFB)-like cells, which persistently induce deposition of ECM and liver fibrosis. Contraction of MFB contributes to increased portal resistance during liver fibrosis that presumably is reversible before the thickened septae, intrahepatic shunts, and lobular distortion of cirrhosis develop, leading to fixed increases in portal pressure.
Transforming growth factor (TGF)-β is a central regulator in chronic liver disease contributing to fibrogenesis through inflammation (Dooley and Ten Dijke, 2012). On the other hand, platelet-derived growth factor (PDGF) through tyrosine kinase receptor is the most powerful mitogen for HSC, and also promotes liver fibrosis (Pinzani, 1999). Within inflammatory microenvironment, both TGF-β and PDGF are secreted by platelets and Kupffer cells (Pinzani and Macias-Barragan, 2010). A significant increase in TGF-β expression is observed in the activated HSC and MFB, thus indicating that TGF-β acts as an autocrine positive regulator for ECM production. Responsiveness of ECM production to TGF-β is transient in the process of tissue repair such as liver regeneration after acute liver injury (Date et al., 1998, 2000), thus suggesting that some regulatory mechanisms for the TGF-β signal are present in the activated HSC. In contrast, persistent TGF-β signal associated with the accelerated ECM accumulation is a common finding in human chronic liver diseases of different etiologies (Kisseleva and Brenner, 2008), indicating that MFB lose their negative regulation for ECM accumulation.
Progress over the past 10 years has disclosed important details of how the TGF-β family elicits its responses (Heldin et al., 1997; Wrana, 2000; Shi and Massague, 2003; Guo and Wang, 2009). Smads, central mediators conveying signals from receptors for TGF-β superfamily members to the nucleus, are modular proteins with conserved Mad-homology (MH)1, intermediate linker, and MH2 domains (Shi and Massague, 2003). In cell-signaling pathways, various transcription factors are phosphorylated at multiple sites by upstream kinases. Catalytically active TGF-β type I receptor (TβRI) phosphorylates COOH-tail serine residues of receptor-activated Smad (R-Smad), which include Smad2 and the highly similar protein Smad3 (Wrana, 2000). Mitogenic signals alternatively cause phosphorylation of R-Smad at specific sites in their middle linker regions (Kretzschmar et al., 1999; Matsuura et al., 2004; Mori et al., 2004; Kamaraju and Roberts, 2005; Sekimoto et al., 2007; Matsuzaki et al., 2009). After phosphorylated R-Smad rapidly oligomerize with Smad4, this complex translocates to the nucleus, where it regulates transcription of target genes. Antagonistic Smad7 acts in opposition to R-Smads and Smad4, forming stable associations with the activated type I receptors, and preventing the phosphorylation of R-Smads (Hayashi et al., 1997; Nakao et al., 1997). The transcription of the Smad7 gene is regulated by TGF-β signaling through direct binding of Smad3 and Smad4 to the Smad7 promoter (Nagarajan et al., 1999; von Gersdorff et al., 2000). Therefore, Smad7 functions in a negative-feedback loop to terminate or reduce the strength of the signal.
Monitoring phosphorylation status of signaling molecules is a key step in dissecting their pathways. In Smad signaling, phosphorylation of not only COOH-tail but also linker regions of R-Smad is likely to be important in regulating Smad activity under physiologic and pathologic conditions (Liu, 2006). In this review, we first examine Smad signaling specificity derives from spatial and temporal dynamics of domain-specific R-Smad phosphorylation in response to TGF-β and/or PDGF. We next consider how TGF-β signal together with PDGF promotes collagen synthesis in the activated HSC after acute liver injury. Then, we examine constitutive fibrogenic TGF-β signal in MFB during chronic liver injury. Finally, we consider differential regulation of ECM accumulation between HSC and MFB.
Canonical TGF-β Signal Transduction
Transforming growth factor-β initiates signaling by binding to type I and type II receptor serine/threonine kinase on the cell surface. R-Smads are phosphorylated by the activated TβRI on the C-terminal SXS motif (Derynck and Miyazono, 2008). The activated R-Smads form the complex with the common partner Smad4. The complex translocates to the nucleus, where they regulate target gene expression by interaction with other transcription factors, co-activators, and co-repressors (Heldin et al., 1997; Wrana, 2000; Shi and Massague, 2003; Guo and Wang, 2009). This pathway is regulated by several autoinhibitory feedback loops. In particular, Smad7 interacts stably with activated TβRI to inhibit COOH-tail phosphorylation of R-Smads (Nakao et al., 1997).
PDGF Signal Transduction
Platelet-derived growth factor is not only a potent mitogen but also a fibrogenic growth factor known to synergize with TGF-β (Pinzani and Macias-Barragan, 2010). Signaling by PDGF begins by interaction with trans-membrane receptor tyrosine kinases (Heldin and Westermark, 1999). Multiple signaling pathways originating from the receptors have been identified. The most prominent pathways are mediated by members of the mitogen-activated protein kinase (MAPK) family, which includes the extracellular signal-regulated protein kinase (ERK) pathway and two stress-activated protein kinase (SAPK) pathways: the c-Jun N-terminal kinase (JNK) and the p38 pathway (Robinson and Cobb, 1997). MAPK is capable of phosphorylating many transcription factors, which are important in initiating cell proliferation and migration.
Multiple Levels of Crosstalk Exist between TGF-β and JNK Signaling Pathways
Transforming growth factor-β elicits signaling responses through non-Smad pathways that are generally considered as important effecter pathways for Ras/MAPK in response to PDGF (Derynck and Zhang, 2003). Notably, JNK activates AP-1 consisting of c-Jun that cooperates with nuclear Smads in mediating TGF-β-induced transcriptional responses (Derynck and Zhang, 2003). Thus, Smad-mediated signaling pathway is controlled by or functions in conjunction with other non-Smad transcription factors governed by JNK.
Although COOH-tail phosphorylation by TβRI is a key event in R-Smad activation, additional phosphorylation can positively and negatively regulate R-Smads pathway. Smad2 and Smad3 contain two conserved polypeptide segments, the MH1 and MH2 domains, joined by a less conserved linker region (Shi and Massague, 2003). The linker domain undergoes regulatory phosphorylation by JNK and cyclin-dependent kinase (CDK) pathways (Matsuura et al., 2004; Mori et al., 2004).
In one set of influential experiments, JNK phosphorylates Smad3 outside its COOH-tail phosphorylation site, and enhances activation and nuclear translocation of Smad3 (Engel et al., 1999). Furthermore, activation of MAPK kinase 1 (MEKK-1), an activator of JNK and ERK MAPK, leads to phosphorylation and activation of Smad2 (Brown et al., 1999). This mechanism may explain the ability of fluid shear stress or some growth factors to activate Smad2 (de Caestecker et al., 1998; Matsuzaki et al., 2009). We further identify phosphorylation sites for JNK in the linker regions of Smad2 and Smad3 (Sekimoto et al., 2007). Thus, multiple levels of crosstalk exist between TGF-β and JNK signaling pathways.
Multiple Smad Phosphoisoforms Exist
JNK simultaneously activates linker-phosphorylated Smad and non-Smad signaling, with both usually operating in parallel. Biologic significance of linker-phosphorylated Smad pathway is therefore difficult to assess in isolation. Antibodies (Abs) reactive with structurally related phosphorylated peptides are emerging as valuable tools for determining phosphorylation sites in vivo, and for investigating their distinct signals via phosphorylated domains. We therefore generated the domain-specific Abs, which selectively distinguish between the phosphorylated linker regions and the C-terminal regions of Smad2 and Smad3. Using the Abs, we have investigated for linker-phosphorylated R-Smads pathway, with a particular focus on how JNK modulates TβRI signaling through Smad linker phosphorylation.
Domain-specific phospho-R-Smad Abs have allowed us to reveal that TβRI and JNK/CDK4 differentially phosphorylate R-Smads to create three phosphorylated forms (phosphoisoforms): COOH-terminally phosphorylated R-Smad (pSmad2C and pSmad3C), linker-phosphorylated R-Smad (pSmad2L and pSmad3L), and dually phosphorylated R-Smad (pSmad2L/C and pSmad3L/C; Matsuzaki, 2006, 2012; Sapkota et al., 2006; Wrighton et al., 2009). Except for pSmad2L with cytoplasmic localization (Kretzschmar et al., 1999; Mori et al., 2004), the other phosphoisoforms are localized to cell nuclei (Furukawa et al., 2003; Mori et al., 2004; Yamagata et al., 2005; Yoshida et al., 2005; Matsuzaki et al., 2007; Sekimoto et al., 2007; Alarcon et al., 2009; Matsuzaki, 2009; Murata et al., 2009; Nagata et al., 2009; Kawamata et al., 2011). Linker phosphorylation can modify COOH-terminally phosphorylated R-Smad signaling (Kretzschmar et al., 1999; Matsuura et al., 2004; Mori et al., 2004; Ho et al., 2005; Sekimoto et al., 2007; Matsuzaki, 2009).
Differential localization of kinases and phosphatases in the cytoplasm or nucleus raises the intriguing possibility of different temporal dynamics for cytoplasmic or nuclear R-Smad phosphoisoforms, and adds to the repertoire of signaling responses that determine cell-fate decisions (Matsuzaki, 2011). Immunohistochemical and immunofluorescence analyses using specific Abs in human tissues can examine clinical significance of context-dependent and cell type-specific signaling mediated by R-Smad phosphoisoforms, by comparison of tissue/cellular localization of these phosphoisoforms in various pathologic specimens.
Differential Phospho-Smad Signaling between HSC and MFB in vitro
Liu (2006) group previously reported that Smad3 is phosphorylated by CDK4 in vivo and in vitro. CDK4-mediated phosphorylation of Smad3 at its linker region inhibits its transcriptional activity and the anti-proliferative activity of TGF-β in fibroblasts (Matsuura et al., 2004; Wang et al., 2009). Importantly, COOH-tail phosphorylation of Smad3 is necessary for TGF-β-induced phosphorylation of Smad3 at its linker region in fibroblasts (Wang et al., 2009; Matsuura et al., 2010). Consistent with these observations concerning Smad3, we have confirmed that the nuclear cyclin D1/CDK4 complex of fibroblasts activated by TGF-β and PDGF signaling directly phosphorylates the linker segment of pSmad2C to produce pSmad2L/C (Matsuzaki et al., 2009). The expression of c-Myc in fibroblasts is initially repressed by TGF-β, but subsequent cyclin D1/CDK4 undergoes a complete functional change to stimulate c-Myc (Matsuzaki et al., 2009). TGF-β inhibits cell growth by down-regulating the c-Myc via the pSmad2C and pSmad3C pathways (Figure 2A, left). However, signaling activated by TGF-β and PDGF can enhance fibroblast growth by up-regulating c-Myc via the CDK4-dependent pSmad2L/C and pSmad3L/C pathways in cell nuclei (Figure 2A, right). Moreover, Hayashida et al. (2003) reported that pSmad3L/C increases collagen I synthesis in human mesangial cells. We further reported that co-treatment of primary cultured HSC with TGF-β and PDGF enhances transcription of collagen I gene via pSmad3L/C pathway (Furukawa et al., 2003; Yoshida et al., 2005). Importantly, TGF-β-mediated Smad7 in HSC terminates the fibrogenic signals via the pSmad2L/C and pSmad3L/C pathways (Tahashi et al., 2002; Yoshida et al., 2005).
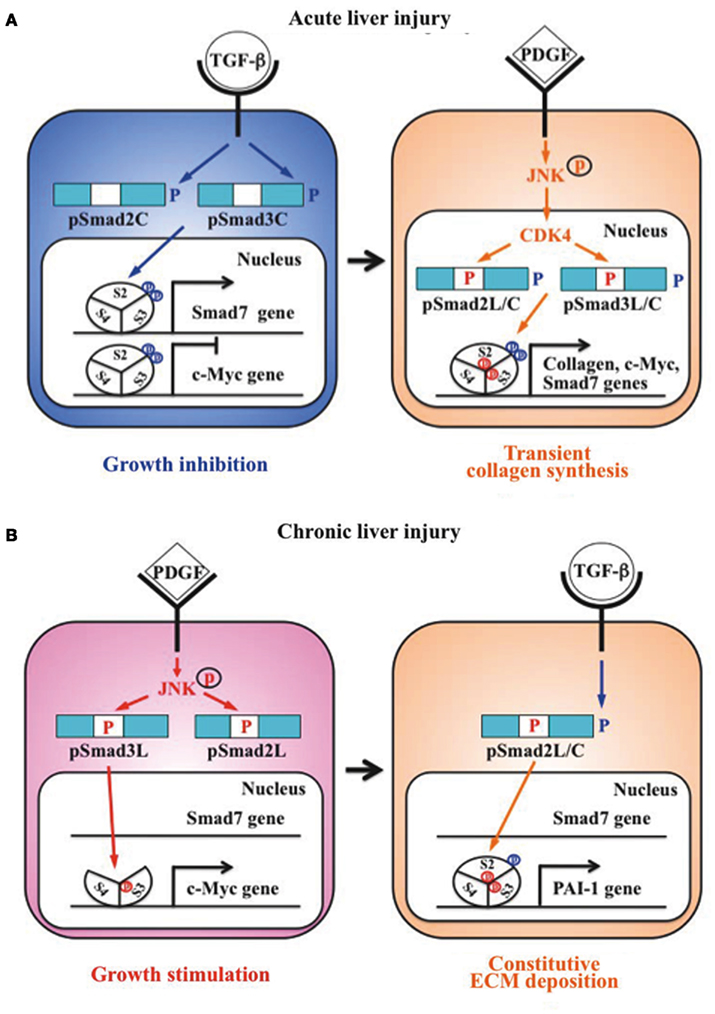
Figure 2. Differential regulation of fibrogenic phospho-Smad signaling between HSC during acute liver injury and MFB during chronic liver injury. (A) Phospho-Smad signaling involved in tissue repair. TGF-β inhibits HSC growth by down-regulating c-Myc expression by pSmad2C and pSmad3C pathways (left); TGF-β signaling in turn enhances HSC growth and collagen synthesis via the CDK4-dependent pSmad2L/C and pSmad3L/C pathways induced by PDGF signal (right). However, Smad7 induced by pSmad3L/C signal terminates the fibrogenic phospho-Smad signaling. This negative-feedback mechanism of the fibrogenic TGF-β/PDGF signal results in a transient collagen synthesis in the activated HSC, which may thus contribute to tissue repair. (B) Phospho-Smad signaling involved in fibrogenesis. In MFB, PDGF activates JNK, which phosphorylates Smad2L and Smad3L (left). The JNK-mediated Smad3L phosphorylation leads to hetero-complex of Smad3 with Smad4 in the nucleus where the complex stimulates MFB growth by upregulation of c-Myc transcription. After COOH-tail phosphorylation of cytoplasmic pSmad2L by TGF-β signal, pSmad2L/C translocates to the nucleus where it binds to the pSmad3L and Smad4 complex, which then stimulates plasminogen activator inhibitor (PAI)-1 transcription (right). In contrast of Smad7 induction in HSC via pSmad3C pathway, pSmad3L cannot induce Smad7 in MFB (left). Under a low level of Smad7, the fibrogenic phospho-Smad signaling can constitutively promote ECM deposition by MFB, which may eventually develop into accelerated liver fibrosis.
Primary cultured HSC are activated into MFB-like cells after being cultured on plastic dishes. The morphologic features of the cell line closely resembled a MFB-like morphology characterized by spindle shape, cell enlargement, and reduction of the size of intracellular vacuoles (Rockey et al., 1993). In MFB, PDGF induces pSmad3L via the activated JNK pathway (Yoshida et al., 2005). The JNK-mediated Smad3L phosphorylation leads to hetero-complex of Smad3 with Smad4 in the nucleus (Mori et al., 2004; Sekimoto et al., 2007). On the other hand, activated JNK retains most Smad2 protein in the cytoplasm (Kretzschmar et al., 1999; Sekimoto et al., 2007). Smad2 can accumulate in the nucleus only if its C-terminus is phosphorylated under conditions of sustained linker phosphorylation by JNK. After COOH-tail phosphorylation of cytoplasmic pSmad2L by TβRI, pSmad2L/C undergoes translocation to the nucleus where it binds to the pSmad3L and Smad4 complex (Furukawa et al., 2003; Matsuzaki et al., 2009; Figure 2B, right), which then in turn stimulates plasminogen activator inhibitor (PAI)-1 transcription (Furukawa et al., 2003). The level of PAI-1 is significantly elevated in fibrotic liver, and PAI-1 overexpression contributes to excessive accumulation of collagen and other ECM proteins, while lack of PAI-1 protects liver fibrosis in response to injury-related profibrotic TGF-β signals (Hu et al., 2009). In contrast to Smad7 induction in HSC via pSmad3C pathway, pSmad3L cannot induce Smad7 transcript in MFB (Tahashi et al., 2002; Tahashi et al., unpublished data). Collectively, fibrogenic TGF-β signal can constitutively accelerate ECM accumulation in MFB by upregulation of PAI-1 protein under a low level of Smad7.
Differential Phospho-Smad Signaling between the Activated HSC during Acute Liver Injury and MFB during Chronic Liver Injury
Signaling by TGF-β was indispensable for ECM synthesis during remodeling of liver disease (Moriya et al., 2011). As TGF-β is secreted in a biologically inactive form, the important step in regulating its biological activity is the conversion of the latent form into the active one. The matricellular protein thrombospondin-1 (TSP-1) was first shown as a component of the α-granule in platelets and can act as a major activator of latent TGF-β (Mosher, 1990; Crawford et al., 1998). Hayashi et al. (2011) recently identified TSP-1 as an inhibitory element in regulating liver regeneration via TGF-β1 activation. Their group also intensively investigates new therapeutic strategies targeting local TGF-β activation in fibrogenesis.
To clarify the differential TGF-β signals between in acute and chronic liver injuries, we previously analyzed the sequential expressions of TGF-β and its receptors in HSC and MFB during acute and chronic CCl4 intoxication (Date et al., 1998, 2000). The finding, however, merely shows an elevated TGF-β expression in both cases, and no differences in the expression of their receptors between HSC and MFB. This implies that a modification of postreceptor TGF-β signal in MFB is a more important step for the process of liver fibrosis during chronic liver injury.
To elucidate how cytostatic TGF-β signaling in HSC takes on collagen-producing features within inflammatory microenvironments during acute liver injury, we focus on the Smad pathway investigating localization of pSmad2L/C and pSmad3L/C in chemically injured rat livers. Nuclear localization of pSmad2L/C and pSmad3L/C is seen in the activated HSC (Yoshida et al., 2005). In particular, strong Smad2/3 phosphorylation at the COOH-tail and threonine residues in the linker regions is observed in the activated HSC (Yoshida et al., 2005). pSmad2L/C and pSmad3L/C signaling may mobilize HSC from the space of Disse to sites of damage, where the activated HSC contribute to tissue repair by producing large amounts of collagens. In HSC after acute liver injury, TβRI activated by endogenous TGF-β signal phosphorylates Smad3C, further up-regulating Smad7 transcription (Figure 2A, right). Subsequently, Smad7 terminates the fibrogenic TGF-β signal mediated by pSmad2L/C and pSmad3L/C pathways. Smad7 could be involved in tight restriction of R-Smads activation in HSC, and regulates the intensity and duration of the TGF-β responses during acute liver injury (Dooley et al., 2000; Tahashi et al., 2002).
The above regulatory mechanism, which avoids unlimited accumulation of ECM proteins that promote liver fibrosis, is disrupted in MFB during chronic liver injury. During transdifferentiation from HSC to MFB in culture, pSmad3C-mediated signal decreases while the pSmad3L pathway predominates (Furukawa et al., 2003; Figure 2B left). The observations fully support the finding of pSmad3L rather than pSmad3C in nuclei of α-smooth muscle actin (SMA)-immunoreactive MFB in portal tracts of chronically HCV-infected liver specimens (Matsuzaki et al., 2007). The presence of α-SMA is associated with transdifferentiation of HSC into scar-forming MFB, an event that is considered pivotal in the fibrogenic response (Pinzani and Macias-Barragan, 2010). In contrast to a transient increase in Smad7 in the activated HSC after acute liver injury, Smad7 remains at a low level in MFB throughout chronic liver injury (Tahashi et al., 2002). Because Smad7 cannot be induced by the pSmad3L pathway (unpublished data), the lack of Smad7 induction in MFB during chronic liver disease might lead to constitutive fibrogenic TGF-β (Dooley et al., 2000; Stopa et al., 2000; Tahashi et al., 2002). Accordingly, Smad7 overexpression results in less accumulation of interstitial collagens and improves liver fibrosis (Dooley et al., 2003). Moreover, IFN-γ displays antifibrotic effects by upregulation of Smad7 expression (Weng et al., 2009).
Conclusion and Future Perspectives
Because TGF-β involves a variety of physiologic processes such as liver regeneration, unraveling the molecular mechanisms of the TGF-β signal in a pathologic condition is critical to our understanding of its role in disease and the development of its therapies (Bataller and Brenner, 2001). This review summarizes Smad phosphoisoform-mediated signals in HSC, showing differences between in acute and chronic liver injuries. After acute liver injury, TGF-β and PDGF synergistically enhance collagen synthesis by activated HSC via the pSmad2L/C and pSmad3L/C pathways. In recovery stage of acute liver injury, Smad7 induced by TGF-β negatively regulates the fibrogenic TGF-β signaling. In contrast, PDGF-mediated pSmad3L cannot induce Smad7 in MFB even if high concentration of TGF-β is observed in chronic liver diseases. Under a low level of Smad7, MFB can constitutively exhibit the mitogenic pSmad3L and fibrogenic pSmad2L/C signaling, thereby accelerating liver fibrosis.
Transforming growth factor-β activates both Smad and non-Smad signals, which crosstalk with JNK pathway at multiple levels to provide context-dependent outcome (Derynck and Akhurst, 2007). Hence, context-specific effects in HSC can be generated by combined assemblies and spatiotemporal dynamics of R-Smad phosphoisoforms and non-Smad pathway activation, changes of interacting protein partners, and availability of repressors/activators. Although our understanding of the molecular mechanism of liver fibrosis in MFB has significantly advanced during the past decade, much work is needed to define the transcriptional regulatory networks in a context-specific way.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alarcon, C., Zaromytidou, A. I., Xi, Q., Gao, S., Yu, J., Fujisawa, S., Barlas, A., Miller, A. N., Manova-Todorova, K., Macias, M. J., Sapkota, G., Pan, D., and Massagué, J. (2009). Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-β pathways. Cell 139, 757–769.
Bataller, R., and Brenner, D. A. (2001). Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 21, 437–451.
Brenner, D. A. (2009). Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 120, 361–368.
Brown, J. D., DiChiara, M. R., Anderson, K. R., Gimbrone, M. A. Jr., and Topper, J. N. (1999). MEKK-1, a component of the stress (stress-activated protein kinase/c-Jun N-terminal kinase) pathway, can selectively activate Smad2-mediated transcriptional activation in endothelial cells. J. Biol. Chem. 274, 8797–8805.
Crawford, S. E., Stellmach, V., Murphy-Ullrich, J. E., Ribeiro, S. M., Lawler, J., Hynes, R. O., Bovin, G. P., and Bouck, N. (1998). Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 93, 1159–1170.
Date, M., Matsuzaki, K., Matsushita, M., Sakitani, K., Shibano, K., Okajima, A., Yamamoto, C., Ogata, N., Okumura, T., Seki, T., Kubota, Y., Kan, M., McKeehan, W. L., and Inoue, K. (1998). Differential expression of transforming growth factor-β and its receptors in hepatocytes and nonparenchymal cells of rat liver after CCl4 administration. J. Hepatol. 28, 572–581.
Date, M., Matsuzaki, K., Matsushita, M., Tahashi, Y., Furukawa, F., and Inoue, K. (2000). Modulation of transforming growth factor β function in hepatocytes and hepatic stellate cells in rat liver injury. Gut 46, 719–724.
de Caestecker, M. P., Parks, W. T., Frank, C. J., Castagnino, P., Bottaro, D. P., Roberts, A. B., and Lechleider, R. J. (1998). Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 12, 1587–1592.
Derynck, R., and Akhurst, R. J. (2007). Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 9, 1000–1004.
Derynck, R., and Miyazono, K. (2008). The TGF-β Signaling. New York: Cold Spring Harbor Laboratory Press.
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584.
Dooley, S., Delvoux, B., Lahme, B., Mangasser-Stephan, K., and Gressner, A. M. (2000). Modulation of transforming growth factor β response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31, 1094–1106.
Dooley, S., Hamzavi, J., Breitkopf, K., Wiercinska, E., Said, H. M., Lorenzen, J., Ten Dijke, P., and Gressner, A. M. (2003). Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125, 178–191.
Dooley, S., and Ten Dijke, P. (2012). TGF-β in progression of liver disease. Cell Tissue Res. 347, 245–256.
Engel, M. E., McDonnell, M. A., Law, B. K., and Moses, H. L. (1999). Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 274, 37413–37420.
Friedman, S. L. (2010). Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 7, 425–436.
Furukawa, F., Matsuzaki, K., Mori, S., Tahashi, Y., Yoshida, K., Sugano, Y., Yamagata, H., Matsushita, M., Seki, T., Inagaki, Y., Nishizawa, M., Fujisawa, J., and Inoue, K. (2003). p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 38, 879–889.
Guo, X., and Wang, X. F. (2009). Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 19, 71–88.
Hayashi, H., Abdollah, S., Qiu, Y., Cai, J., Xu, Y.-Y., Grinnell, B. W., Richardson, M. A., Topper, J. N., Gimbrone, M. A. Jr., Wrana, J. L., and Falb, D. (1997). The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 89, 1165–1173.
Hayashi, H., Sakai, K., Baba, H., and Sakai, T. (2011). Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy via TGF-β1 activation in mice. Hepatology doi:10.1002/hep.24800 [Epub ahead of print].
Hayashida, T., Decaestecker, M., and Schnaper, H. W. (2003). Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 17, 1576–1578.
Heldin, C. H., Miyazono, K., and ten Dijke, P. (1997). TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471.
Heldin, C. H., and Westermark, B. (1999). Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316.
Ho, J., Cocolakis, E., Dumas, V. M., Posner, B. I., Laporte, S. A., and Lebrun, J. J. (2005). The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFβ signal transduction. EMBO J. 24, 3247–3258.
Hu, P. F., Chen, H., Zhong, W., Lin, Y., Zhang, X., Chen, Y. X., and Xie, W. F. (2009). Adenovirus-mediated transfer of siRNA against PAI-1 mRNA ameliorates hepatic fibrosis in rats. J. Hepatol. 51, 102–113.
Inagaki, Y., and Okazaki, I. (2007). Emerging insights into transforming growth factor β Smad signal in hepatic fibrogenesis. Gut 56, 284–292.
Kamaraju, A. K., and Roberts, A. B. (2005). Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 280, 1024–1036.
Kawamata, S., Matsuzaki, K., Murata, M., Seki, T., Matsuoka, K., Iwao, Y., Hibi, T., and Okazaki, K. (2011). Oncogenic Smad3 signaling induced by chronic inflammation is an early event in ulcerative colitis-associated carcinogenesis. Inflamm. Bowel Dis. 17, 683–695.
Kisseleva, T., and Brenner, D. A. (2008). Mechanisms of fibrogenesis. Exp. Biol. Med. (Maywood) 233, 109–122.
Kretzschmar, M., Doody, J., Timokhina, I., and Massague, J. (1999). A mechanism of repression of TGFβ /Smad signaling by oncogenic Ras. Genes Dev. 13, 804–816.
Liu, F. (2006). Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 17, 9–17.
Matsuura, I., Chiang, K. N., Lai, C. Y., He, D., Wang, G., Ramkumar, R., Uchida, T., Ryo, A., Lu, K., and Liu, F. (2010). Pin1 promotes transforming growth factor-β-induced migration and invasion. J. Biol. Chem. 285, 1754–1764.
Matsuura, I., Denissova, N. G., Wang, G., He, D., Long, J., and Liu, F. (2004). Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 430, 226–231.
Matsuzaki, K. (2006). Smad3 phosphoisoform-mediated signaling during sporadic human colorectal carcinogenesis. Histol. Histopathol. 21, 645–662.
Matsuzaki, K. (2009). Modulation of TGF-β signaling during progression of chronic liver diseases. Front. Biosci. 14, 2923–2934.
Matsuzaki, K. (2011). Smad phosphoisoform signaling specificity: the right place at the right time. Carcinogenesis 32, 1578–1588.
Matsuzaki, K. (2012). Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 347, 225–243.
Matsuzaki, K., Kitano, C., Murata, M., Sekimoto, G., Yoshida, K., Uemura, Y., Seki, T., Taketani, S., Fujisawa, J., and Okazaki, K. (2009). Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-β signal in later stages of human colorectal cancer. Cancer Res. 69, 5321–5330.
Matsuzaki, K., Murata, M., Yoshida, K., Sekimoto, G., Uemura, Y., Sakaida, N., Kaibori, M., Kamiyama, Y., Nishizawa, M., Fujisawa, J., Okazaki, K., and Seki, T. (2007). Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 46, 48–57.
Mori, S., Matsuzaki, K., Yoshida, K., Furukawa, F., Tahashi, Y., Yamagata, H., Sekimoto, G., Seki, T., Matsui, H., Nishizawa, M., Fujisawa, J., and Okazaki, K. (2004). TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 23, 7416–7429.
Moriya, K., Bae, E., Honda, K., Sakai, K., Sakaguchi, T., Tsujimoto, I., Kamisoyama, H., Keene, D. R., Sasaki, T., and Sakai, T. (2011). A fibronectin-independent mechanism of collagen fibrillogenesis in adult liver remodeling. Gastroenterology 140, 1653–1663.
Murata, M., Matsuzaki, K., Yoshida, K., Sekimoto, G., Tahashi, Y., Mori, S., Uemura, Y., Sakaida, N., Fujisawa, J., Seki, T., Kobayashi, K., Yokote, K., Koike, K., and Okazaki, K. (2009). Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 49, 1203–1217.
Nagarajan, R. P., Zhang, J., Li, W., and Chen, Y. (1999). Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J. Biol. Chem. 274, 33412–33418.
Nagata, H., Hatano, E., Tada, M., Murata, M., Kitamura, K., Asechi, H., Narita, M., Yanagida, A., Tamaki, N., Yagi, S., Ikai, I., Matsuzaki, K., and Uemoto, S. (2009). Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 49, 1944–1953.
Nakao, A., Afrakhte, M., Moren, A., Nakayama, T., Christian, J. L., Heuchel, R., Itoh, S., Kawabata, M., Heldin, N. E., Heldin, C. H., and ten Dijke, P. (1997). Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635.
Pinzani, M., and Macias-Barragan, J. (2010). Update on the pathophysiology of liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 4, 459–472.
Robinson, M. J., and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9, 180–186.
Rockey, D. C., Housset, C. N., and Friedman, S. L. (1993). Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J. Clin. Invest. 92, 1795–1804.
Sapkota, G., Knockaert, M., Alarcon, C., Montalvo, E., Brivanlou, A. H., and Massague, J. (2006). Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 281, 40412–40419.
Sekimoto, G., Matsuzaki, K., Yoshida, K., Mori, S., Murata, M., Seki, T., Matsui, H., Fujisawa, J., and Okazaki, K. (2007). Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 67, 5090–5096.
Shi, Y., and Massague, J. (2003). Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700.
Stopa, M., Anhuf, D., Terstegen, L., Gatsios, P., Gressner, A. M., and Dooley, S. (2000). Participation of Smad2, Smad3, and Smad4 in transforming growth factor β (TGF-β)-induced activation of Smad7. THE TGF-beta response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J. Biol. Chem. 275, 29308–29317.
Tahashi, Y., Matsuzaki, K., Date, M., Yoshida, K., Furukawa, F., Sugano, Y., Matsushita, M., Himeno, Y., Inagaki, Y., and Inoue, K. (2002). Differential regulation of TGF-β signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 35, 49–61.
von Gersdorff, G., Susztak, K., Rezvani, F., Bitzer, M., Liang, D., and Böttinger, E. P. (2000). Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor β. J. Biol. Chem. 275, 11320–11326.
Wang, G., Matsuura, I., He, D., and Liu, F. (2009). Transforming growth factor-β-inducible phosphorylation of Smad3. J. Biol. Chem. 284, 9663–9673.
Weng, H. L., Liu, Y., Chen, J. L., Huang, T., Xu, L. J., Godoy, P., Hu, J. H., Zhou, C., Stickel, F., Marx, A., Bohle, R. M., Zimmer, V., Lammert, F., Mueller, S., Gigou, M., Samuel, D., Mertens, P. R., Singer, M. V., Seitz, H. K., and Dooley, S. (2009). The etiology of liver damage imparts cytokines transforming growth factor β1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 50, 230–243.
Wrighton, K. H., Lin, X., and Feng, X. H. (2009). Phospho-control of TGF-β superfamily signaling. Cell Res. 19, 8–20.
Yamagata, H., Matsuzaki, K., Mori, S., Yoshida, K., Tahashi, Y., Furukawa, F., Sekimoto, G., Watanabe, T., Uemura, Y., Sakaida, N., Yoshioka, K., Kamiyama, Y., Seki, T., and Okazaki, K. (2005). Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res. 65, 157–165.
Yoshida, K., Matsuzaki, K., Mori, S., Tahashi, Y., Yamagata, H., Furukawa, F., Seki, T., Nishizawa, M., Fujisawa, J., and Okazaki, K. (2005). Transforming growth factor-β and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 166, 1029–1039.
Keywords: HSC, MFB, TGF-β, Smad, liver fibrosis
Citation: Yoshida K and Matsuzaki K (2012) Differential regulation of TGF-β/Smad signaling in hepatic stellate cells between acute and chronic liver injuries. Front. Physio. 3:53. doi: 10.3389/fphys.2012.00053
Received: 13 January 2012; Paper pending published: 22 January 2012;
Accepted: 26 February 2012; Published online: 19 March 2012.
Edited by:
Honglei Weng, University of Heidelberg, GermanyCopyright: © 2012 Yoshida and Matsuzaki. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Katsunori Yoshida, Department of Gastroenterology and Hepatology, Kansai Medical University, 10-15 Fumizonocho, Moriguchi, Osaka 570-8506, Japan. e-mail: yoshidka@takii.kmu.ac.jp