 Min Li
Min Li Xiaoying Li1
Xiaoying Li1 Huijie Zhang
Huijie Zhang Yan Lu
Yan Lu- 1Department of Endocrinology and Metabolism, Zhongshan Hospital, Fudan University, Shanghai, China
- 2Department of Endocrinology and Metabolism, Nanfang Hospital, Southern Medical University, Guangzhou, China
Metformin has been the first-line drug treatment for hyperglycemia and insulin resistance for over 50 years. However, the molecular basis of its therapeutic role remained incompletely understood. Recent advances demonstrate that metformin could exert its glucose-lowering effect by multiple mechanisms, including activation of 5′-AMP-activated protein kinase, decreasing production of cyclic AMP, suppressing mitochondrial complex I of the electron transport chain, targeting glycerophosphate dehydrogenase, and altering the gut microbiome. In addition, epidemiological and clinical observation studies suggest that metformin reduced cancer risk in patients with type 2 diabetes and improved survival outcome of human cancers. Experimental studies have shown that this drug can inhibit cancer cell viability, growth, and proliferation through inhibiting mTORC1 signaling and mitochondrial complex I, suggesting that it may be a promising drug candidate for malignancy. Here, we summarize recent progress in studies of metformin in type 2 diabetes and tumorigenesis, which provides novel insight on the therapeutic development of human diseases.
Introduction
Metformin, a derivative of guanidine, has been used in the treatment of hyperglycemia and type 2 diabetes mellitus (T2DM) for over 50 years (American Diabetes Association, 2014). Metformin, phenformin, and buformin are derivatives of guanidine, which was extracted from the plant isoamylene in the 1920s (Bailey and Day, 1989). Phenformin and buformin have been withdrawn in the early 1970s because of their higher risk of cardiac mortality and lactic acidosis (Luft et al., 1978), whereas the use of metformin has been expanded from T2DM to polycystic ovary disease, diabetic nephropathy, gestational diabetes, T2DM-associated cardiovascular complications (Viollet et al., 2012), due to its superior safety profile.
Metformin specifically suppresses hepatic gluconeogenesis without increasing the burden of pancreatic β cells to enhance insulin secretion or promoting adipocyte differentiation to induce weight gain (Inzucchi et al., 1998). However, the exact molecular mechanisms of its glucose-lowering effects remain unclear. Besides, metformin has gained attention for its pleiotropic effects. Importantly, accumulation of numerous epidemiological studies indicates the preventive and therapeutic effects of metformin on many types of human cancers (Morales and Morris, 2015). Herein, we summarized the action and molecular mechanisms through which metformin inhibits hepatic gluconeogenesis and tumorigenesis, which may help to suggest directions for future investigation.
Molecular Mechanisms of Anti-Diabetic Effects
In the past decade, several mechanisms have been identified for the action of metformin in hepatic gluconeogenesis and glucose production (Figure 1). An important breakthrough was that metformin could activate adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) (Zhou et al., 2001), a master regulator of various metabolic pathways (Hardie et al., 2012; Lin and Hardie, 2018), by increasing its phosphorylation at Thr-172. Through screening of a compound library containing more than 10,000 molecules, compound C was discovered as an AMPK inhibitor and attenuated metformin’s effect in hepatocytes (Zhou et al., 2001), indicating that activation of AMPK is essential for its inhibitory effects on glucose production in hepatocytes. A subsequent study by Shaw et al. (2005) reported that deletion of liver kinase B1 (LKB1), the upstream kinase that phosphorylates and activates AMPK, led to a nearly complete loss of AMPK activity in the liver of adult mice. Loss of LKB1 blocked the therapeutic effects of metformin, suggesting that metformin treatment of mice increased AMPK activity in the liver and lowered blood glucose levels in an LKB1-dependent manner (Shaw et al., 2005). Besides, some studies demonstrated that metformin treatment increases cellular levels of AMP through suppressing complex I of the mitochondrial electron transport chain. This inhibition resulted in a reduced cellular ATP concentrations and an elevated AMP levels (Batandier et al., 2006). Moreover, low concentrations of metformin was shown to promote the formation of the AMPK αβγ complex through augmenting phosphorylation by LKB1 and antagonizing dephosphorylation by PP2C, leading to the phosphorylation of the AMPK α catalytic subunit at Thr-172 (Meng et al., 2015). Moreover, one recent study revealed that metformin activates AMPK through the lysosomal pathway, consisting of AXIN/LKB1-v-ATPase-Ragulator pathway (Zhang et al., 2016). Therefore, the mechanisms for metformin to activate AMPK remain obscure and controversial. On the other hand, the molecular mechanism underlying the AMPK-mediated inhibition of gluconeogenesis remained elusive. A study from Choi et al. showed that metformin suppresses hepatic glucose production and expression of gluconeogenic genes (PEPCK and G6Pase) through AMPK-dependent upregulation of small heterodimer partner (SHP), a transcriptional co-repressor (Kim et al., 2008). SHP could interact with and repress the transcriptional activity of hepatocyte nuclear factor 4α (HNF4α), forkhead box protein O1 (FoxO1), and forkhead box protein A2 (FoxA2), which are critical in the transcriptional regulation of gluconeogenic genes (Rines et al., 2016). Another study demonstrated that metformin inhibits hepatic gluconeogenesis through phosphorylation of CREB binding protein (CBP) at serine 436 via AMPK-PKCι/λ, leading to the dissociation of the CREB-CBP-CRTC2 transcription complex and down-regulation of gluconeogenic genes (He et al., 2009). In addition, AMPK could phosphorylate acetyl-CoA carboxylase 1 (ACC1) and ACC2 to inhibit the conversion of acetyl-CoA to malonyl-CoA. As a result, AMPK activation by metformin results in reduced liver lipogenesis and hepatosteatosis, contributing to improved insulin resistance and hyperglycemia (Fullerton et al., 2013; Ford et al., 2015; Boudaba et al., 2018). Together, all these studies highlight the importance of AMPK signaling in the anti-diabetic action of metformin (Rena et al., 2017).
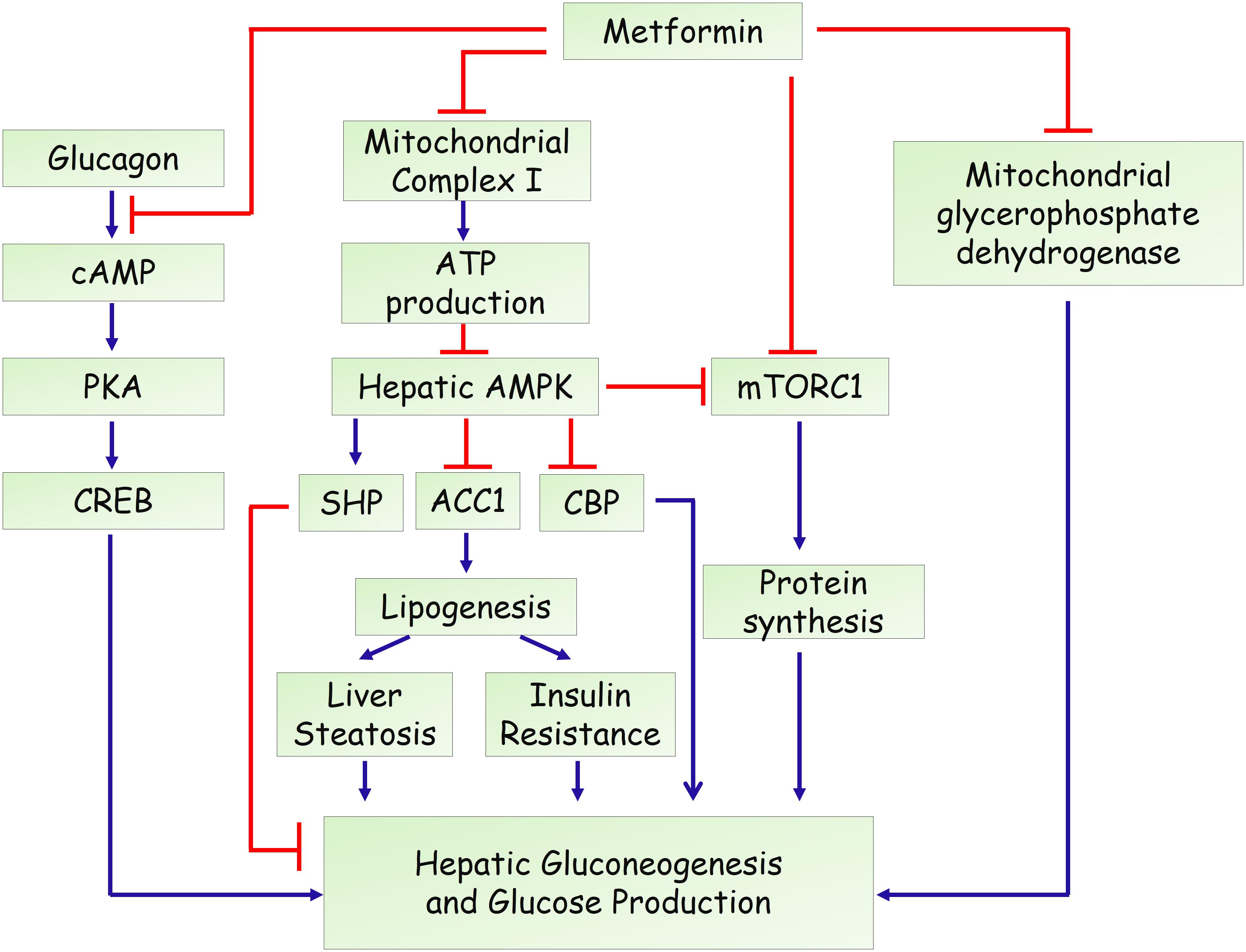
FIGURE 1. Proposed actions of metformin in the hepatic gluconeogenesis.
The controversy arised in 2010 when Foretz et al. (2010) showed that metformin could inhibit hepatic gluconeogenesis in mice lack of either AMPKα1α2 catalytic isoforms or LKB1. They found that the hypoglycemic effect of metformin was unaltered in liver AMPK deficient mice, compared with wild-type mice (Foretz et al., 2010). Consistently, reduced expression of gluconeogenic genes by metformin was also comparable in the wild-type, AMPKα1α2 deficient, and LKB1 deficient hepatocytes, further confirming that neither AMPK nor LKB1 are required for metformin-mediated suppression of hepatic gluconeogenesis (Foretz et al., 2010). However, a recent study questioned the high or supra-pharmacological concentrations of metformin in Foretz’s study and found that low concentrations of metformin (60–80 μM in the portal vein of animals) suppress glucose production via an AMPK dependent mechanism (Cao et al., 2014). In agreement, Howell et al. (2017) showed that low doses of metformin inhibit the mammalian target of rapamycin complex I (mTORC1) through AMPK and higher doses act through alternative mechanisms. Therefore, it is speculated that the mechanisms of metformin might be associated with its concentrations (doses) (He and Wondisford, 2015).
On the other hand, Cokorinos et al. (2017) demonstrated that direct pharmacological activation of AMPK by small-molecule AMPK activators in liver is not sufficient for acute glucose lowering in obese mice. Thus, an important question raised by these work is that how metformin could lower blood glucose or hepatic glucose production in the absence of AMPK. Foretz et al. (2010) proposed that the action of metformin is attribute to decreased cellular ATP concentrations and increased AMP/ATP ratio. Besides, Miller et al. (2013) reported that metformin could antagonize the role of glucagon, to reduce blood glucose levels. They found that treatment of metformin could increase levels of AMP and related nucleotides to suppress adenylate cyclase and protein kinase A activity, abolish CREB phosphorylation, and block glucagon-stimulated glucose output in hepatocytes (Miller et al., 2013). Madiraju et al. (2014) demonstrated that metformin could suppress the mitochondrial glycerophosphate dehydrogenase (mGPD), leading to reduced conversion of lactate and glycerol to glucose. Furthermore, recent studies implicates that the gastrointestinal tract may be involved in the antidiabetic effect of metformin (Bailey et al., 2008). It has been shown that preabsorptive metformin could activate duodenal mucosal AMPK to inhibit hepatic gluconeogenesis and improve hyperglycemia in high-fat-diet-induced obese rodents (Duca et al., 2015; Jensen et al., 2016). Duodenal infection of an adenovirus containing the dominant negative AMPK largely attenuated the glucose lowering effect of intraduodenal metformin (Duca et al., 2015). Moreover, metformin upregulates the expression levels of sodium glucose cotransporter-1 (SGLT1) in upper small intestine, partly by increasing the abundance of Lactobacillus (Bauer et al., 2018). In addition, two studies using T2DM patients further implicates the gut microbiota as an important site of metformin action (Forslund et al., 2015; Wu et al., 2017). Importantly, a randomized, placebo-controlled, double-blind study in newly diagnosed T2DM subjects showed that metformin had rapid effects on the composition and function of the gut microbiota (Wu et al., 2017). Animal experiments further confirmed that transfer of the metformin-altered microbiota to germ-free mice could improve glucose metabolism (Wu et al., 2017), suggesting that altered gut microbiota contributes to the therapeutic effects of the drug.
Molecular Mechanisms of Antineoplastic Effects
Dilman et al. (1978) found that daily oral administration of phenformin suppressed dimethylbenzathracene-induced mammary tumor development in rats. They further reported that breast cancer patients taking phenformin had an improvement in metabolic parameters and immunologic status (Dilman et al., 1982). In recent years, lots of epidemiological studies looked into the preventive and therapeutic actions of metformin on many types of human cancers. The first report was a case–control study showing a decreased risk of developing cancer in T2DM patients taking metformin (Evans et al., 2005), which was further confirmed by subsequent meta-analysis using 18 observational studies in liver, colon, and pancreatic cancers (Franciosi et al., 2013). In addition to its preventive action, the beneficial effect of metformin on improvement of overall survival outcomes or reduction in mortality was also observed in liver, pancreatic, colorectal, and breast cancer (Zhang et al., 2013; Morales and Morris, 2015), suggesting that it can also serve as a potential anti-tumor agent (Jiralerspong et al., 2009). For instance, a study involving 1,013 breast cancer patients showed that the HER-2 positive rate was lower in the metformin-treated group than in the nonmetformin-treated group (Hou et al., 2013). Besides, metformin-treated group was associated with better clinical outcomes and lower mortality risk (Hou et al., 2013).
Although the use of metformin is still limited to T2DM, insulin resistance and hyperglycemia, its effect in non-diabetic cancer patients was also observed. It was reported that metformin inhibited colonic epithelial proliferation and reduced rectal aberrant crypt foci in non-diabetic patients with colorectal cancer (Hosono et al., 2010). Besides, Hadad et al. (2011, 2015) performed a pre-operative trial, which provides support for anti-proliferative effects of metformin in non-diabetic breast cancer humans. In addition, recent in vitro and in vivo studies indicate that metformin can enhance the effects of other anti-cancer drug, such as cisplatin, vincristine, 5-fluorouracil, and doxorubicin (Iliopoulos et al., 2011; Miranda et al., 2014; Yi et al., 2017; Candido et al., 2018), suggesting metformin can act as part of combinatorial therapy to decrease the chemotherapy dose in cancer patients.
Hyperinsulinemia represents a risk factor for several types of human malignancy and induces adverse prognosis (Pollak, 2012; Garg et al., 2014). Therefore, systemic effects related to reduced blood glucose levels, improved insulin resistance and decreased pro-inflammatory cytokines, are involved in the complexity of the roles of metformin on tumorigenesis (Pernicova and Korbonits, 2014). Besides, a direct action of metformin in cancer cells needs more attention. Likewise, the anti-diabetic actions, initial studies showed that LKB1-dependent and AMPK-dependent growth inhibitor was responsible for the antineoplastic effect of metformin (Figure 2) (Zakikhani et al., 2006; Dowling et al., 2007). Knockdown of AMPK α1 subunit by small interfering RNA rescued breast and ovarian cancer cells from the inhibitory effect of metformin (Zakikhani et al., 2006). AMPK activation induces phosphorylation of p53 on Ser15, and this phosphorylation is required to initiate AMPK-dependent cell-cycle arrest (Jones et al., 2005). Activation of AMPK by metformin also promotes phosphorylation of human MDMX on Ser342, which inhibits p53 ubiquitylation and stabilizes p53 (He et al., 2014). However, a subsequent study found that the antiproliferative role of metformin is not mediated by AMPK in prostate cancer cells and proposed that inhibition of mTOR represents an alternative pathway for metformin action (Ben Sahra et al., 2011). mTOR is a catalytic subunit of two multiprotein complexes, mTORC1 and mTORC2, which integrate both intracellular and extracellular stimuli and act as a key regulator of cell growth (Laplante and Sabatini, 2012; Saxton and Sabatini, 2017). mTOR inhibition could disturb protein synthesis, and thereby suppress tumor cell proliferation. Metformin was shown to suppress the activation of mTOR through AMPK-dependent and -independent mechanisms. AMPK-dependent suppression of mTORC1 activity is attributed to activation of tuberous sclerosis complex 1 (TSC1) and TSC2, which form an mTOR-inhibiting complex (Inoki et al., 2003). Moreover, AMPK could directly phosphorylate Raptor, the mTOR binding partner protein, which is required for the inhibition of mTORC1 induced by energy stress (Gwinn et al., 2008). In addition, Kalender et al. (2010) reported that metformin can inhibit mTORC1 signaling through Ras-related GTPase, independent of AMPK and TSC1/2. In addition to AMPK and mTOR, metformin has been shown to affect other oncogenic signaling pathways. Li and colleagues reported that metformin suppresses the proliferation and growth of osteosarcoma and renal cell carcinoma cells by suppressing Akt phosphorylation, which was associated with increased phosphatase and tensin (PTEN) expression (Kalogirou et al., 2016; Li et al., 2018). Besides, metformin could activate the MEK/ERK signaling pathway to promote leukemia cell differentiation and apoptosis (Huai et al., 2012). Moreover, metformin inhibits activation of NF-κB and Stat3 signalings in cancer stem cells, resulting in a reduced inflammatory response and attenuated tumor growth (Hirsch et al., 2013).
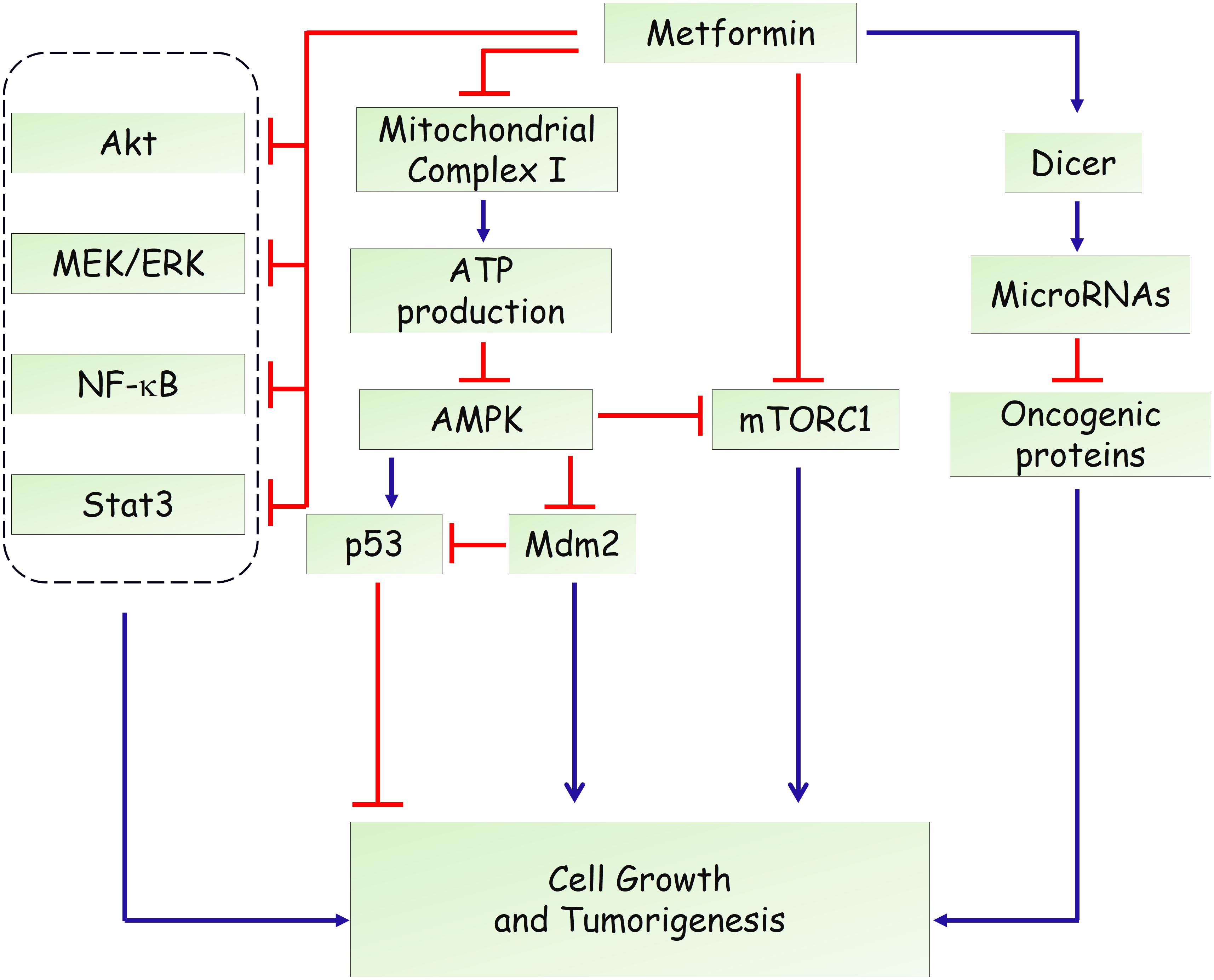
FIGURE 2. Potential mechanisms of metformin in anti-tumorigenesis.
Furthermore, modulation of microRNA expression has been proposed to underlie the anticancer actions of metformin. It has been reported that metformin treatment could induce the expression of DICER, an enzyme that is crucial in regulating microRNA biogenesis (Blandino et al., 2012). Downregulation of DICER has been shown to represent an intrinsically oncogenic event and predicts poor survival of several types of cancers (Karube et al., 2005; Merritt et al., 2008; Martello et al., 2010). Forced overexpression of DICER recapitulated the antineoplastic role of metformin in vitro and in vivo. Besides, the effects of metformin are substantially impaired in DICER-deficient tumor cells (Blandino et al., 2012), suggesting that upregulation of DICER is required for its actions. As a result, metformin treatment affected expression levels of many microRNAs such as miR-21, miR-26a, miR-33a, miR-140-5p, miR-142-3p, miR-181a, miR-192, miR-193b, R-20mi0, miR-205, miR-222, let-7a, and let-7c, which further modulates several target genes in metabolic or preoncogenic pathways (Pulito et al., 2014; Zhou et al., 2015).
Moreover, a recent study showed that the environment drastically altered sensitivity to metformin (Gui et al., 2016). They demonstrated that complex I supports proliferation by regenerating nicotinamide adenine dinucleotide (NAD) and metformin’s anti-proliferative effect is due to loss of NAD/NADH homeostasis and inhibition of aspartate biosynthesis (Gui et al., 2016). In agreement, through an integrative metabolomics analysis of metformin action in ovarian cancer, Liu et al. (2016) showed that metformin could target central carbon metabolism, suggesting mitochondrial requirements for the effects of metformin on cancer cells. In addition, through genetic screening in C. elegans, Wu et al. (2016) identified two metformin response elements: the nuclear pore complex (NPC) and acylCoA dehydrogenase family member-10 (ACAD10). They demonstrated that metformin inhibited cell growth by inhibiting mitochondrial respiratory capacity, which restrains transit of the RagA-RagC GTPase heterodimer through the NPC (Wu et al., 2016). Together, these findings not only provide precise indications of metformin in cancer but also uncover new insights into mitochondrial metabolism.
Overall, the anti-proliferative effects of metformin share common mechanisms with its anti-diabetic action, including activation of AMPK signaling, inhibition of mTOR signaling, targeting mitochondria complex I (Figures 1, 2). Although the detailed reason remains poorly understood, we speculate that a unified mechanism might exist in metformin-treated normal cells and cancer cells, such as the alteration of cellular energy state. In addition, several studies also demonstrated that use of metformin is not associated with reduced incidence or improved outcome in certain types of human cancers (Mamtani et al., 2014; Suissa and Azoulay, 2014; Kordes et al., 2015). Therefore, the therapeutic effect of metformin might be cell- or tissue-specific, which needs to be determined in future studies.
Conclusion
Metformin has been widely used in the treatment of T2DM and related metabolic diseases. However, as reviewed here, both AMPK-dependent and AMPK-independent pathways have been proposed to explain the glucose-lowering and anti-tumor effect of metformin (Figures 1, 2). Besides, although the liver is considered as the primary site of metformin pharmacodynamics, the gut is also recognized an important site for its anti-diabetic and anti-tumor effects (Duca et al., 2015; Paleari et al., 2018). In addition, recent studies demonstrated that metformin might affect metabolite profiles in patients with type 2 diabetes or tumor cells (Zhang et al., 2014; He et al., 2015; Xu et al., 2015). All these knowledge, we hope, will help to fully understand the mechanistic action of metformin, which may propel the development of novel potential therapeutic targets in treating human diseases.
Author Contributions
ML, XL, HZ, and YL drafted the manuscript. YL and HZ handled funding and supervision. All authors reviewed the manuscript.
Funding
This study was supported by grants from Natural Science Foundation of China (Nos. 81570769 and 81570785), Shanghai Rising-Star Program by Science and Technology Commission of Shanghai Municipality (17QA1400800), and Chenguang Program supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (15CG11).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
American Diabetes Association (2014). Standards of medical care in diabetes-2014. Diabetes Care 37(Suppl. 1), S14–S80. doi: 10.2337/dc14-S014
Bailey, C. J., and Day, C. (1989). Traditional plant medicines as treatments for diabetes. Diabetes Care 12, 553–564. doi: 10.2337/diacare.12.8.553
Bailey, C. J., Wilcock, C., and Scarpello, J. H. (2008). Metformin and the intestine. Diabetologia 51, 1552–1553. doi: 10.1007/s00125-008-1053-5
Batandier, C., Guigas, B., Detaille, D., El-Mir, M. Y., Fontaine, E., Rigoulet, M., et al. (2006). The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 38, 33–42. doi: 10.1007/s10863-006-9003-8
Bauer, P. V., Duca, F. A., Waise, T. M. Z., Rasmussen, B. A., Abraham, M. A., Dranse, H. J., et al. (2018). Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab. 27, 101–117. doi: 10.1016/j.cmet.2017.09.019
Ben Sahra, I., Regazzetti, C., Robert, G., Laurent, K., Le Marchand-Brustel, Y., Auberger, P., et al. (2011). Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 71, 4366–4372. doi: 10.1158/0008-5472.CAN-10-1769
Blandino, G., Valerio, M., Cioce, M., Mori, F., Casadei, L., Pulito, C., et al. (2012). Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 3:865. doi: 10.1038/ncomms1859
Boudaba, N., Marion, A., Huet, C., Pierre, R., Viollet, B., and Foretz, M. (2018). AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209. doi: 10.1016/j.ebiom.2018.01.008
Candido, S., Abrams, S. L., Steelman, L., Lertpiriyapong, K., Martelli, A. M., Cocco, L., et al. (2018). Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 68, 13–30. doi: 10.1016/j.jbior.2018.02.002
Cao, J., Meng, S., Chang, E., Beckwith-Fickas, K., Xiong, L., Cole, R. N., et al. (2014). Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 289, 20435–20446. doi: 10.1074/jbc.M114.567271
Cokorinos, E. C., Delmore, J., Reyes, A. R., Albuquerque, B., Kjøbsted, R., Jørgensen, N. O., et al. (2017). Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 25, 1147–1159. doi: 10.1016/j.cmet.2017.04.010
Dilman, V. M., Berstein, L. M., Ostroumova, M. N., Fedorov, S. N., Poroshina, T. E., Tsyrlina, E. V., et al. (1982). Metabolic immunodepression and metabolic immunotherapy: an attempt of improvement in immunologic response in breast cancer patients by correction of metabolic disturbances. Oncology 39, 13–19. doi: 10.1159/000225596
Dilman, V. M., Berstein, L. M., Zabezhinski, M. A., Alexandrov, V. A., Bobrov, J. F., and Pliss, G. B. (1978). Inhibition of DMBA-induced carcinogenesis by phenformin in the mammary gland of rats. Arch. Geschwulstforsch. 48, 1–8.
Dowling, R. J., Zakikhani, M., Fantus, I. G., Pollak, M., and Sonenberg, N. (2007). Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67, 10804–10812. doi: 10.1158/0008-5472.CAN-07-2310
Duca, F. A., Côté, C. D., Rasmussen, B. A., Zadeh-Tahmasebi, M., Rutter, G. A., Filippi, B. M., et al. (2015). Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 21, 506–511. doi: 10.1038/nm.3787
Evans, J. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R., and Morris, A. D. (2005). Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305. doi: 10.1136/bmj.38415.708634.F7
Ford, R. J., Fullerton, M. D., Pinkosky, S. L., Day, E. A., Scott, J. W., Oakhill, J. S., et al. (2015). Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 468, 125–132. doi: 10.1042/BJ20150125
Foretz, M., Hébrard, S., Leclerc, J., Zarrinpashneh, E., Soty, M., Mithieux, G., et al. (2010). Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369. doi: 10.1172/JCI40671
Forslund, K., Hildebrand, F., Nielsen, T., Falony, G., Le Chatelier, E., Sunagawa, S., et al. (2015). Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266. doi: 10.1038/nature15766
Franciosi, M., Lucisano, G., Lapice, E., Strippoli, G. F., Pellegrini, F., and Nicolucci, A. (2013). Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One 8:e71583. doi: 10.1371/journal.pone.0071583
Fullerton, M. D., Galic, S., Marcinko, K., Sikkema, S., Pulinilkunnil, T., Chen, Z. P., et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 1649–1654. doi: 10.1038/nm.3372
Garg, S. K., Maurer, H., Reed, K., and Selagamsetty, R. (2014). Diabetes and cancer: two diseases with obesity as a common risk factor. Diabetes Obes. Metab. 16, 97–110. doi: 10.1111/dom.12124
Gui, D. Y., Sullivan, L. B., Luengo, A., Hosios, A. M., Bush, L. N., Gitego, N., et al. (2016). Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 24, 716–727. doi: 10.1016/j.cmet.2016.09.006
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hadad, S., Iwamoto, T., Jordan, L., Purdie, C., Bray, S., Baker, L., et al. (2011). Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 128, 783–794. doi: 10.1007/s10549-011-1612-1
Hadad, S. M., Coates, P., Jordan, L. B., Dowling, R. J., Chang, M. C., Done, S. J., et al. (2015). Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res. Treat. 150, 149–155. doi: 10.1007/s10549-015-3307-5
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
He, G., Zhang, Y. W., Lee, J. H., Zeng, S. X., Wang, Y. V., Luo, Z., et al. (2014). AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell. Biol. 34, 148–157. doi: 10.1128/MCB.00670-13
He, J., Wang, K., Zheng, N., Qiu, Y., Xie, G., Su, M., et al. (2015). Metformin suppressed the proliferation of LoVo cells and induced a time-dependent metabolic and transcriptional alteration. Sci. Rep. 5:17423. doi: 10.1038/srep17423
He, L., Sabet, A., Djedjos, S., Miller, R., Sun, X., Hussain, M. A., et al. (2009). Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137, 635–646. doi: 10.1016/j.cell.2009.03.016
He, L., and Wondisford, F. E. (2015). Metformin action: concentrations matter. Cell Metab. 21, 159–162. doi: 10.1016/j.cmet.2015.01.003
Hirsch, H. A., Iliopoulos, D., and Struhl, K. (2013). Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. U.S.A. 110, 972–977. doi: 10.1073/pnas.1221055110
Hosono, K., Endo, H., Takahashi, H., Sugiyama, M., Sakai, E., Uchiyama, T., et al. (2010). Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev. Res. 3, 1077–1083. doi: 10.1158/1940-6207.CAPR-10-0186
Hou, G., Zhang, S., Zhang, X., Wang, P., Hao, X., and Zhang, J. (2013). Clinical pathological characteristics and prognostic analysis of 1,013 breast cancer patients with diabetes. Breast Cancer Res. Treat. 137, 807–816. doi: 10.1007/s10549-012-2404-y
Howell, J. J., Hellberg, K., Turner, M., Talbott, G., Kolar, M. J., Ross, D. S., et al. (2017). Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 25, 463–471. doi: 10.1016/j.cmet.2016.12.009
Huai, L., Wang, C., Zhang, C., Li, Q., Chen, Y., Jia, Y., et al. (2012). Metformin induces differentiation in acute promyelocytic leukemia by activating the MEK/ERK signaling pathway. Biochem. Biophys. Res. Commun. 422, 398–404. doi: 10.1016/j.bbrc.2012.05.001
Iliopoulos, D., Hirsch, H. A., and Struhl, K. (2011). Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 71, 3196–3201. doi: 10.1158/0008-5472.CAN-10-3471
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/S0092-8674(03)00929-2
Inzucchi, S. E., Maggs, D. G., Spollett, G. R., Page, S. L., Rife, F. S., Walton, V., et al. (1998). Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N. Engl. J. Med. 338, 867–872. doi: 10.1056/NEJM199803263381303
Jensen, J. B., Sundelin, E. I., Jakobsen, S., Gormsen, L. C., Munk, O. L., Frøkiær, J., et al. (2016). [11C]-Labeled metformin distribution in the liver and small intestine using dynamic positron emission tomography in mice demonstrates tissue-specific transporter dependency. Diabetes Metab. Res. Rev. 65, 1724–1730. doi: 10.2337/db16-0032
Jiralerspong, S., Palla, S. L., Giordano, S. H., Meric-Bernstam, F., Liedtke, C., Barnett, C. M., et al. (2009). Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 27, 3297–3302. doi: 10.1200/JCO.2009.19.6410
Jones, R. G., Plas, D. R., Kubek, S., Buzzai, M., Mu, J., and Xu, Y. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293. doi: 10.1016/j.molcel.2005.03.027
Kalender, A., Selvaraj, A., Kim, S. Y., Gulati, P., Brûlé, S., Viollet, B., et al. (2010). Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 11, 390–401. doi: 10.1016/j.cmet.2010.03.014
Kalogirou, C., Schäfer, D., Krebs, M., Kurz, F., Schneider, A., Riedmiller, H., et al. (2016). Metformin-derived growth inhibition in renal cell carcinoma depends on miR-21-mediated PTEN expression. Urol. Int. 96, 106–115. doi: 10.1159/000441011
Karube, Y., Tanaka, H., Osada, H., Tomida, S., Tatematsu, Y., Yanagisawa, K., et al. (2005). Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 96, 111–115. doi: 10.1111/j.1349-7006.2005.00015.x
Kim, Y. D., Park, K. G., Lee, Y. S., Park, Y. Y., Kim, D. K., Nedumaran, B., et al. (2008). Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes Metab. Res. Rev. 57, 306–314. doi: 10.2337/db07-0381
Kordes, S., Pollak, M. N., Zwinderman, A. H., Mathôt, R. A., Weterman, M. J., Beeker, A., et al. (2015). Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 16, 839–847. doi: 10.1016/S1470-2045(15)00027-3
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Li, Z., Wang, L., Luo, N., Zhao, Y., Li, J., Chen, Q., et al. (2018). Metformin inhibits the proliferation and metastasis of osteosarcoma cells by suppressing the phosphorylation of Akt. Oncol. Lett. 15, 7948–7954. doi: 10.3892/ol.2018.8297
Lin, S. C., and Hardie, D. G. (2018). AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313. doi: 10.1016/j.cmet.2017.10.009
Liu, X., Romero, I. L., Litchfield, L. M., Lengyel, E., and Locasale, J. W. (2016). Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 24, 728–739. doi: 10.1016/j.cmet.2016.09.005
Luft, D., Schmülling, R. M., and Eggstein, M. (1978). Lactic acidosis in biguanide-treated diabetics: a review of 330 cases. Diabetologia 14, 75–87. doi: 10.1007/BF01263444
Madiraju, A. K., Erion, D. M., Rahimi, Y., Zhang, X. M., Braddock, D. T., Albright, R. A., et al. (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546. doi: 10.1038/nature13270
Mamtani, R., Pfanzelter, N., Haynes, K., Finkelman, B. S., Wang, X., Keefe, S. M., et al. (2014). Incidence of bladder cancer in patients with type 2 diabetes treated with metformin or sulfonylureas. Diabetes Care 37, 1910–1917. doi: 10.2337/dc13-1489
Martello, G., Rosato, A., Ferrari, F., Manfrin, A., Cordenonsi, M., Dupont, S., et al. (2010). A MicroRNA targeting dicer for metastasis control. Cell 141, 1195–1207. doi: 10.1016/j.cell.2010.05.017
Meng, S., Cao, J., He, Q., Xiong, L., Chang, E., Radovick, S., et al. (2015). Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J. Biol. Chem. 290, 3793–3802. doi: 10.1074/jbc.M114.604421
Merritt, W. M., Lin, Y. G., Han, L. Y., Kamat, A. A., Spannuth, W. A., Schmandt, R., et al. (2008). Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 359, 2641–2650. doi: 10.1056/NEJMoa0803785
Miller, R. A., Chu, Q., Xie, J., Foretz, M., Viollet, B., and Birnbaum, M. J. (2013). Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 256–260. doi: 10.1038/nature11808
Miranda, V. C., Barroso-Sousa, R., Glasberg, J., and Riechelmann, R. P. (2014). Exploring the role of metformin in anticancer treatments: a systematic review. Drugs Today 50, 623–640. doi: 10.1358/dot.2014.50.9.2229920
Morales, D. R., and Morris, A. D. (2015). Metformin in cancer treatment and prevention. Annu. Rev. Med. 66, 17–29. doi: 10.1146/annurev-med-062613-093128
Paleari, L., Burhenne, J., Weiss, J., Foersch, S., Roth, W., Parodi, A., et al. (2018). High accumulation of metformin in colonic tissue of subjects with diabetes or the metabolic syndrome. Gastroenterology 154, 1543–1545. doi: 10.1053/j.gastro.2017.12.040
Pernicova, I., and Korbonits, M. (2014). Metformin–mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 10, 143–156. doi: 10.1038/nrendo.2013.256
Pollak, M. N. (2012). Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2, 778–790. doi: 10.1158/2159-8290.CD-12-0263
Pulito, C., Donzelli, S., Muti, P., Puzzo, L., Strano, S., and Blandino, G. (2014). microRNAs and cancer metabolism reprogramming: the paradigm of metformin. Ann. Transl. Med. 2:58. doi: 10.3978/j.issn.2305-5839.2014.06.03
Rena, G., Hardie, D. G., and Pearson, E. R. (2017). The mechanisms of action of metformin. Diabetologia 60, 1577–1585. doi: 10.1007/s00125-017-4342-z
Rines, A. K., Sharabi, K., Tavares, C. D., and Puigserver, P. (2016). Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 15, 786–804. doi: 10.1038/nrd.2016.151
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Shaw, R. J., Lamia, K. A., Vasquez, D., Koo, S. H., Bardeesy, N., and Depinho, R. A. (2005). The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646. doi: 10.1126/science.1120781
Suissa, S., and Azoulay, L. (2014). Metformin and cancer: mounting evidence against an association. Diabetes Care 37, 1786–1788. doi: 10.2337/dc14-0500
Viollet, B., Guigas, B., Sanz Garcia, N., Leclerc, J., Foretz, M., and Andreelli, F. (2012). Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. 122, 253–270. doi: 10.1042/CS20110386
Wu, H., Esteve, E., Tremaroli, V., Khan, M. T., Caesar, R., Mannerås-Holm, L., et al. (2017). Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858. doi: 10.1038/nm.4345
Wu, L., Zhou, B., Oshiro-Rapley, N., Li, M., Paulo, J. A., Webster, C. M., et al. (2016). An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell 167, 1705.e13–1718.e13. doi: 10.1016/j.cell.2016.11.055
Xu, T., Brandmaier, S., Messias, A. C., Herder, C., Draisma, H. H., Demirkan, A., et al. (2015). Effects of metformin on metabolite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care 38, 1858–1867. doi: 10.2337/dc15-0658
Yi, Y., Gao, L., Wu, M., Ao, J., Zhang, C., Wang, X., et al. (2017). Metformin sensitizes leukemia cells to vincristine via activation of AMP-activated protein kinase. J. Cancer 8, 2636–2642. doi: 10.7150/jca.19873
Zakikhani, M., Dowling, R., Fantus, I. G., Sonenberg, N., and Pollak, M. (2006). Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66, 10269–10273. doi: 10.1158/0008-5472.CAN-06-1500
Zhang, C. S., Li, M., Ma, T., Zong, Y., Cui, J., Feng, J. W., et al. (2016). Metformin activates AMPK through the lysosomal pathway. Cell Metab. 24, 521–522. doi: 10.1016/j.cmet.2016.09.003
Zhang, P., Li, H., Tan, X., Chen, L., and Wang, S. (2013). Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. 37, 207–218. doi: 10.1016/j.canep.2012.12.009
Zhang, Y., Hu, C., Hong, J., Zeng, J., Lai, S., Lv, A., et al. (2014). Lipid profiling reveals different therapeutic effects of metformin and glipizide in patients with type 2 diabetes and coronary artery disease. Diabetes Care 37, 2804–2812. doi: 10.2337/dc14-0090
Zhou, G., Myers, R., Li, Y., Chen, Y., Shen, X., Fenyk-Melody, J., et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174. doi: 10.1172/JCI13505
Keywords: metformin, type 2 diabetes, gluconeogenesis, hepatic glucose production, cancer, cell proliferation, AMPK, mTOR
Citation: Li M, Li X, Zhang H and Lu Y (2018) Molecular Mechanisms of Metformin for Diabetes and Cancer Treatment. Front. Physiol. 9:1039. doi: 10.3389/fphys.2018.01039
Received: 13 April 2018; Accepted: 12 July 2018;
Published: 31 July 2018.
Edited by:
Luigi Iuliano, Sapienza Università di Roma, ItalyReviewed by:
Benoit Viollet, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceAlessandro Laviano, Sapienza Università di Roma, Italy
Copyright © 2018 Li, Li, Zhang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijie Zhang, huijiezhang2005@126.com Yan Lu, rjluyan@126.com