 Hatem Maamoun1
Hatem Maamoun1 Tarek Benameur
Tarek Benameur Gianfranco Pintus
Gianfranco Pintus Shankar Munusamy
Shankar Munusamy Abdelali Agouni
Abdelali Agouni- 1Department of Medical Biochemistry and Molecular Biology, Faculty of Medicine, Ain Shams University, Cairo, Egypt
- 2College of Medicine, King Faisal University, Al-Ahsa, Saudi Arabia
- 3Department of Biomedical Sciences, College of Health Sciences, Qatar University, Doha, Qatar
- 4Department of Pharmaceutical and Administrative Sciences, College of Pharmacy and Health Sciences, Drake University, Des Moines, IA, United States
- 5Department of Pharmaceutical Sciences, College of Pharmacy, Qatar University, Doha, Qatar
Type-2 diabetes prevalence is continuing to rise worldwide due to physical inactivity and obesity epidemic. Diabetes and fluctuations of blood sugar are related to multiple micro- and macrovascular complications, that are attributed to oxidative stress, endoplasmic reticulum (ER) activation and inflammatory processes, which lead to endothelial dysfunction characterized, among other features, by reduced availability of nitric oxide (NO) and aberrant angiogenic capacity. Several enzymatic anti-oxidant and anti-inflammatory agents have been found to play protective roles against oxidative stress and its downstream signaling pathways. Of particular interest, heme oxygenase (HO) isoforms, specifically HO-1, have attracted much attention as major cytoprotective players in conditions associated with inflammation and oxidative stress. HO operates as a key rate-limiting enzyme in the process of degradation of the iron-containing molecule, heme, yielding the following byproducts: carbon monoxide (CO), iron, and biliverdin. Because HO-1 induction was linked to pro-oxidant states, it has been regarded as a marker of oxidative stress; however, accumulating evidence has established multiple cytoprotective roles of the enzyme in metabolic and cardiovascular disorders. The cytoprotective effects of HO-1 depend on several cellular mechanisms including the generation of bilirubin, an anti-oxidant molecule, from the degradation of heme; the induction of ferritin, a strong chelator of free iron; and the release of CO, that displays multiple anti-inflammatory and anti-apoptotic actions. The current review article describes the major molecular mechanisms contributing to endothelial dysfunction and altered angiogenesis in diabetes with a special focus on the interplay between oxidative stress and ER stress response. The review summarizes the key cytoprotective roles of HO-1 against hyperglycemia-induced endothelial dysfunction and aberrant angiogenesis and discusses the major underlying cellular mechanisms associated with its protective effects.
Introduction
Endothelial dysfunction is considered as the structural alteration and functional impairment of vascular endothelial layer, characterized by a reduction in NO bioavailability and aberrant angiogenesis (Lerman and Burnett, 1992; Creager et al., 2003; Jamwal and Sharma, 2018). Endothelial dysfunction is a key player in the pathological onset of various cardiovascular complications associated with diabetes, whether affecting microvasculature including retinopathy, nephropathy, and neuropathy or macrovasculature such as ischemic heart disease and ischemic stroke (Ebrahimian et al., 2006). There is much scientific evidence showing that hyperglycemia, a characteristic manifestation of diabetes, is implicated in the development of endothelial dysfunction. Other molecular mechanisms involved in vascular endothelial perturbations that ensue in such a metabolic disorder include disruption of a large array of metabolic pathways within the endothelial cell leading to endoplasmic reticulum (ER) stress, oxidative stress, inflammation and apoptosis (Cimellaro et al., 2016; Incalza et al., 2018).
Heme oxygenase (HO) is a cytoprotective enzyme, which contributes to maintaining a healthy vascular endothelium. HO operates as a key rate-limiting enzyme in the process of degradation of the iron-containing molecule, heme, yielding the following byproducts: carbon monoxide (CO), iron, and biliverdin (Kikuchi et al., 2005; Ryter et al., 2006; Rochette et al., 2018). There are two main HO isoforms known (HO-1, HO-2), in addition to a putative third one (HO-3) (Nitti et al., 2017). Among the three isoforms, the role of HO-1 as a major protective enzyme is best documented. Its anti-oxidant, anti-apoptotic (Wang et al., 2010; Mishra and Ndisang, 2014; Tiwari and Ndisang, 2014), and anti-inflammatory (Hayashi et al., 1999; Lee and Chau, 2002; Morse et al., 2003; Ndisang and Jadhav, 2009) effects have attracted much interest in literature. The cytoprotective effects of HO-1 depend on multiple cellular processes including the generation of bilirubin, an anti-oxidant molecule, from the degradation of heme; the induction of ferritin, a strong chelator of free molecular iron; and the liberation of CO, responsible for HO-1 major anti-inflammatory and anti-apoptotic effects (Durante, 2003; Tiwari and Ndisang, 2014). However, the anti-inflammatory and anti-apoptotic effects of CO are only observed if CO is generated in low levels. Large amounts of CO can have lethal consequences because of the strong affinity of CO binding to the heme in hemoglobin and mitochondrial proteins. CO is a stable non-radical small gas molecule that is weakly soluble in water. It belongs to the family of gasotransmitters which emerged as important signaling molecules. CO signals inside the cell in several manners. For instance, CO stimulates guanylyl cyclase to form cGMP and regulates the action of some transcription factors (Mustafa et al., 2009; Garcia-Mata and Lamattina, 2013).
HO-1 is induced by a variety of pro-oxidant agents and stimuli, such as ultraviolet rays, heavy metals, inflammatory cytokines and iron-containing molecule, heme (Li et al., 2011). HO-1 is now recognized for playing anti-oxidative and cytoprotective roles both in vitro and in vivo. For example, mice deficient for HO-1 were found to spontaneously develop iron depots in kidneys and livers, tissue damage, chronic inflammation and oxidative structure modification of macromolecules (e.g., proteins, DNA) (Poss and Tonegawa, 1997a,b). Similar to animal models, the first reported human case of HO-1 deficiency involved significant iron tissue depots, growth deficiency, anemia and high susceptibility to reactive oxygen species (ROS)-mediated damage (Yachie et al., 1999). At the molecular level, it is believed that HO-1 protects vessel wall from pathological remodeling and endothelial cell dysfunction (Duckers et al., 2001; Durante, 2003; Awede et al., 2010; Hyvelin et al., 2010). Several methodologies were employed to induce HO-1 in the vessels with the use of pharmacological inducers being one of the most promising approaches (Awede et al., 2010; Hyvelin et al., 2010). Heme itself and its man-made analogs are strong pharmacological inducers of HO-1 and were found to protect against the development of cardiovascular diseases in several studies both in vitro and in vivo (Awede et al., 2010; Hyvelin et al., 2010; Li et al., 2011). A study conducted by Yang et al. (2015) where the serum of rats exposed to cigarette smoke was used to induce oxidative stress in human umbilical vein endothelial cells (HUVECs), has shown a significant decrease in endogenous production of ROS following the induction of HO-1 by hemin (Yang et al., 2015). Maamoun et al. (2017) found that the pharmacological induction of HO-1 using Cobalt-protoporphyrin (CoPP) reduced ROS production in HUVECs exposed to intermittent high glucose.
With regards to the anti-inflammatory effects of HO-1, Chang et al. (2014) have shown that the treatment of HUVECs with iodine contrast medium caused anti-proliferative and inflammatory reactions, and enhanced the expression of intercellular adhesion molecule (ICAM)-1 and adhesion molecules receptors while cells co-incubated with the HO-1 inducer were completely protected (Chang et al., 2014). The cytoprotective role of HO-1 has also been illustrated in cancer cells, where one study has demonstrated that the upregulation of HO-1 in renal cancer cells promoted their survival capacity via the induction of the expression of pro-survival molecule Bcl-xL and decreased expression of Beclin-1 and LC3B-II, that are involved in the process of autophagy, an effect that has been reversed by HO-1 knockdown (Banerjee et al., 2012). Furthermore, in vascular cells, it has been found that HO-1 induction protected HUVECs from high glucose mediated cell death through the reduction of caspases 3 and 7 activation (Maamoun et al., 2017).
In the current article, we have reviewed the major mechanisms contributing to endothelial dysfunction, the key initial step in the onset of atherosclerotic process, in the context of diabetes and hyperglycemia. Furthermore, we have reviewed the cytoprotective roles of HO-1 against diabetes- and hyperglycemia-induced endothelial dysfunction and aberrant angiogenesis and discussed the major underlying molecular mechanisms associated with these protective effects with special emphasis on signaling pathways related to oxidative stress and ER stress response.
Endothelial Dysfunction and Hyperglycemia: Key Molecular Disturbances
The endothelium is a single cell layer that forms the interface between blood stream and adjacent tissues. Over the recent decades the complexity of this selectively permeable barrier and its important contribution to controlling vascular homeostasis have been established (Michiels, 2003; Khazaei et al., 2008; Jamwal and Sharma, 2018). The endothelium allows the selective passage of certain substances such as nutrients through the vessel wall to the adjacent tissues. The endothelium is recognized as an endocrine organ that is able to produce and secrete many hormones and mediators which are crucial for the optimal functioning of the vasculature such as factors regulating vascular tone, coagulation, immune response and growth of adjacent vascular cells (Khazaei et al., 2008; Jamwal and Sharma, 2018). In normal physiological conditions, vascular endothelial cells are exposed to plasma blood sugar levels between 3.8 and 6.1 mmol/L. Exposure of endothelial cells to glucose levels of 11.1 mmol/L and above is regarded as a diabetic condition [the national institute for health and care excellence (NICE) guidelines, 2015]. However, unlike in vivo conditions, in cell culture, the definition of high glucose varies considerably according to the cell model used, depending on glucose levels in culture medium where the cells are being selected and harbored. Two examples that demonstrate such variability are the endothelial cell line EA.hy926 and HUVECs, where the former is propagated in culture medium containing 25 mM glucose, whereas the latter is routinely grown in a culture medium containing 5.5 mM of glucose (Maamoun et al., 2017). As a result, 25 mM glucose would be considered to be a high concentration of glucose for HUVECs, but is a standard level in EA.hy626 endothelial cell culture medium. Alterations occur in both the micro- and macrovasculature in response to hyperglycemia. An abnormally high glucose concentration can disrupt the balance within the endothelial cell and a state of ‘endothelial cell dysfunction’ results. Endothelial cell dysfunction is defined by the presence of one or more of the following characteristics: (I) low bioavailability of NO, (II) impaired endothelium-dependent relaxation, (III) weak fibrinolytic capability, (IV) excess of production of growth factors, adhesion molecules and pro-inflammatory molecules (e.g., cytokines), oxidative stress, and aberrant angiogenesis (Cade, 2008).
Hyperglycemia-Mediated Oxidative Stress and Endothelial Injury
Macrovascular and microvascular complications have been shown to be mainly or partly dependent on hyperglycemia (Zoungas et al., 2012). Hyperglycemia can induce vascular endothelial damage through different pathways: (I) enhanced polyol activity, causing sorbitol and fructose accumulation; (II) enhanced production of advanced glycation end products (AGEs); (III) activation of mitogenic protein kinase C (PKC); (IV) heightened hexosamine flux pathway and (V) a decrease of body anti-oxidant defenses (Brownlee, 2001; Kornfeld et al., 2015). There is considerable evidence that these biochemical pathways can be induced by excessive generation of ROS, leading to increased oxidative stress in a positive feedback loop (Son, 2012). Oxidative stress occurs when the concentrations of ROS exceed those of anti-oxidant neutralizing species, such as glutathione (GSH) and HO. ROS are a heterogeneous population of molecules including free radicals, such as hydroxyl radical (OH-), superoxide anion (O2-), peroxyl (RO2), and hydroperoxyl (HRO2-), and non-charged species, such as hydrogen peroxide (H2O2) and hydrochloric acid (HCl) (Pieper et al., 1997). Under intracellular hyperglycemic conditions, excessive production of ROS develops through several mechanisms, the most important of which is excessive activation of mitochondrial electron transport chain. Other sources of ROS include glucose-induced activation of NADPH oxidase (NOX) and xanthine oxidase (Nishikawa et al., 2000). This excessive ROS production inflicts DNA damage resulting in poly ADP ribose polymerase (PARP)-1 activation, which in turn inhibits the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (G3PDH) resulting in the pile-up of upstream glycolytic metabolites, with the resulting influx into polyol pathway, hexosamine pathway, diacylglycerol (DAG) and PKC pathway, as well as generation of AGEs (Du et al., 2003) (Figure 1).
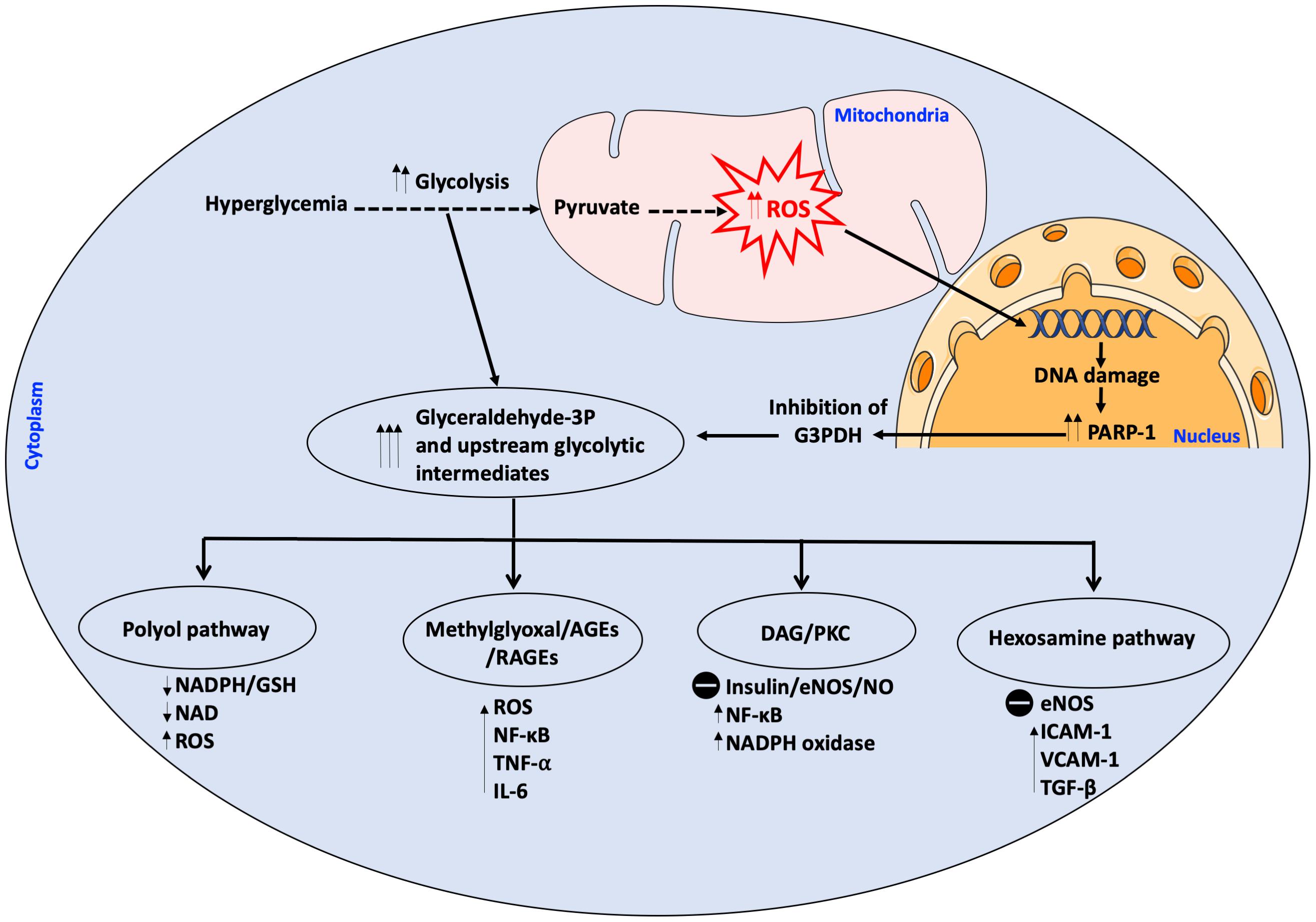
Figure 1. Intracellular hyperglycemia results in increased production of mitochondrial ROS. ROS cause DNA damage releasing PARP-1 which inhibits G3PDH. This results in the accumulation of upstream glycolytic intermediates. Other pathways are activated as a result of the accumulation of glycolytic intermediates upstream of G3P which have detrimental effects on the cell; these pathways are; (1) Polyol pathway which depletes the cell of its antioxidant GSH system due to scavenging of NADPH, thereby augmenting ROS production, (2) Methylglyoxal which is a precursor to AGEs/RAGEs which activate pro-inflammatory and pro-oxidant molecules, (3) DAG/PKC which impairs insulin signaling and eNOS activity, activates pro-inflammatory and pro-oxidant molecules, (4) Hexosamine pathway with the resultant activation of pro-inflammatory molecules, inhibition of eNOS at the Akt activation site and activation of TGF-β with increase in ROS. All these pathways increase ROS in a positive feedback loop. AGEs, advanced glycation end products; DAG, diacylglycerol; eNOS, endothelial NO synthase; G3PDH, Glyceraldehyde 3-phosphate dehydrogenase; GSH, Glutathione; ICAM-1, intracellular adhesion molecule-I; IL-6, interleukin 6; NAD, nicotinamide adenine dinucleotide; NADP, NAD phosphate; NF-κB, nuclear factor kappa B; NO, nitric oxide; PARP-1, poly ADP ribose polymerase 1; PKC, protein kinase C; RAGE, AGEs receptor; ROS, reactive oxygen species; TGF-β, transforming growth factor β; TNF-α, tumor necrosis factor α; VCAM-1, vascular cell adhesion molecule 1. The symbol  means inhibition.
means inhibition.
Glucose, through the polyol pathway, is converted into polyalcohol sorbitol by aldose reductase utilizing NADPH as a cofactor, which in turn reduces intracellular concentrations of NADPH resulting in the inhibition of glutathione/peroxidase anti-oxidant system, which is dependent on NADPH as a cofactor, and consequently induces overproduction of H2O2 and ROS in general. Moreover, ROS accumulation inhibits glucose-6-phosphate dehydrogenase (G6PDH), which is the rate limiting enzyme for pentose shunt pathway that is fundamental to the maintenance of reducing equivalents for the glutathione/peroxidase system anti-oxidant system, amplifying the oxidative stress (Figure 2). Furthermore, sorbitol is oxidized to fructose by sorbitol dehydrogenase using NAD as a co-factor, increasing the intracellular ratio of NADH/NAD+, exacerbating the inhibition of G3PDH which is dependent on NAD as a cofactor; thus amplifying the accumulation of glyceraldehyde-3-phosphate (G3P) with the consequent flux into triose phosphate pathway via triose phosphate isomerase (TPI) yielding dihydroxyacetone phosphate (DHAP). Higher levels of triose phosphate promote the formation methylglyoxal and DAG. Methylglyoxal is a precursor of AGEs, which are formed by binding of methylglyoxal to the free amino groups of intracellular and extracellular proteins (Allaman et al., 2015; Maessen et al., 2015) (Figure 3).
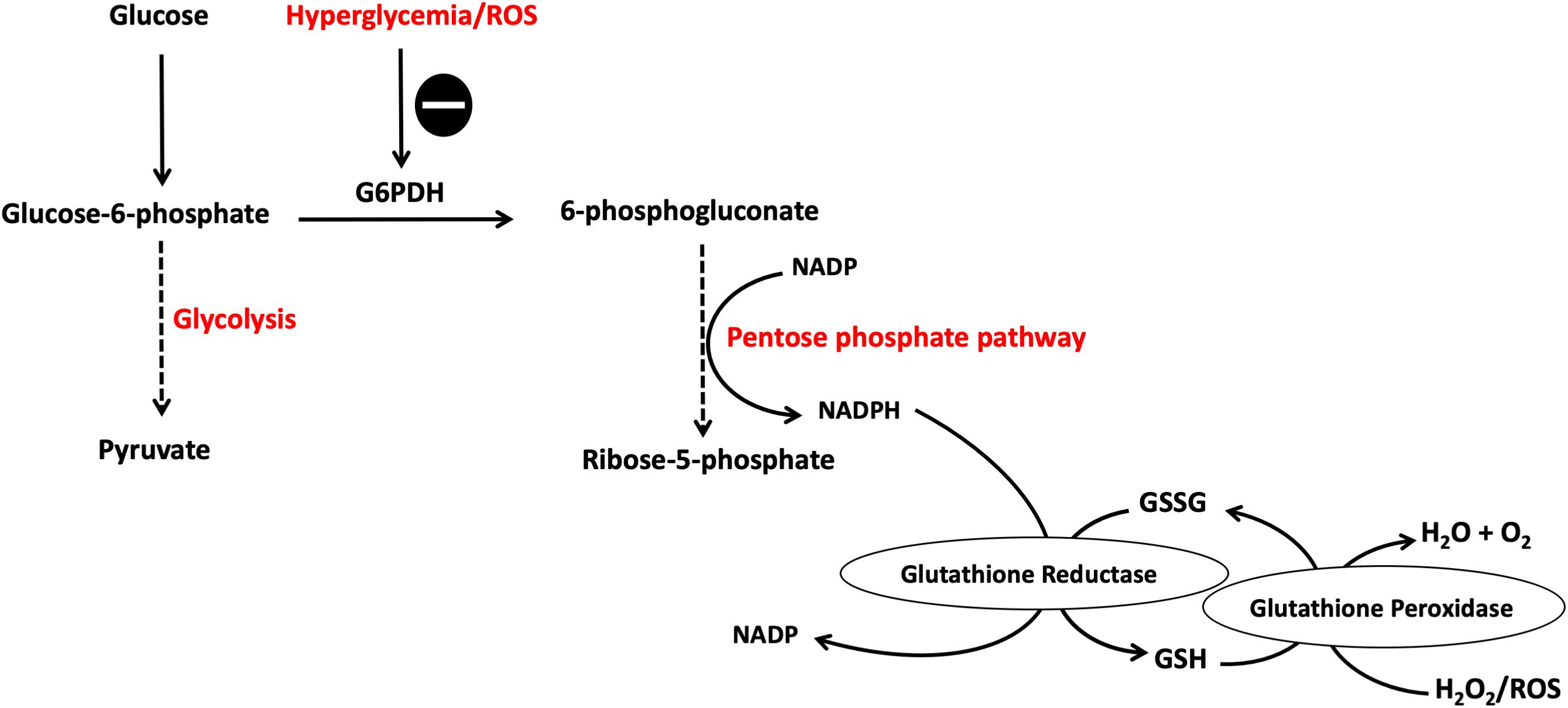
Figure 2. Pentose phosphate pathway (PPP) is inhibited by hyperglycemia and ROS. Hyperglycemia-induced ROS cause the inhibition of G6PDH which is the rate limiting enzyme for this pathway. This results in reduced NADPH production, a cofactor fundamental in the GSH/peroxidase antioxidant system, with the consequent buildup of more ROS causing a vicious circle of oxidative stress induction. G6PDH, glucose-6-phosphate dehydrogenase; GSH, glutathione; NADPH, reduced NAD phosphate. The symbol means inhibition.
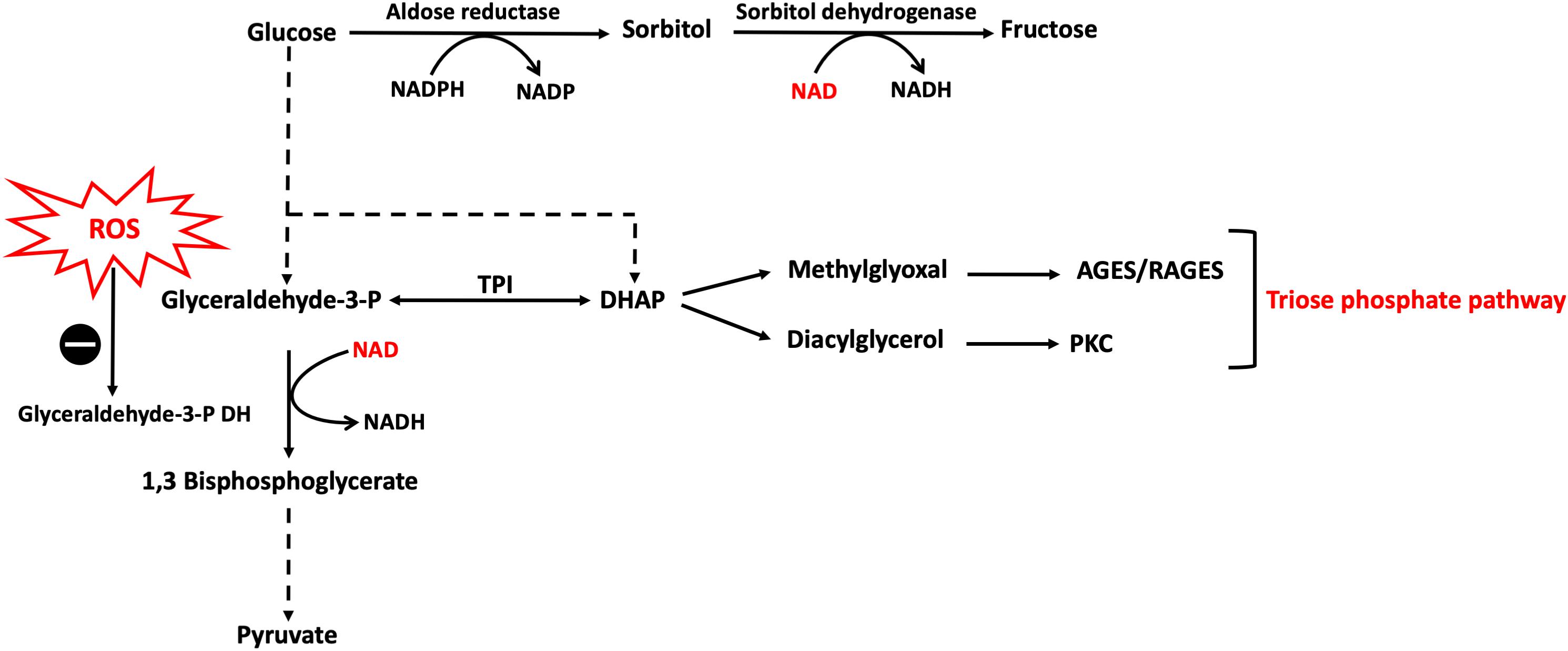
Figure 3. Polyol and triose phosphate pathways activation caused by hyperglycemia-induced ROS. Hyperglycemia results in an excess of ROS production which inhibits G3PDH with the consequent accumulation of glycolytic intermediates upstream to G3P leading to increase flux into other pathways, namely polyol and triose phosphate. In polyol pathway, NAD reducing equivalent is consumed by sorbitol dehydrogenase reaction leading to reductions in NAD levels inside the cell which further confounds the inhibition of G3PDH. DHAP, dihydroxyacetone phosphate; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; NAD, nicotinamide adenine dinucleotide; TPI, triose phosphate isomerase. The symbo means inhibition.
DAG activates certain PKC isoforms by binding to their membrane-bound receptors. Consequently, active PKC isoforms may contribute to hyperglycemia-induced vascular endothelial damage by inhibiting insulin-stimulated endothelial NO synthase (eNOS) expression and NO production in endothelial cells, increasing thus the activity of nuclear factor kappa B (NF-κB) and the pro-oxidant enzyme NOX (Giri et al., 2018). Under hyperglycemic conditions, PKC-α and PKC-γ, were reported to be able to enhance NOX activity (Venugopal et al., 2002; Dasu et al., 2008). PKC-β, a molecular mediator of hypertrophic responses, was also found to be selectively over-activated in vascular and cardiac cells of animal models of diabetes. Liu et al. (2012) observed that the selective inhibition of PKC-β, improved left ventricular function and structure in a diabetes animal model of streptozotocin-injected rats. More recently, it was reported that the activation of PKC-α in the intestine of streptozotocin-induced diabetic mice contributed to the enhanced uptake of iron leading to iron loading that contributes to diabetic complications (Zhao et al., 2018). Furthermore, the inhibition of PKC-ζ was shown recently to improve insulin sensitivity and uptake of glucose in rat adipocytes rendered insulin-resistant by incubating them in high glucose and high insulin concentrations (Lu et al., 2018).
Hyperglycemia-induced ROS overproduction is also caused by the interaction of AGEs with their receptors (RAGEs) (Rochette et al., 2014). This interaction generates intracellular ROS and activates NF-κB, which modulates the expression of a many genes associated with inflammation and vascular remodeling, including interleukin (IL)-6, tumor necrosis factor (TNF)-α, ICAM-1, vascular cell adhesion molecule (VCAM)-1 and monocyte chemotactic protein-1 (MCP-1) (Bierhaus et al., 2001; Younce et al., 2010). Furthermore, AGEs which are present in the extracellular matrix can decrease the availability of NO, reducing thereby endothelium-dependent vasodilation (Xu B. et al., 2005). Finally, the activation of the hexosamine pathway contributes to the vascular damage induced by hyperglycemia owing to its capacity to disturb multiple cellular processes, including signal transduction, gene transcription, cell survival, and proteasome-mediated degradation (Love and Hanover, 2005). The activation of the hexosamine pathway can inhibit eNOS activity by impairing insulin receptor activation and signal transduction and increasing ROS production (Rajapakse et al., 2009a,b).
ER Stress and Endothelial Dysfunction in Diabetes
In addition to hyperglycemia-induced oxidative stress, ER stress was also linked to various features of endothelial dysfunction in diabetics with decreased NO bioavailability and aberrant angiogenesis being two prominent features (Sheikh-Ali et al., 2010). Aberrant angiogenesis may be either an excessive angiogenic response (e.g., retinopathy, nephropathy) or a deficiency in angiogenesis (e.g., disturbed wound healing, reduced sprouting of collateral vessels, neuropathy, ischemia and peripheral vascular disease) (Kota et al., 2012). Under normal physiological conditions, the ER plays a central role in protein synthesis and co-translational protein modification through the folding of newly synthesized proteins by native disulfide bond formation to attain the final stable conformational state prior to being directed to their final destination whether it is to be secretory or membrane bound proteins. The homeostasis of ER is put at stress when the influx of the newly synthesized misfolded or unfolded polypeptide chains exceeds the repair and refolding capacity of the ER; this condition is commonly referred to as ER stress response (Walter and Ron, 2011).
Endoplasmic reticulum stress response develops both in type-1 or type-2 diabetes mellitus due to the presence of hyperglycemia that increases the demand for the synthesis of enzymatic machinery necessary to carry out complete glucose oxidation through glycolysis, tricarboxylic acid cycle (TCA) and oxidative phosphorylation by mitochondrial electron transport chain (Flamment et al., 2012; Hu et al., 2017). This increase in protein load on the ER increases the chances of accumulation of unfolded or poorly folded proteins inside the ER by the formation of incorrect or non-native disulfide bonds due to mispairing of cysteine residues in the protein being synthesized (Lisa et al., 2012). When this occurs, the cell triggers the unfolded protein response (UPR), which aims to resolve the problem by three different mechanisms working synergistically together: (I) Enhancement of gene expression of molecular chaperones to support and improve the correct folding of proteins; (II) Activation of ER-associated degradation machinery (ERAD) to get rid of aberrant proteins; and (III) Attenuation of translation rate to slow down the entry of new proteins to the ER lumen (Flamment et al., 2012). The molecular system involved in such response includes three membrane-bound proteins that orchestrate the whole process, namely inositol requiring enzyme (IRE)-1α, protein kinase RNA-like ER kinase (PERK) and activating transcription factor (ATF)-6, which under basal conditions are kept in an inactive state by binding to 78 kDa glucose-regulated protein (GRP)-78 or binding immunoglobulin protein (BiP); however, when there is an increase of misfolded proteins, GRP78/BiP preferentially binds to them hence relieving those ER effectors from their inhibition state. The dissociation of GRP78 from the three effectors is followed by the dimerization and hence activation of both IRE-1α and PERK, and the transfer of ATF-6 to the Golgi body for proteolytic processing activation before the final active ATF-6 translocates to the nucleus and stimulates the transcription of target genes. The UPR is a physiological pro-survival process which attempts to restore normal homeostatic state of the ER within the cell; however, if the stress condition remains unresolved, these three effectors involved in the UPR can also activate multiple inflammatory responses and eventually may lead to cell death through the activation of several pro-apoptotic sub-pathways (Gardner and Walter, 2011; Walter and Ron, 2011; Hassler et al., 2012; Gardner et al., 2013) (Figure 4).
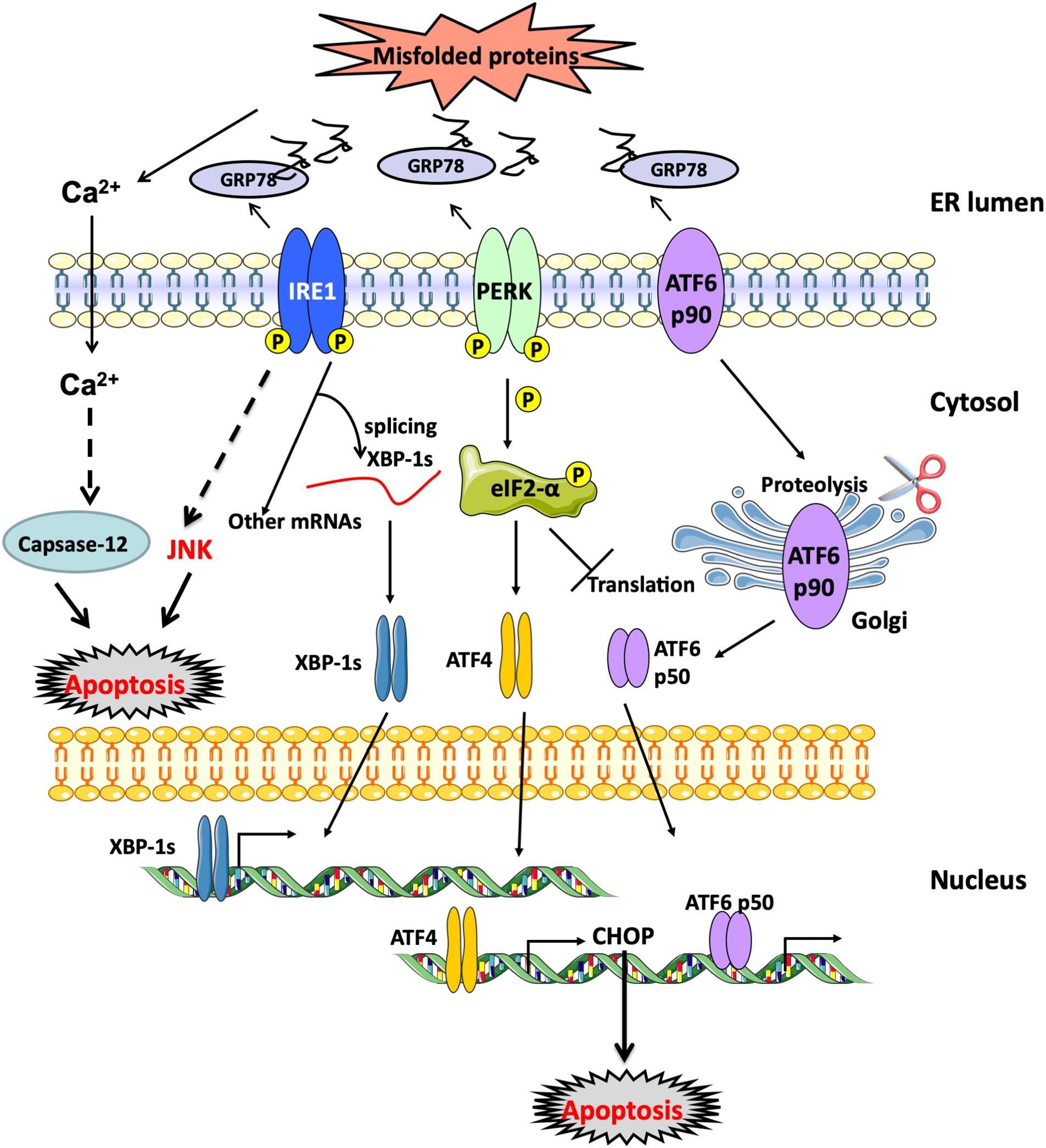
Figure 4. UPR response activation. ER membrane-bound proteins, PERK, ATF-6 and IRE-1α, are activated when unfolded proteins accumulate within the ER lumen and GRP78/BiP dissociates from them. Downstream effects of PERK involve the phosphorylation of eIF-2α resulting in the attenuation of protein translation with the preferential increase in ATF-4 expression. ATF-6 (p90) translocates to the Golgi body where it undergoes proteolytic cleavage. Active cleaved ATF-6 (p50) translocates then to the nucleus and stimulates the expression of molecular chaperones to aid in the folding process. Finally, active IRE-1α through its endoribonuclease activity undergoes intron splicing of XBP-1 mRNA to synthesize the active spliced XBP-1s which binds to the promoter regions of several genes involved in the synthesis of chemical chaperones and ERAD proteins to restore normal protein homeostasis as part of the UPR rescue response. ATF-6, activating transcription factor 6; BiP, binding immunoglobulin protein; eIF-2α, Eukaryotic translation initiation factor 2α; ERAD, endoplasmic-reticulum-associated protein degradation; GRP78, 78-kDa glucose regulated protein; IRE-1α, inositol-requiring enzyme 1α; PERK, PKR-like ER kinase; XBP-1, X-box binding protein 1.
Previous studies have demonstrated the role of ER stress response in endothelial cell inflammation and cell death in vascular diseases associated with diabetes. Work by Chen et al. (2012) demonstrated that the ATF-4-mediated activation of signal transducer and activator of transcription 3 (STAT3) is responsible for inflammation, vascular endothelial damage and loss of angiogenic capacity in streptozotocin-induced wild-type mouse model of type-1 diabetes, while ATF-4 knockout mice were protected. Other in vitro studies have also shown that the blockade of STAT3 reduced high glucose-mediated ER stress response activation, suggesting a crosstalk between ER stress and inflammatory signaling pathways. Moreover, another study of diabetic cardiovascular complications in streptozotocin-induced mouse model of diabetes found that the activation of the epidermal growth factor receptor (EGFR) tyrosine kinase signaling pathway participated in the induction of ER stress response and microvascular dysfunction in these mice. Enhanced EGFR phosphorylation was associated with the activation of the PERK/eukaryotic initiation factor (eIF)-2α/ATF-4 axis. The pharmacologic blockade of the kinase activity of EGFR ameliorated endothelium-dependent relaxation and increased NO production (Galan et al., 2012). Previously, Agouni et al. (2011) investigated in a mouse model of diet-induced obesity the impact of whole-body alleviation of ER stress response through the specific liver-deletion of protein tyrosine phosphatase (PTP)-1B, a protein located on the luminal surface of the ER that was shown to be implicated in the activation of ER stress (Owen et al., 2013), on endothelial cell function. Authors reported that the alleviation of ER stress response improved endothelium-dependent vasodilation in mouse aortas and improved vascular NO bioavailability (Agouni et al., 2014).
The role of ER stress in ischemia-induced neovascularization was demonstrated in type-2 diabetes genetic mouse model (db/db). The chemical chaperones, tauroursodeoxycholic acid (TUDCA) and 4-phenylbutyric acid (4-PBA), known to alleviate ER stress, improved hind-limb ischemia after femoral artery ligation. These small molecules reduced the expression of ATF-4 and C/EBP homologous protein (CHOP), enhanced blood flow recovery following ligation, and increased the expression of pro-angiogenic factors (Amin et al., 2012). There is also growing evidence that ER stress response might play an important role in promoting angiogenesis, and therefore may act as a novel mediator of the angiogenic process. This evidence implicates these pathways in the upregulation of angiogenic factors such as vascular endothelial growth factor (VEGF)-A, fibroblast growth factor (FGF), IL-8, and endothelin (ET)-1 (Paridaens et al., 2014). Notably, it was shown that ER stress-mediated overexpression of VEGF-A was dependent on the IRE-1α/X-box binding protein (XBP)-1, PERK/ATF-4, as well as ATF-6 arms of ER stress response. The upregulation of VEGF-A expression by UPR effectors was also reported in diabetic nephropathies and retinopathies, indicating the involvement of UPR and ER stress responses in the enhancement of angiogenesis beyond the physiological conditions (Ghosh et al., 2010). However, Maamoun et al. (2017) have reported more recently that the chemical chaperone, 4-PBA prevented intermittent high glucose-induced impaired angiogenic capacity in HUVECs characterized by a weaker ability to form tube-like structures on a matrigel matrix and a reduced the expression of VEGF-A. Nonetheless, the exact molecular mechanisms involved in the switch between pro- and anti-angiogenic stimulation by ER stress response are not fully understood.
Endoplasmic reticulum stress also plays a key role in the onset of atherosclerosis in diabetes, a major consequence of endothelial dysfunction. Several independent risk factors for cardiovascular diseases, including hyperglycemia (Werstuck et al., 2006), hyper-homocysteinemia (Werstuck et al., 2001), obesity (Ozcan et al., 2004; Agouni et al., 2011), and dyslipidemia have been associated with ER stress, indicating that it may be a converging molecular link for atherogenesis (Cunha et al., 2008). The activation of UPR response has been observed at all stages of atherogenesis (Zhou et al., 2005). ER stress inducers can promote lipid tissue accumulation by activating the sterol regulatory element binding proteins (SREBP), which control the transcriptional regulation of lipid synthesis and transport in endothelial cells (Colgan et al., 2007). ER stress inducers also activate NF-κB, which promotes the expression of pro-inflammatory and pro-coagulant genes contributing thus to atherogenesis (Jiang et al., 2003). Moreover, ER stress effector IRE-1α causes the activation of c-Jun N-terminal kinases (JNK) which impairs insulin signaling reducing thus insulin-stimulated eNOS activity with subsequent reduction of NO production (Andreozzi et al., 2007). ER stress has also been shown in vivo and in vitro to activate pro-apoptotic caspases and cell death of human endothelial cells (Xu C. et al., 2005; Maamoun et al., 2017).
There is also evidence suggesting a crosstalk between ER stress and oxidative stress. Maintaining ER balance is intimately associated with oxidative state of the cell. In contrast to cytosol, the ER lumen is an oxidizing environment characterized by an elevated ratio of oxidized to reduced glutathione (GSSG/GSH) which encourages the proper native disulfide bond formation (van der Vlies et al., 2003). During disulfide bond-dependent protein folding, proteins are oxidized to form disulfide bonds by protein disulfide isomerase which allows proper polypeptide rearrangement to reach their final native conformational state. Any mispairing of cysteine residues would result in the formation of non-native disulfide bonds that prevent proteins from achieving their native optimal conformation, a condition that results when there is an abnormally high request for protein synthesis as in the case of hyperglycemia. In addition, hyperglycemia-induced AGEs accumulation as well as hexosamine and polyol pathways activation can impair the correct protein folding, leading to the activation of UPR and ER stress responses (Chakravarthi and Bulleid, 2004).
In normal conditions, during protein folding, electrons are transferred from targeted cysteine residues in the polypeptide chain being synthesized forming native disulfide bonds to bring the protein structure to its final conformational state. This reaction is catalyzed by two crucial enzymes, namely protein disulfide isomerase (PDI) and ER oxidoreductase (ERO)-1α, which aid in this electron transfer from the target cysteine residues to molecular oxygen, generating H2O2; however, in the presence of a high protein folding load, as in hyperglycemia, there is an increase in non-native disulfide bond formation, which results in GSH consumption as a protective mechanism. This results in GSH depletion contributing thus to excessive ROS generation and consequent development of oxidative stress (van der Vlies et al., 2003) (Figure 5). Moreover, hyperglycemia-induced oxidative stress is suggested to contribute to the inactivation of disulfide isomerases resulting in the buildup of unfolded proteins in addition to the misfolded ones (Tu and Weissman, 2004). Since protein folding is a highly energy-dependent process, protein misfolding-induced ATP depletion leads to more glucose consumption to promote mitochondrial oxidative phosphorylation to increase ATP synthesis and consequently increasing ROS production to pathological levels. As a result of unfolded/misfolded protein accumulation, Ca2+ leak ensues from the ER lumen into the cytosol and increased intracellular Ca2+ levels further stimulate mitochondrial ROS production. In fact, Ca2+ increases the permeability of inner mitochondrial membrane and blocks the electron transport chain at the level of complex III, thereby causing electron leak to molecular O2, thus exacerbating the ongoing ROS production (Gorlach et al., 2006) (Figure 6). Taken together, there is substantial evidence that both ER stress and oxidative stress are concomitantly induced in hyperglycemic conditions and they play an important role in the perturbations that ensue in the vascular endothelium leading to the development of endothelial dysfunction in diabetes.

Figure 5. Schematic representation showing ER protein folding as a source of ROS. During ER stress response, there is accumulation of misfolded proteins within the ER where non-native disulfide bonds form resulting in the depletion of glutathione (GSH) that is trying to correct the aberrant bonds. IRE-1α, inositol-requiring enzyme 1 α; PDI, protein disulfide isomerase.
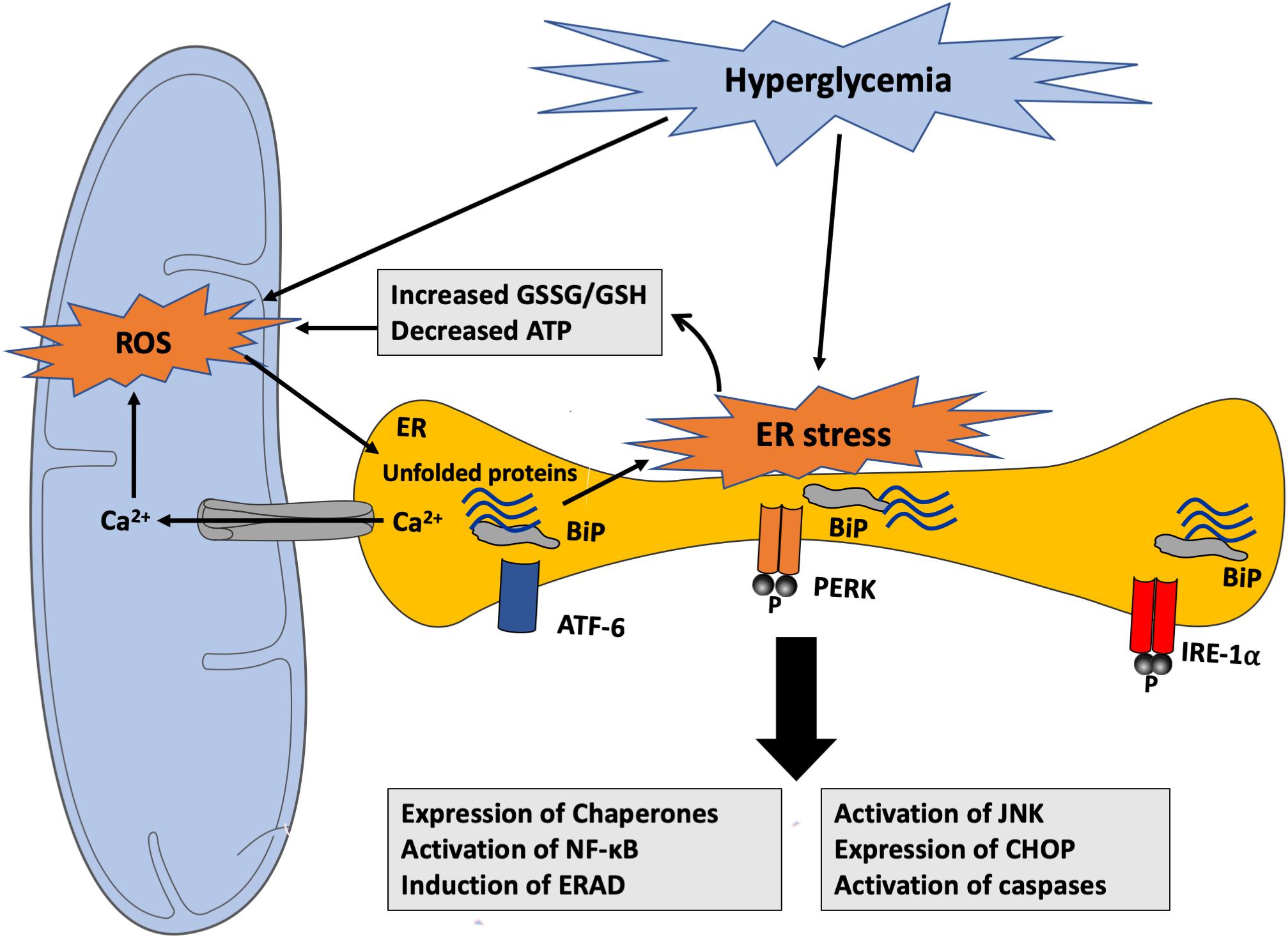
Figure 6. Hyperglycemia-induced ROS generation and activation of ER stress response. Hyperglycemia-generated ROS can promote the accumulation of unfolded proteins within the ER contributing to ER stress response through different mechanisms. ER dysfunction causes GSH depletion, Ca2+ leak from the ER lumen into the cytosol and into the mitochondria, and ATP depletion that can impair its structural and functional integrity and stimulate mitochondrial ROS production. ROS, in turn, can alter protein folding, thereby inducing unfolded protein accumulation within the ER lumen. ATF-6, activating transcription factor 6; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; ERAD, endoplasmic-reticulum-associated protein degradation; GSH, Glutathione; GSSG, oxidized Glutathione; IRE-1α, inositol-requiring enzyme 1α; JNK, c-Jun N-terminal kinases; NF-κB, nuclear factor kappa B; PERK, PKR-like ER kinase; ROS, reactive oxygen species.
Mitochondrial Dysfunction and Endothelial Dysfunction in Diabetes
Much evidence suggests that mitochondrial dysfunction is a major source for hyperglycemia-mediated generation of ROS through the electron transport chain, which contributes to oxidative damage pathways as discussed above, namely the polyol, triose phosphate, hexosamine, and AGEs pathways (Nishikawa et al., 2000). Normally, NADH and FADH2 released from the TCA cycle translocate from the mitochondrial matrix to the inner mitochondrial membrane, where they will donate free electrons to the series of mitochondrial membrane-bound complexes. Free electrons are transferred through mitochondrial complexes I, II, III and cytochrome c (IV), until they are finally transferred to molecular oxygen to form water. The electron transfer across these complexes generates a gradient of protons in the mitochondrial intermembrane space, generating thus a proton potential gradient across the inner mitochondrial membrane, which activates ATP synthesis. When electrons are moving from complex III to complex IV, ROS radicals are generated in small amounts during normal oxidative phosphorylation process; these ROS radicals are then captured by enzymatic anti-oxidants such as glutathione (GSH) and superoxide dismutase (SOD) isoforms. Under hyperglycemic conditions, higher levels of glucose-derived pyruvate and acetyl-CoA enter the TCA cycle increasing thus the generation of NADH and FADH2 that, in turn, transfer free electrons into the electron transport chain and consequently massively increase proton gradient through the mitochondrial membrane. This abnormally high proton gradient blocks complex III, forcing free electrons to move back to coenzyme Q10, which then transfers these electrons to molecular oxygen in a process known as electron leak, thereby generating O2- anions (Rolo and Palmeira, 2006). As mentioned above, in normal physiological conditions there is still some mitochondrial ROS being generated during the electron migration at complex IV; however, it is within the capacity of the glutathione/peroxidase system and SOD to scavenge it and prevent buildup of oxidative stress (Figure 7).
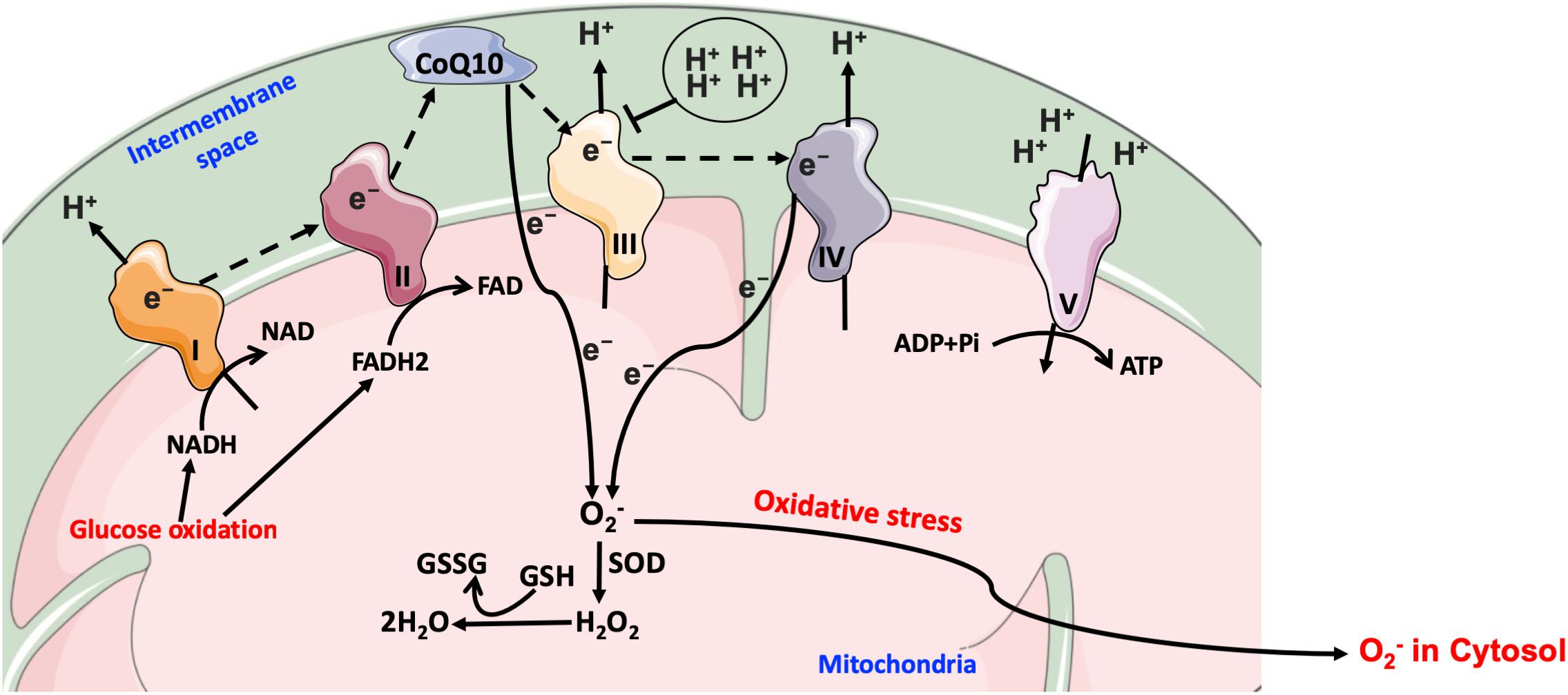
Figure 7. Mitochondrial electron transport chain in hyperglycemia. Under hyperglycemic conditions, higher levels of glucose-derived pyruvate increase the flux of NADH and FADH2 into the electron transport chain and consequently increase the proton gradient across the mitochondrial membrane. The transfer of free electrons to complex III is blocked, leading the electrons to be donated to coenzyme Q10, which then transfers electrons to molecular oxygen, thereby generating O2-. CoQ10, coenzyme Q10; e-, electrons; O2-, superoxide; GSH, glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; Pi, inorganic phosphate; SOD, O2- dismutase.
NADPH Oxidase (NOX) Activation and Endothelial Dysfunction in Diabetes
In addition to the mitochondrial dysfunction being a major source of hyperglycemia-induced ROS overproduction, studies have shown there are other principle sources that include: NOX and uncoupled eNOS (Inoguchi et al., 2000; Volpe et al., 2018). The NOX family of enzymes were identified first in phagocytic cells, neutrophils and macrophages. They are the only family of enzymes with a primary function to generate ROS (H2O2, O2-) by catalyzing the electron transfer from cytosolic NADPH to molecular O2 across plasma and lysosomal membranes (Thannickal and Fanburg, 2000; Salazar, 2018). There is a total of seven isoforms identified so far which are abundantly expressed in phagocytic cells. These isoforms include, NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2. Four of these are expressed by endothelial cells, NOX1, NOX2, NOX4, and NOX5 (Salazar, 2018). In endothelial cells, NOXs are believed to produce moderate levels of ROS which are needed for redox signaling during normal cell metabolism process. However, in certain pathological states, NOX expression and activity in the vasculature was found to be increased, thus contributing to excess release of ROS.
NOX isoforms consist of a catalytic subunit “NOX,” which catalyzes the transfer of free electrons from cytosolic NADPH to molecular oxygen. Specifically, for NOX-1, 2, and 4, additional regulatory subunits are also required for a complete activation of the complex. These subunits include p22phox, p47phox, p67phox, and p40phox. By contrast to the other isoforms, NOX-5 is a unique polypeptide subunit which is regulated by Ca2+. One additional difference between these isoforms is that while NOX-1, 2, and 5 generate superoxide O2-, NOX-4 generates the non-charged radical H2O2 (Drummond and Sobey, 2014). The subunit p47phox is pivotal for NOX activation and is frequently named the ‘organizer subunit’ since it is crucial for the formation of an active NOX complex. It resides within the cytosol in resting state; however, upon its phosphorylation at several serine residues, it undergoes conformational changes to expose its intramolecular SH-3 domain which interacts with the cytosolic activator p67phox subunit, and chaperones it to the membrane-bound subunits, NOX and p22phox, to finally form the active NOX complex (Schulz et al., 2011; Drummond and Sobey, 2014).
Hyperglycemia, free fatty acids, and oxidized low-density lipoprotein (LDL) have been shown to enhance endothelial NOX activity (Shen, 2010). Vessels isolated from diabetic subjects exhibit increased O2- production and higher expression levels of several NOX subunits (p22phox, p47phox, and p67phox), suggesting that NOX isoforms are more active in diabetes (Pitocco et al., 2010). The excess of O2- can rapidly react with NO to form ONOO- which, in turn, oxidizes tetrahydrobiopterin (BH4), thus making it unavailable for interaction with eNOS. In the presence of low amounts of BH4, eNOS becomes uncoupled and donates free electrons to O2 instead of L-arginine, producing therefore O2- instead of NO. Clinical studies demonstrated that BH4 supplementation improved endothelium-dependent vasodilation in diabetic patients, highlighting the important role of uncoupled eNOS in endothelial cell dysfunction in diabetes (Son, 2012). Collectively, hyperglycemia is tightly associated with NOX activation, mitochondrial dysfunction, and ER stress response. These cell responses are unequivocally major mechanisms underpinning ROS overproduction and oxidative stress-mediated endothelial cell damage in diabetes.
Endothelial Dysfunction and Altered Angiogenic Capacity in Diabetes: a Focal Role for NO and ROS
Blunted eNOS activity and/or protein expression and subsequent decreased NO bioavailability is a hallmark of endothelial dysfunction associated with diabetes (Hirata et al., 2010). Protein expression of eNOS is tightly controlled by several molecular processes, which include transcriptional and post-transcriptional regulation of mRNA expression (Searles, 2006), and post-translational regulation of the protein structure and function where the role of Akt activity is crucial in enhancing the phosphorylation of eNOS at Ser1177 causing its activation (Shen et al., 2006). In early stages of hyperglycemia-induced cellular stress, high glucose-induced ROS production alters Ca2+ homeostasis leading to Ca2+ leaking from ER stores into the cytosol (Ding and Triggle, 2010; Triggle and Ding, 2010). As a result, intra-ER Ca2+ levels fall below a critical level interfering thus with the activity of many ER chaperones which depend on Ca2+ availability inside the ER lumen and hence interfere with proper protein folding leading to activation of UPR and eventually apoptosis (Morgan et al., 2008). Furthermore, the resulting increase in cytosolic Ca2+ induces the activation of eNOS which can produce high amounts of NO that can then quickly react with O2- to form ONOO-, thereby oxidizing BH4 and eventually leading to eNOS uncoupling and a reduction in the bioavailability of NO (Ding and Triggle, 2010). In advanced stages, when ER stress reaches a point where it cannot be resolved, the cell must activate pro-apoptotic signals. Endothelial cells incubated in high glucose, activate IRE-1α -mediated cell death response following a disruption Ca2+ homeostasis due to an increase in the levels of ROS (Bishara and Ding, 2010).
The activation IRE-1α stimulates JNK and p38 mitogen-activated protein kinases (MAPK) pathways triggering thus the downstream pro-apoptotic mediators, apoptosis signal-regulating kinase (ASK)-1 and caspase-12, via CHOP activation (Xu C. et al., 2005). ASK-1 can cause NO deficiency by inhibiting eNOS through reduced phosphorylation at Ser1177 site (Yamashita et al., 2007). A study conducted by Galan et al. (2014) demonstrated in coronary artery endothelial cells that were subjected to high glucose or ER stress inducer, tunicamycin, the role of NOX in the suppression of eNOS promotor region and decreased phosphorylation. These effects were restored by a chemical chaperone, 4-PBA, that helped resolve ER stress. They also reproduced similar results in vivo by showing a significant reduction in endothelium-dependent and independent relaxation in control mice compared with p47phox-deficient mice, indicating the involvement of p47phox subunit of NOX complex as a molecular link for ER stress-induced reduction in NO production and the consequent vascular endothelial dysfunction (Galan et al., 2014). More recently, it has been reported that high glucose-induced ER stress caused an increase in the phosphorylation of p47phox at Ser345 (Maamoun et al., 2017). This critical phosphorylation was found to precede the activation of NOX (Perisic et al., 2004). Nevertheless, regardless of the exact mechanisms involved in the inhibition of eNOS, it is generally accepted that high glucose induces a reduction in NO production and bioavailability and hence contributes to endothelial dysfunction.
Micro- and macrovascular beds are altered in diabetes by various changes in angiogenic mechanisms (Simons, 2005). However, from a vascular standpoint, diabetes is a paradoxical disease (Costa and Soares, 2013). Excessive angiogenesis contributes to the onset of both diabetic retinopathy (Wilkinson-Berka, 2004) and nephropathy (Osterby and Nyberg, 1987). Insufficient angiogenesis, however, plays a critical role in several pathological states including impaired wound healing process, weak neovascularization of coronary collaterals, embryonic vasculopathy during gestational diabetes, and acute transplant rejection in recipients suffering from diabetes (Galiano et al., 2004). Furthermore, diabetic neuropathy is a complication linked with reduced nutritive blood flow secondary to diabetes. Altered arteriogenesis, which is referred to as the process of remodeling (such as increase in diameter) of arteries, has also been widely observed in diabetics (Abaci et al., 1999). The weak mobilization of endothelial progenitor cells (EPC) from bone marrow and their defective function are also key features of diabetes that can cause aberrant neovascularization and hence contribute to the increase of cardiovascular risk (Tepper et al., 2002).
Angiogenesis is a process of formation of new capillary networks in response to reduced oxygen concentration (hypoxia) or pro-angiogenic molecular signals. This mechanism implicates the release of pro-angiogenic factors from both hypoxic endothelial cells and the their surrounding pericytes, which stimulate endothelial cell proliferation and sprouting of new vessels from existing ones (for review see Stapor et al., 2014). This is different from arteriogenesis, a mechanism involving the growth and development of existing arterioles following an acute vascular occlusion (Folkman and Shing, 1992). There are several equally important hypotheses for the mechanisms underpinning disturbances in angiogenesis in diabetes, including a downregulation of VEGF-A signaling response, alterations in inflammatory signals, activation of ER stress response, and accumulation of AGEs initiating oxidative stress and over-production of ROS (Tamarat et al., 2003). Accumulating data indicate that the impact of ROS on the vasculature depends essentially on the oxidative level of the environment. However, the precise amount of ROS needed to cause vascular dysfunction is difficult to predict. Under basal conditions, low ROS levels, especially hydrogen peroxide, can work as intracellular secondary messengers to modulate the action of pro-angiogenic factors such as VEGF-A; however, higher levels of ROS can conversely disturb neovascularization process (Griendling and FitzGerald, 2003a,b).
Several ROS species, including O2-, H2O2, OH- radicals, lipid peroxides, and ONOO-, have been recognized to play major roles in vascular biology controlling the angiogenic process (Tojo et al., 2005). Ebrahimian et al. (2006) reported that diabetes-mediated oxidative stress impaired post-ischemic neovascularization. Authors found that the reduction of ROS production restored key pro-angiogenic signaling pathways (e.g., VEGF-A). Nishikawa et al. (2000) proposed that high glucose-mediated mitochondrial production of ROS can stimulate several molecular mechanisms involved in aberrant angiogenesis. Other studies indicated that O2- release by NOX complex plays a key role in VEGF expression and angiogenesis in a mouse model for diabetic ischemic vascular disease. In addition, increased expression of NADPH subunit NOX2 (gp91phox) has been found to correlate with increases in ROS and decreased VEGF-A in ischemic diabetic retinopathy (Al-Shabrawey et al., 2008). Furthermore, high levels of O2- production by NOX are likely to scavenge NO or cause eNOS uncoupling and hence reduce NO bioavailability (Guzik et al., 2002). These changes in NO signaling pathway may also contribute to the regulation of post-ischemic angiogenic process because of the well-documented role of NO as a key modulator of bone marrow mononuclear cell (MNC) mobilization, differentiation and homing (Aicher et al., 2003). Tepper et al. (2002) reported that the phosphorylation and activation of p38 MAPK by diabetes-induced ROS production leads to impaired differentiation of bone marrow MNC into EPC in vitro and impaired their pro-angiogenic capacity in vivo. Diabetes has also been shown to activate p38 MAPK in vascular wall via signaling pathways that may involve PKC (Igarashi et al., 1999). The activation of p38 MAPK is known to downregulate EPC proliferation and differentiation which may further contribute to impaired angiogenesis in diabetes. Not only has development of oxidative stress with increased production of ROS been reported in in vitro and in vivo diabetic models of impaired angiogenesis, but also the anti-oxidant defense capacity is reduced which can also contribute to diabetes-induced oxidative stress (Wohaieb and Godin, 1987). Previous reports showed that chronic exposure of HUVECs to high glucose caused suppression of kelch-like ECH-associated protein (Keap)-1/erythroid 2–related factor (NrF)-2 pathway which is the major modulator of cytoprotective responses to stresses mediated by ROS including the HO system. The suppression of Keap1/NrF-2 correlated with impaired migration and tube-like formation capacity of HUVECs (Chen et al., 2015) (Figure 8).
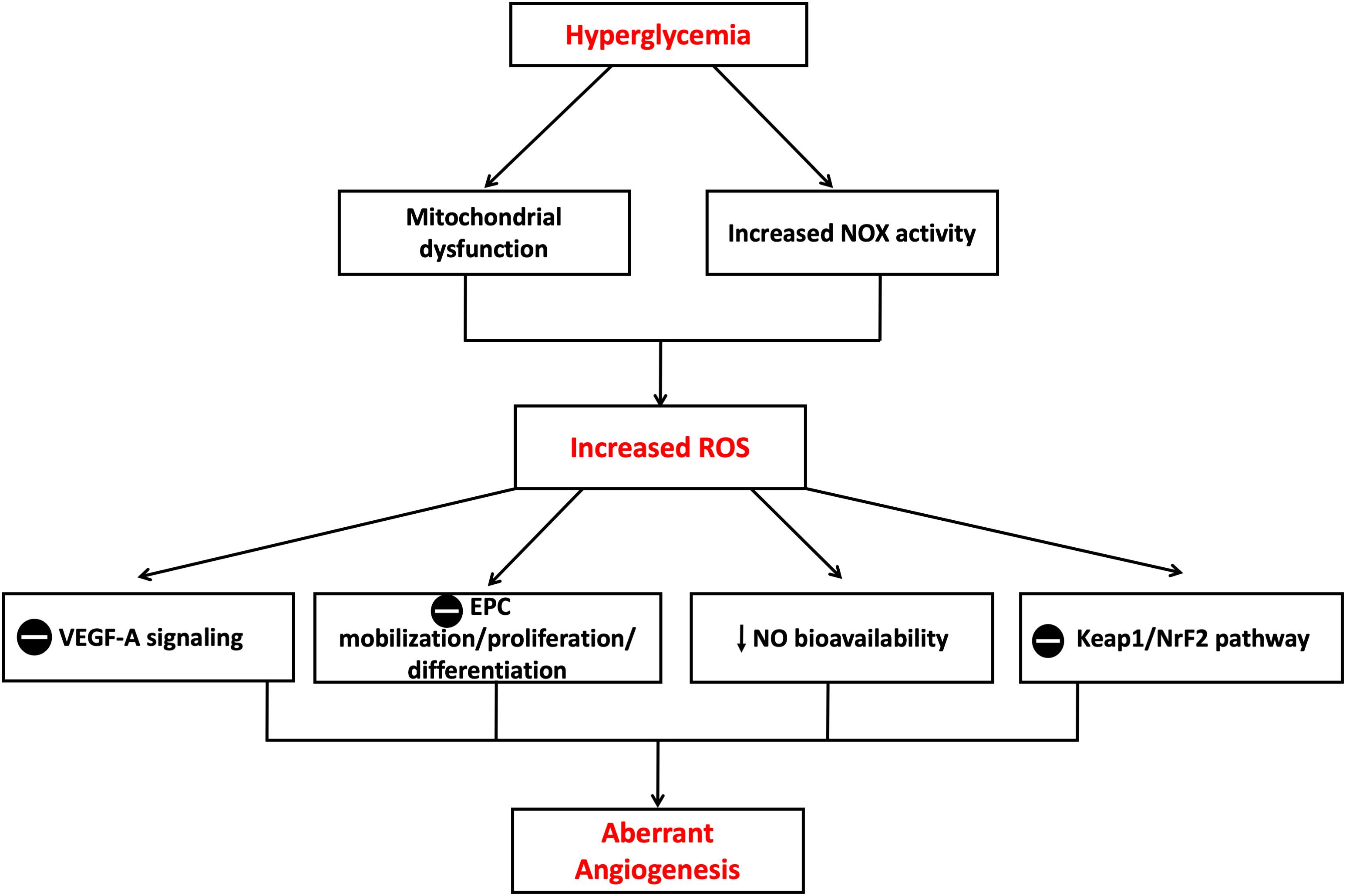
Figure 8. Mechanisms of aberrant angiogenesis induced in hyperglycemia. Increased ROS production in hyperglycemic environment both in vitro and in vivo is believed to cause insult on the angiogenic process. Theories proposed include suppression of VEGF-A signaling, suppression of EPC mobilization, proliferation and differentiation, reduction in NO bioavailability and suppression of Keap1/NrF-2 pathway with resultant suppression in HO-1 expression. EPC, Endothelial progenitor cell; Keap1, Kelch-like ECH-associated protein 1; NrF-2, nuclear factor erythroid 2–related factor 2; VEGF-A, Vascular endothelial growth factor A. The symbol means inhibition.
HO-1 and Its Protective Effects Against Diabetes-Mediated Endothelial Disturbances
Heme oxygenases are a group of anti-oxidant enzymes that play a pivotal role controlling intracellular levels of heme by mediating the biodegradation of heme to release the following byproducts, Fe2+, CO, and biliverdin in mammalian cells (Kikuchi et al., 2005; Ryter et al., 2006; Abraham and Kappas, 2008; Rochette et al., 2018). Biliverdin, a water-soluble molecule, is then metabolized by biliverdin reductase into bilirubin. In diabetes and other cardiovascular disorders such as hypertension, the excess of free heme leads to the formation ROS species, which in turn contribute to the onset of endothelial dysfunction. The HO system, by metabolizing the excess heme, can generate various byproducts, which may initiate different cytoprotective cardiovascular effects: (I) Pro-survival effect on endothelial cells; (II) Reduced inflammation and oxidative stress in vasculature; (III) Improved vascular homeostasis and control of the vascular tone regulation; and (IV) Enhanced neovascularization. Bilirubin and biliverdin are the two main active byproducts of HO action and are considered as the strongest scavengers of ROS in the body; however, CO can have anti-apoptotic and anti-inflammatory roles via the activation of guanylyl cyclase (Stocker et al., 1987; Nitti et al., 2017). CO is capable of suppressing the release of key inflammatory molecules such as of TNF-α and IL-1β and stimulating the synthesis of the human cytokine-synthesis inhibitory factor (CSIF or IL-10) (Otterbein et al., 2000). The last byproduct of heme degradation is free iron, which despite participating in Fenton reaction, which generates the very reactive OH- radical, also activates Fe/ATPase pump, which pumps free iron outside of the cell and enhances protein expression of ferritin which can then scavenge free iron and exert therefore beneficial effects (Crichton et al., 2002).
Three HO isoforms have been reported in humans: HO-1 (32 KDa), which is the inducible form, and two other isoforms with constitutive expression [HO-2 (36 KDa) and a putative HO-3 (33 KDa)] (Nitti et al., 2017). The inducible HO-1, is expressed at very small amounts in normal tissues, except the liver and spleen, where it plays a key role in the elimination process of damaged and aged red blood cells (Gottlieb et al., 2012). Protein expression of HO-1 can be enhanced by many stimuli, which can induce ROS production. Among the pharmacological and chemical stimuli known to induce HO-1, there is heme itself [a byproduct of HO-1 action), heavy metals (e.g., cobalt], inflammatory cytokines, ultraviolet radiation, bacterial membrane component lipopolysaccharide (LPS), hyperglycemia, H2O2, cell growth factors, NO, and CO (Ryter et al., 2006). On the other hand, the expression of HO-2 is not induced by the factors inducing HO-1. However, HO-2 is constitutively expressed in high levels in certain tissues such as the brain, testis, smooth muscle cells and endothelial cells (Chen et al., 2003). In addition to maintaining heme homeostasis, basal amounts of HO-2 also play a key role in anti-oxidant cellular responses by regulating the activation of extracellular SOD, Akt, and ASK-1, through the modulation of heme degradation rate into its biological byproducts. Furthermore, HO-2 plays a pivotal role in sensing levels of oxygen and in cellular response to hypoxia (Munoz-Sanchez and Chanez-Cardenas, 2014). The protective role of HO-2 in hypoxia was demonstrated in endothelial cells. HUVECs and aortic endothelial cells exposed to hypoxia were found to exhibit a stable HO-2 protein expression despite a significant decline in its mRNA expression and a decrease in general protein translation. An increased interaction of HO-2 transcript with polysomes was observed during hypoxic events, leading to a selective activation of HO-2 protein translation, which maintained protein expression levels of HO-2. The maintenance of steady levels of HO-2 protected endothelial cells from apoptosis during hypoxic episodes (He et al., 2010). Finally, HO-3 is encoded by a pseudogene derived from the transcript of HO-2, but so far has only been identified in rats (McCoubrey et al., 1997).
The modulation of HO-1 expression involves a complex molecular interplay between different regulatory mechanisms. MAPK, phosphatidyl inositol 3-kinase (PI-3K)/Akt, protein kinases (PKA, PKC, and PKG) (Stocker and Perrella, 2006), NrF-2, activator protein (AP)-1, NF-κB, cyclic adenosine monophosphate-response element-binding (CREB) protein, and activation transcription factor (ATF)-2 are signaling pathways that have been found to play a role in gene regulation of HO-1 expression (Kim H.P. et al., 2011; Kim Y.M. et al., 2011). Of particular interest, in response to oxidative damage, NrF-2 controls the transcriptional induction of HO-1. NrF-2 is maintained in inactive form in the cytosol by interacting with protein Keap1, which prevents the nuclear translocation of NrF-2 and enhances proteasome-mediated degradation of NrF-2. Following induction of an oxidative stress state, NrF-2 is activated after its dissociation from Keap1; active NrF-2 translocates then to the nucleus and triggers the transcription of HO-1 gene. In support of the role of NrF-2 in controlling HO-1 expression, previous reports have found that fibroblasts from mice deficient in NrF-2 express low levels of HO-1 (Leung et al., 2003; Liu et al., 2007).
Several heavy metal protoporphyrins induce the expression of HO-1; however, Cobalt protoporphyrin (CoPP) is regarded as probably the most potent. CoPP indirectly modulates the production of two factors involved in regulating the expression of HO-1, NrF-2 and transcription regulator protein Bach1. Bach1 is a basic leucine zipper transcription factor that forms heterodimers with proteins from the Maf family and repress the transcription of the HO-1 gene. CoPP enhances the degradation of Bach1 protein. NrF-2 is also a member of the family of basic leucine zipper transcription factors, which is involved in protection against oxidative stress through its antioxidant response element (ARE)-directed induction of HO-1. CoPP also induces the overexpression of NrF-2 by decreasing its degradation, and being a basic leucine zipper transcription factor, the NrF-2 dimerizes with the Maf family transcription factors and together these heterodimers bind to ARE of the HO-1 promotor region resulting in enhancement in expression (Shan et al., 2006). Liu et al. (2013) also reported that CoPP increases the expression of the transcription factor forkhead box protein O1 (FOXO1), which results in transcriptional upregulation of HO-1 gene (Liu et al., 2013).
Besides this mRNA transcriptional activation of HO-1, it was found that HO-1 is also subjected to a post-transcriptional regulatory process which aims at reducing the expression of HO-1 to prevent that its overexpression becomes constitutive. This negative regulation of HO-1 expression is under the control of two microRNA (miR) molecules, miR-217 and miR-377, which work together to reduce the mRNA levels of HO-1 and hence reduce its protein expression. Knocking down these two microRNA molecules abrogated the attenuation of HO-1 expression (Beckman et al., 2011). Lin P.H. et al. (2008) showed that HO-1 expression is also regulated by a post-translational mechanism under the action of the ubiquitin/proteasome system. These findings were further supported by the increased HO-1 expression in the presence of proteasome inhibitor, MG-132, in a model of heme-induced oxidative damage (Chen and Regan, 2005).
Role of HO-1 in Vascular Inflammation
It has been established that HO-1 is able to suppress inflammatory responses by the concomitant generation of CO and bilirubin, two anti-inflammatory molecules, and the removal of pro-inflammatory agent, heme. CO and bilirubin are reported to blunt both innate and adaptive immune responses by modulating the activation and functions of multiple cell types involved in inflammation such as immune cells, endothelial cells and platelets, which ultimately results in reduced recruitment and tissue infiltration of immune cells (Durante, 2011). The anti-inflammatory role of HO-1 was determined in HO-1 knockout mice. In these mice, HO-1 deficiency led to higher release of pro-inflammatory molecules (Kapturczak et al., 2004). In patients undergoing a surgery, higher levels of HO-1 were associated with lower levels of pro-inflammatory cytokine IL-6 (Taha et al., 2010). HO-1 was found to lower leukocyte rolling, adhesion, and transmigration to the sub-intimal space, by blunting the expression and function of surface adhesion molecules. In contrast, the inhibition of HO-1 caused the overexpression and over activation of adhesion molecules, activating thus leukocyte recruitment (Belcher et al., 2006). Multiple preclinical and clinical studies have been performed in many inflammatory vascular diseases to understand the impact of HO-1 overexpression on the pathogenesis and progression of the inflammatory process. Much evidence shows that the induction of HO-1 in the vessel wall inhibited the transmigration of monocytes to the sub-intimal space facilitated by oxidized low-density lipoproteins (LDL), leading to reduced atherosclerotic lesion buildup in mice lacking LDL receptor, a widely used model for atherosclerosis (Ishikawa et al., 2001). Circulating amounts of bilirubin in the general public were found to negatively correlate with the incidence of ischemic accidents (Mayer, 2000). This is not surprising knowing that bilirubin was found to decrease endothelial activation and dysfunction (Kawamura et al., 2005).
Role of HO-1 in Angiogenesis
HO-1 and its gas byproduct CO have been recognized for their strong pro-angiogenic roles. Several pro-angiogenic molecules, including VEGF, were found to mediate their effects via the induction of HO-1. However, depending on the situation, the pro-angiogenic properties of HO-1 could be considered as beneficial or deleterious. For instance, favoring angiogenesis is critical for many physiological responses such as wound healing and neovascularization following an ischemic episode. However, activation of angiogenesis driven by HO-1 may be deleterious in other conditions such neoplastic states (Dulak et al., 2008).
In investigating the role of HO-1 in promoting angiogenesis, Zhao et al. (2011) found that partial deficiency of maternal HO-1 caused defects in the feto-maternal interface, structural defects in placental vasculature and deficiency in spiral artery remodeling. These alterations were not related to the fetal genetic background and were only dependent on the maternal genetic status of HO-1. Moreover, restoring blood flow and the reestablishment of optimal oxygen concentrations to an injury site are also achieved by a proper angiogenic process. The wound-healing sequence of events consists of multiple steps that all require an intact angiogenesis process (Bauer et al., 2005). Growth factors (e.g., VEGF), chemokines, and hypoxia-inducible factors (HIF) all contribute to the orchestration of the multi-step wound healing procedure (Tepper et al., 2005). The deletion of HO-1 in mice led to an impaired wound healing process compared to wild-type littermates. This was partly because of an impaired EPC mobilization capacity linked to capillary formation at the site of injury (Deshane et al., 2007). Furthermore, it has been noted that the natural activation of HO-1 expression in wounded skin areas was weaker and slower in diabetic mice compared to controls. In these animal models of diabetes, the local adenoviral-mediated delivery of HO-1, improved the wound healing process (Grochot-Przeczek et al., 2009). Furthermore, HO-1 was found to confer a cytoprotective role in ischemic cardiac tissue through the activation of protein expression of pro-angiogenic factors in infarcted areas of cardiac tissue (Zeng et al., 2010). Lin H.H. et al. (2008) observed that the transfer of the HO-1 gene to infarcted heart tissue confers protection, at least partly, through the activation of VEGF-A expression and the subsequent activation of angiogenesis. In addition, the overexpression of HO-1 in mesenchymal stem cells implanted in infarcted hearts, markedly reduced apoptosis and fibrosis in cardiac tissue and improved cardiac function and remodeling (Jeon et al., 2007). Recently, Maamoun et al. (2017) have reported that the incubation of HUVECs in intermittent high glucose conditions for 5 consecutive days caused the activation of ER stress response and an impaired capacity of cells to form tube-like structures on a 3-dimensional matrigel matrix. This effect was associated with a decrease in the mRNA and protein expression of VEGF-A, indicating reduced angiogenesis capacity in these cells. Authors found that the pharmacological induction of HO-1 with CoPP reduced ER stress response and prevented the effects of high glucose-mediated ER stress on angiogenic capacity of cells, further stressing the pro-angiogenic effects of HO-1 in endothelial cells (Maamoun et al., 2017).
It is crucial to also highlight that despite the evidence in literature in support of the beneficial role of HO-1 in angiogenesis control, some reports identified a negative impact of HO-1 in tumoral neovascularization. Some human tumors, such as renal cell carcinoma and prostate cancer, exhibit elevated expression levels of HO-1 (Goodman et al., 1997). HO-1 could therefore contribute to tumor cell growth, survival and progression and ultimately participate in the resistance to drug therapy (Nowis et al., 2006). In these conditions, the inhibition of HO-1 reduced tumor development in murine cancer models indicating a potential therapeutic role for HO-1 inhibition in certain cancer types (Dulak et al., 2008). However, there are also some reports that show HO-1 may play an anti-angiogenic role in certain cancer situations. In prostatic cancer cells (PC3), Ferrando et al. (2011) found that the overexpression of HO-1 caused the downregulation of several pro-inflammatory and angiogenic factors (NF-κB, VEGF-A, VEGF-C). To model the in vivo angiogenic process, the authors intradermally inoculated PC3 cells, that had been transfected with a stable HO-1 DNA construct, into immunodeficient mice and found that these cells produced tumors that were less richly vascularized than controls (Ferrando et al., 2011). However, in other conditions driven by excessive angiogenesis such as diabetic retinopathy (Wilkinson-Berka, 2004) and nephropathy (Osterby and Nyberg, 1987), HO-1 induction was still reported to be protective, indicating the complexity of the physiological actions of HO-1. For instance, Fan et al. (2012) observed in streptozotocin-injected diabetic rats pre-injected with hemin, a HO-1 inducer, that retinal ganglion cells were less prone to apoptosis compared to controls (not pre-treated with hemin), suggesting that HO-1 induction in rat retinas protected against diabetes-mediated neurodegeneration. Furthermore, the expression levels of VEGF, a strong pro-angiogenic factor which contributes to exaggerated angiogenesis, was also attenuated in retinas from streptozotocin-injected rats treated with hemin (Fan et al., 2012).
Taken together, data outlined here identify HO-1 as a key modulator of the angiogenic process in different pathophysiological conditions. Therefore, HO-1 induction could be a potential therapeutic target for the inhibition of angiogenesis in carcinogenic conditions, as opposed to being a therapeutic target for the induction of angiogenesis in diabetic cardiovascular complications. These studies also highlight the complexity of signaling pathways underpinning the action of HO-1 in angiogenesis.
Role of HO-1 in Oxidative Stress
The most well-known properties of HO-1 are its anti-oxidant functions (Hayashi et al., 1999). Of particular note, the work conducted by Ishikawa et al. (2012) demonstrated that mice lacking the gene of HO exhibited a weaker anti-oxidant capacity and an extensive lipid peroxidation. The anti-oxidant capacity of HO-1 is due to its byproducts biliverdin and CO. Bilirubin strongly scavenges several oxygen free radicals including singlet oxygen, O2-, ONOO- and RO2 radicals. Moreover, unlike GSH, bilirubin, because of its lipophilic nature, is closely associated with cell membranes, and hence protects them against lipid damage by peroxidation. Bilirubin was found to work better as an O2- scavenger and inhibitor of ONOO--mediated protein nitration than biliverdin (Jansen et al., 2010). In addition to its ROS scavenging properties, some studies proposed that the protective actions of bilirubin are also partly related to its capacity to inhibit inducible NOS (iNOS), which eventually leads to less production of the highly reactive and potent ONOO- free radical (Wang et al., 2004). CO also mediates part of the global anti-oxidant actions of HO-1 (Srisook et al., 2006). CO was reported to block ONOO--mediated protein nitration in endothelial cells through heightened intracellular GSH levels (Li et al., 2007). The anti-oxidant role of HO-1 was further demonstrated recently in HUVECs treated with intermittent high glucose. Authors reported that the induction of HO-1 using CoPP prevented high glucose-mediated concomitant increase in superoxide production and reduction in NO release, indicating an improved NO bioavailability. These focal effects were associated with a decrease in ER stress response activation, reduced phosphorylation of NOX regulatory subunit p47phox at Ser345, and an enhanced phosphorylation of eNOS at its activatory site (Ser1177) (Maamoun et al., 2017).
There is accumulating evidence that oxidative stress contributes to the early stages as well as the maintenance and progression of diabetic cardiovascular complications. Therefore, HO-1, owing to its strong anti-oxidant properties, may be utilized as a viable and promising therapeutic target in these conditions. The protective actions of HO-1 induction have been demonstrated in animal models of diabetic cardiomyopathy and vasculopathy (Li et al., 2012). Moreover, in vitro studies have demonstrated the highly potent anti-oxidant effect of bilirubin with increased protection of vascular endothelial cells, where even concentrations of bilirubin in the nanomolar range protected cells against oxidative stress damage both through direct scavenging of ROS radicals and indirectly by blocking the activity of NOX complex (Abraham and Kappas, 2008).
Role of HO-1 in Apoptosis
A high percentage of apoptotic endothelial cells was observed in animal models of diabetes-mediated vascular injury (Negi et al., 2011). Clinical data also suggest the involvement of apoptosis in diabetic vascular diseases where the expression of pro-apoptotic molecules was high while in contrast the expression of anti-apoptotic ones was low (Arya et al., 2011). HO-1 mediates its anti-apoptotic action through multiple molecular mechanisms including the inhibition of oxidative stress, ER stress and inflammation. NF-κB was shown to control cellular apoptosis in addition to its well-known role in inflammation. HO-1 induction by CoPP was found to reduce the expression of IL-6, a gene regulated by NF-κB, and CHOP in response to stressful stimuli such as ischemia-reperfusion injury, with the consequent inhibition of apoptosis (Ben-Ari et al., 2013). Other mechanisms suggested for the anti-apoptotic effects of HO-1 include blunting the apoptotic signaling pathway mediated by the interaction of TNF-α with its receptor (Jaeschke and Woolbright, 2013). These anti-apoptotic actions of HO-1 may be driven by the release of CO, which was shown to protect against apoptosis in many cell types, including endothelial cells, fibroblasts, hepatocytes and pancreatic β-cells. Brouard et al. (2000) were able to show that HO-1 inhibits apoptosis, namely by generating CO. The inhibition of HO-1 activity by tin protoporphyrin (SnPPIX) failed to prevent endothelial cell apoptosis. The direct effect of CO on apoptosis was confirmed by showing that exposing endothelial cells to exogenous CO, while inhibiting HO-1 activity by SnPPIX, prevented apoptosis. Moreover, investigators have also explored some of the signaling pathways behind the action of HO-1/CO, and showed that it is partially mediated by the activation of p38 MAPK signaling transduction pathway. This was further supported by the observation that the selective blockade of p38 MAPK by the over-expression of a dominant negative mutant resulted in the abolishment of the anti-apoptotic effect of HO-1 (Brouard et al., 2000). In addition, as previously discussed, in diabetic vascular disease, oxidative stress occurs in endothelial cells which subsequently leads to apoptosis. Hence, HO-1 by merely exerting its anti-oxidant effects is reported to protect these cells from apoptosis (Schmeichel et al., 2003). Other studies focused on the role of bilirubin in this pro-survival behavior of HO-1. It has been shown that bilirubin was found to mediate its anti-apoptotic role by upregulating extracellular-signal-regulated kinase (ERK) MAPK and PI-3K/Akt/eNOS pathways (Hung et al., 2010).
More recently, pharmacological induction of HO-1 using CoPP was reported to protect endothelial cells from high glucose-mediated apoptosis through the alleviation of ER stress response. Maamoun et al. (2017) observed that the exposure of HUVECs to intermittent high glucose for a duration of 5 days led to cell death, while induction of HO-1 prevented this deleterious manifestation. The cytoprotective effects of HO-1 were associated with a reduction in the activation of PARP-1 and caspases 3 and 7. Furthermore, HO-1 induction in HUVECs prevented the activation of NF-κB and JNK pathways caused by high glucose treatment, indicating reduced cellular inflammation in the presence of HO-1 (Maamoun et al., 2017). While the amount of scientific evidence is still relatively small, some studies suggest HO-1 activation as a novel and promising strategy to improve survival of endothelial cells through HO-1 byproducts, bilirubin and CO, which may delay the development of various diabetic cardiovascular complications.
Conclusion
Crosstalk between oxidative stress and ER stress significantly contributes to the onset and development of endothelial dysfunction and altered angiogenesis and therefore represent major therapeutic targets for alleviating micro- and macrovascular complications associated with this metabolic disturbance. HO-1 carries out, anti-oxidant, anti-inflammatory, anti-apoptotic and angiogenic actions, through its by-products CO and bilirubin, and can thus affect therefore multiple cellular pathways involved in endothelial dysfunction, including oxidative stress and ER stress response. As a multi-target molecule affecting major aspects of cardiovascular dysfunction, HO-1 induction represents a potential therapeutic approach for cardiovascular complications associated with diabetes.
Author Contributions
HM and AA wrote the manuscript and constructed the figures. TB, GP, and SM contributed to selected sections and critically revised the manuscript. All authors approved the final version for submission.
Funding
This publication was made possible by a NPRP award (NPRP8-1750-3-360) from the Qatar National Research Fund (a member of Qatar Foundation) and a Qatar University high collaborative grant (QUCG-CPH-2018\2019-2) to AA. The publication of this article was funded by Qatar National Library.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures in this article were partly produced by adapting freely available tools from Servier Medical Art, that are licensed under a Creative Commons Attribution 3.0 Unported License https://creativecommons.org/licenses/by/3.0/. This manuscript was based on the doctoral thesis of HM.
References
Abaci, A., Oguzhan, A., Eryol, N. K., and Ergin, A. (1999). Effect of potential confounding factors on the thrombolysis in myocardial infarction (TIMI) trial frame count and its reproducibility. Circulation 100, 2219–2223. doi: 10.1161/01.CIR.100.22.2219
Abraham, N. G., and Kappas, A. (2008). Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 60, 79–127. doi: 10.1124/pr.107.07104
Agouni, A., Mody, N., Owen, C., Czopek, A., Zimmer, D., Bentires-Alj, M., et al. (2011). Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem. J. 438, 369–378. doi: 10.1042/BJ20110373
Agouni, A., Tual-Chalot, S., Chalopin, M., Duluc, L., Mody, N., Martinez, M. C., et al. (2014). Hepatic protein tyrosine phosphatase 1B (PTP1B) deficiency protects against obesity-induced endothelial dysfunction. Biochem. Pharmacol. 92, 607–617. doi: 10.1016/j.bcp.2014.10.008
Aicher, A., Heeschen, C., Mildner-Rihm, C., Urbich, C., Ihling, C., Technau-Ihling, K., et al. (2003). Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 9, 1370–1376. doi: 10.1038/nm948
Allaman, I., Belanger, M., and Magistretti, P. J. (2015). Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9:23. doi: 10.3389/fnins.2015.00023
Al-Shabrawey, M., Rojas, M., Sanders, T., Behzadian, A., El-Remessy, A., Bartoli, M., et al. (2008). Role of NADPH oxidase in retinal vascular inflammation. Invest. Ophthalmol. Vis. Sci. 49, 3239–3244. doi: 10.1167/iovs.08-1755
Amin, A., Choi, S. K., Galan, M., Kassan, M., Partyka, M., Kadowitz, P., et al. (2012). Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J. Pathol. 227, 165–174. doi: 10.1002/path.3960
Andreozzi, F., Laratta, E., Procopio, C., Hribal, M. L., Sciacqua, A., Perticone, M., et al. (2007). Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol. Cell Biol. 27, 2372–2383. doi: 10.1128/MCB.01340-06
Arya, A. K., Pokharia, D., and Tripathi, K. (2011). Relationship between oxidative stress and apoptotic markers in lymphocytes of diabetic patients with chronic non healing wound. Diabetes Res. Clin. Pract. 94, 377–384. doi: 10.1016/j.diabres.2011.08.004
Awede, B., Lemaire, M. C., Hyvelin, J. M., Halimi, J. M., Bonnet, P., and Eder, V. (2010). Hemin, a carbon monoxide donor, improves systemic vascular compliance by inhibiting the RhoA-Rhokinase pathway in spontaneous hypertensive rats. Eur. J. Pharmacol. 626, 256–261. doi: 10.1016/j.ejphar.2009.09.045
Banerjee, S., Jia, Y., Siburt, C. J., Abraham, B., Wood, F., Bonaventura, C., et al. (2012). Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic. Biol. Med. 53, 1317–1326. doi: 10.1016/j.freeradbiomed.2012.07.023
Bauer, S. M., Bauer, R. J., and Velazquez, O. C. (2005). Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc. Endovascular Surg. 39, 293–306. doi: 10.1177/153857440503900401
Beckman, J. D., Chen, C., Nguyen, J., Thayanithy, V., Subramanian, S., Steer, C. J., et al. (2011). Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J. Biol. Chem. 286, 3194–3202. doi: 10.1074/jbc.M110.148726
Belcher, J. D., Mahaseth, H., Welch, T. E., Otterbein, L. E., Hebbel, R. P., and Vercellotti, G. M. (2006). Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J. Clin. Invest. 116, 808–816. doi: 10.1172/JCI26857
Ben-Ari, Z., Issan, Y., Katz, Y., Sultan, M., Safran, M., Michal, L. S., et al. (2013). Induction of heme oxygenase-1 protects mouse liver from apoptotic ischemia/reperfusion injury. Apoptosis 18, 547–555. doi: 10.1007/s10495-013-0814-x
Bierhaus, A., Schiekofer, S., Schwaninger, M., Andrassy, M., Humpert, P. M., Chen, J., et al. (2001). Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50, 2792–2808. doi: 10.2337/diabetes.50.12.2792
Bishara, N. B., and Ding, H. (2010). Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 298, H171–H178. doi: 10.1152/ajpheart.00699.2009
Brouard, S., Otterbein, L. E., Anrather, J., Tobiasch, E., Bach, F. H., Choi, A. M., et al. (2000). Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 192, 1015–1026. doi: 10.1084/jem.192.7.1015
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820. doi: 10.1038/414813a
Cade, W. T. (2008). Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 88, 1322–1335. doi: 10.2522/ptj.20080008
Chakravarthi, S., and Bulleid, N. J. (2004). Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 279, 39872–39879. doi: 10.1074/jbc.M406912200
Chang, C. F., Liu, X. M., Peyton, K. J., and Durante, W. (2014). Heme oxygenase-1 counteracts contrast media-induced endothelial cell dysfunction. Biochem. Pharmacol. 87, 303–311. doi: 10.1016/j.bcp.2013.11.002
Chen, B., Lu, Y., Chen, Y., and Cheng, J. (2015). The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 225, R83–R99. doi: 10.1530/JOE-14-0662
Chen, J., and Regan, R. F. (2005). Increasing expression of heme oxygenase-1 by proteasome inhibition protects astrocytes from heme-mediated oxidative injury. Curr. Neurovasc. Res. 2, 189–196. doi: 10.2174/1567202054368344
Chen, Y., Wang, J. J., Li, J., Hosoya, K. I., Ratan, R., Townes, T., et al. (2012). Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 55, 2533–2545. doi: 10.1007/s00125-012-2594-1
Chen, Y. H., Yet, S. F., and Perrella, M. A. (2003). Role of heme oxygenase-1 in the regulation of blood pressure and cardiac function. Exp. Biol. Med. 228, 447–453. doi: 10.1177/15353702-0322805-03
Cimellaro, A., Perticone, M., Fiorentino, T. V., Sciacqua, A., and Hribal, M. L. (2016). Role of endoplasmic reticulum stress in endothelial dysfunction. Nutr. Metab. Cardiovasc. Dis. 26, 863–871. doi: 10.1016/j.numecd.2016.05.008
Colgan, S. M., Tang, D., Werstuck, G. H., and Austin, R. C. (2007). Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 39, 1843–1851. doi: 10.1016/j.biocel.2007.05.002
Costa, P. Z., and Soares, R. (2013). Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 92, 1037–1045. doi: 10.1016/j.lfs.2013.04.001
Creager, M. A., Luscher, T. F., Cosentino, F., and Beckman, J. A. (2003). Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Circulation 108, 1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32
Crichton, R. R., Wilmet, S., Legssyer, R., and Ward, R. J. (2002). Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J. Inorg. Biochem. 91, 9–18. doi: 10.1016/S0162-0134(02)00461-0
Cunha, D. A., Hekerman, P., Ladriere, L., Bazarra-Castro, A., Ortis, F., Wakeham, M. C., et al. (2008). Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 121(Pt 14), 2308–2318. doi: 10.1242/jcs.026062
Dasu, M. R., Devaraj, S., Zhao, L., Hwang, D. H., and Jialal, I. (2008). High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes 57, 3090–3098. doi: 10.2337/db08-0564
Deshane, J., Chen, S., Caballero, S., Grochot-Przeczek, A., Was, H., Li Calzi, S., et al. (2007). Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 204, 605–618. doi: 10.1084/jem.20061609
Ding, H., and Triggle, C. R. (2010). Endothelial dysfunction in diabetes: multiple targets for treatment. Pflugers Arch. 459, 977–994. doi: 10.1007/s00424-010-0807-3
Drummond, G. R., and Sobey, C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25, 452–463. doi: 10.1016/j.tem.2014.06.012
Du, X., Matsumura, T., Edelstein, D., Rossetti, L., Zsengeller, Z., Szabo, C., et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest. 112, 1049–1057. doi: 10.1172/JCI18127
Duckers, H. J., Boehm, M., True, A. L., Yet, S. F., San, H., Park, J. L., et al. (2001). Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 7, 693–698. doi: 10.1038/89068
Dulak, J., Deshane, J., Jozkowicz, A., and Agarwal, A. (2008). Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117, 231–241. doi: 10.1161/CIRCULATIONAHA.107.698316
Durante, W. (2003). Heme oxygenase-1 in growth control and its clinical application to vascular disease. J. Cell Physiol. 195, 373–382. doi: 10.1002/jcp.10274
Durante, W. (2011). Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 16, 2372–2388. doi: 10.2741/3860
Ebrahimian, T. G., Heymes, C., You, D., Blanc-Brude, O., Mees, B., Waeckel, L., et al. (2006). NADPH oxidase-derived overproduction of reactive oxygen species impairs postischemic neovascularization in mice with type 1 diabetes. Am. J. Pathol. 169, 719–728. doi: 10.2353/ajpath.2006.060042
Fan, J., Xu, G., Jiang, T., and Qin, Y. (2012). Pharmacologic induction of heme oxygenase-1 plays a protective role in diabetic retinopathy in rats. Invest. Ophthalmol. Vis. Sci. 53, 6541–6556. doi: 10.1167/iovs.11-9241
Ferrando, M., Gueron, G., Elguero, B., Giudice, J., Salles, A., Leskow, F. C., et al. (2011). Heme oxygenase 1 (HO-1) challenges the angiogenic switch in prostate cancer. Angiogenesis 14, 467–479. doi: 10.1007/s10456-011-9230-4
Flamment, M., Hajduch, E., Ferre, P., and Foufelle, F. (2012). New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 23, 381–390. doi: 10.1016/j.tem.2012.06.003
Galan, M., Kassan, M., Choi, S. K., Partyka, M., Trebak, M., Henrion, D., et al. (2012). A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension 60, 71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500
Galan, M., Kassan, M., Kadowitz, P. J., Trebak, M., Belmadani, S., and Matrougui, K. (2014). Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta 1843, 1063–1075. doi: 10.1016/j.bbamcr.2014.02.009
Galiano, R. D., Tepper, O. M., Pelo, C. R., Bhatt, K. A., Callaghan, M., Bastidas, N., et al. (2004). Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 164, 1935–1947. doi: 10.1016/S0002-9440(10)63754-6
Garcia-Mata, C., and Lamattina, L. (2013). Gasotransmitters are emerging as new guard cell signaling molecules and regulators of leaf gas exchange. Plant Sci. 201–202, 66–73. doi: 10.1016/j.plantsci.2012.11.007
Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5:a013169. doi: 10.1101/cshperspect.a013169
Gardner, B. M., and Walter, P. (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894. doi: 10.1126/science.1209126
Ghosh, R., Lipson, K. L., Sargent, K. E., Mercurio, A. M., Hunt, J. S., Ron, D., et al. (2010). Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One 5:e9575. doi: 10.1371/journal.pone.0009575
Giri, B., Dey, S., Das, T., Sarkar, M., Banerjee, J., and Dash, S. K. (2018). Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: an update on glucose toxicity. Biomed. Pharmacother. 107, 306–328. doi: 10.1016/j.biopha.2018.07.157
Goodman, A. I., Choudhury, M., da Silva, J. L., Schwartzman, M. L., and Abraham, N. G. (1997). Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc. Soc. Exp. Biol. Med. 214, 54–61. doi: 10.3181/00379727-214-44069
Gorlach, A., Klappa, P., and Kietzmann, T. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418. doi: 10.1089/ars.2006.8.1391
Gottlieb, Y., Truman, M., Cohen, L. A., Leichtmann-Bardoogo, Y., and Meyron-Holtz, E. G. (2012). Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 97, 1489–1493. doi: 10.3324/haematol.2012.063651
Griendling, K. K., and FitzGerald, G. A. (2003a). Oxidative stress and cardiovascular injury: part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108, 1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB
Griendling, K. K., and FitzGerald, G. A. (2003b). Oxidative stress and cardiovascular injury: part II: animal and human studies. Circulation 108, 2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4
Grochot-Przeczek, A., Lach, R., Mis, J., Skrzypek, K., Gozdecka, M., Sroczynska, P., et al. (2009). Heme oxygenase-1 accelerates cutaneous wound healing in mice. PLoS One 4:e5803. doi: 10.1371/journal.pone.0005803
Guzik, T. J., Mussa, S., Gastaldi, D., Sadowski, J., Ratnatunga, C., Pillai, R., et al. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105, 1656–1662. doi: 10.1161/01.CIR.0000012748.58444.08
Hassler, J., Cao, S. S., and Kaufman, R. J. (2012). IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev. Cell 23, 921–923. doi: 10.1016/j.devcel.2012.10.025
Hayashi, S., Takamiya, R., Yamaguchi, T., Matsumoto, K., Tojo, S. J., Tamatani, T., et al. (1999). Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ. Res. 85, 663–671. doi: 10.1161/01.RES.85.8.663
He, J. Z., Ho, J. J., Gingerich, S., Courtman, D. W., Marsden, P. A., and Ward, M. E. (2010). Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J. Biol. Chem. 285, 9452–9461. doi: 10.1074/jbc.M109.077230
Hirata, Y., Nagata, D., Suzuki, E., Nishimatsu, H., Suzuki, J., and Nagai, R. (2010). Diagnosis and treatment of endothelial dysfunction in cardiovascular disease. Int. Heart J. 51, 1–6. doi: 10.1536/ihj.51.1
Hu, M., Phan, F., Bourron, O., Ferre, P., and Foufelle, F. (2017). Steatosis and NASH in type 2 diabetes. Biochimie 143, 37–41. doi: 10.1016/j.biochi.2017.10.019
Hung, S. Y., Liou, H. C., and Fu, W. M. (2010). The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 58, 321–329. doi: 10.1016/j.neuropharm.2009.11.003
Hyvelin, J. M., Maurel, B., Uzbekov, R., Motterlini, R., and Lermusiaux, P. (2010). Hemin prevents in-stent stenosis in rat and rabbit models by inducing heme-oxygenase-1. J. Vasc. Surg. 51, 417–428. doi: 10.1016/j.jvs.2009.09.004
Igarashi, M., Wakasaki, H., Takahara, N., Ishii, H., Jiang, Z. Y., Yamauchi, T., et al. (1999). Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J. Clin. Invest. 103, 185–195. doi: 10.1172/JCI3326
Incalza, M. A., D’Oria, R., Natalicchio, A., Perrini, S., Laviola, L., and Giorgino, F. (2018). Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 100, 1–19. doi: 10.1016/j.vph.2017.05.005
Inoguchi, T., Li, P., Umeda, F., Yu, H. Y., Kakimoto, M., Imamura, M., et al. (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945. doi: 10.2337/diabetes.49.11.1939
Ishikawa, K., Navab, M., and Lusis, A. J. (2012). Vasculitis, atherosclerosis, and altered HDL composition in heme-oxygenase-1-knockout mice. Int. J. Hypertens. 2012:948203. doi: 10.1155/2012/948203
Ishikawa, K., Sugawara, D., Wang, X., Suzuki, K., Itabe, H., Maruyama, Y., et al. (2001). Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ. Res. 88, 506–512. doi: 10.1161/01.RES.88.5.506
Jaeschke, H., and Woolbright, B. L. (2013). Role of heme oxygenase 1 in TNF/TNF receptor-mediated apoptosis after hepatic ischemia/reperfusion in rats. Shock 39: 380-388, 2013. Shock 40, 75–76. doi: 10.1097/SHK.0b013e3182971d2b
Jamwal, S., and Sharma, S. (2018). Vascular endothelium dysfunction: a conservative target in metabolic disorders. Inflamm. Res. 67, 391–405. doi: 10.1007/s00011-018-1129-8
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., et al. (2010). Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell. Cardiol. 49, 186–195. doi: 10.1016/j.yjmcc.2010.04.011
Jeon, O., Song, S. J., Bhang, S. H., Choi, C. Y., Kim, M. J., and Kim, B. S. (2007). Additive effect of endothelial progenitor cell mobilization and bone marrow mononuclear cell transplantation on angiogenesis in mouse ischemic limbs. J. Biomed. Sci. 14, 323–330. doi: 10.1007/s11373-007-9145-7
Jiang, H. Y., Wek, S. A., McGrath, B. C., Scheuner, D., Kaufman, R. J., Cavener, D. R., et al. (2003). Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell Biol. 23, 5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003
Kapturczak, M. H., Wasserfall, C., Brusko, T., Campbell-Thompson, M., Ellis, T. M., Atkinson, M. A., et al. (2004). Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 165, 1045–1053. doi: 10.1016/S0002-9440(10)63365-2
Kawamura, K., Ishikawa, K., Wada, Y., Kimura, S., Matsumoto, H., Kohro, T., et al. (2005). Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 25, 155–160. doi: 10.1161/01.ATV.0000148405.18071.6a
Khazaei, M., Moien-Afshari, F., and Laher, I. (2008). Vascular endothelial function in health and diseases. Pathophysiology 15, 49–67. doi: 10.1016/j.pathophys.2008.02.002
Kikuchi, G., Yoshida, T., and Noguchi, M. (2005). Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 338, 558–567. doi: 10.1016/j.bbrc.2005.08.020
Kim, H. P., Pae, H. O., Back, S. H., Chung, S. W., Woo, J. M., Son, Y., et al. (2011). Heme oxygenase-1 comes back to endoplasmic reticulum. Biochem. Biophys. Res. Commun. 404, 1–5. doi: 10.1016/j.bbrc.2010.11.067
Kim, Y. M., Pae, H. O., Park, J. E., Lee, Y. C., Woo, J. M., Kim, N. H., et al. (2011). Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 14, 137–167. doi: 10.1089/ars.2010.3153
Kornfeld, O. S., Hwang, S., Disatnik, M. H., Chen, C. H., Qvit, N., and Mochly-Rosen, D. (2015). Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ. Res. 116, 1783–1799. doi: 10.1161/CIRCRESAHA.116.305432
Kota, S. K., Meher, L. K., Jammula, S., Kota, S. K., Krishna, S. V., and Modi, K. D. (2012). Aberrant angiogenesis: the gateway to diabetic complications. Indian J. Endocrinol. Metab. 16, 918–930. doi: 10.4103/2230-8210.102992
Lee, T. S., and Chau, L. Y. (2002). Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 8, 240–246. doi: 10.1038/nm0302-240
Lerman, A., and Burnett, J. C. (1992). Intact and altered endothelium in regulation of vasomotion. Circulation 86(Suppl. 6), III12–III19.
Leung, L., Kwong, M., Hou, S., Lee, C., and Chan, J. Y. (2003). Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 278, 48021–48029. doi: 10.1074/jbc.M308439200
Li, B., Liu, S., Miao, L., and Cai, L. (2012). Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy. Exp. Diabetes Res. 2012:216512. doi: 10.1155/2012/216512
Li, M. H., Jang, J. H., Na, H. K., Cha, Y. N., and Surh, Y. J. (2007). Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J. Biol. Chem. 282, 28577–28586. doi: 10.1074/jbc.M701916200
Li, T., Tian, H., Zhao, Y., An, F., Zhang, L., Zhang, J., et al. (2011). Heme oxygenase-1 inhibits progression and destabilization of vulnerable plaques in a rabbit model of atherosclerosis. Eur. J. Pharmacol. 672, 143–152. doi: 10.1016/j.ejphar.2011.09.188
Lin, H. H., Chen, Y. H., Chang, P. F., Lee, Y. T., Yet, S. F., and Chau, L. Y. (2008). Heme oxygenase-1 promotes neovascularization in ischemic heart by coinduction of VEGF and SDF-1. J. Mol. Cell. Cardiol. 45, 44–55. doi: 10.1016/j.yjmcc.2008.04.011
Lin, P. H., Chiang, M. T., and Chau, L. Y. (2008). Ubiquitin-proteasome system mediates heme oxygenase-1 degradation through endoplasmic reticulum-associated degradation pathway. Biochim. Biophys. Acta 1783, 1826–1834. doi: 10.1016/j.bbamcr.2008.05.008
Lisa, S., Domingo, B., Martinez, J., Gilch, S., Llopis, J. F., Schatzl, H. M., et al. (2012). Failure of prion protein oxidative folding guides the formation of toxic transmembrane forms. J. Biol. Chem. 287, 36693–36701. doi: 10.1074/jbc.M112.398776
Liu, X., Cui, Y., Li, M., Xu, H., Zuo, J., Fang, F., et al. (2013). Cobalt protoporphyrin induces HO-1 expression mediated partially by FOXO1 and reduces mitochondria-derived reactive oxygen species production. PLoS One 8:e80521. doi: 10.1371/journal.pone.0080521
Liu, X. M., Peyton, K. J., Ensenat, D., Wang, H., Hannink, M., Alam, J., et al. (2007). Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 75, 381–389. doi: 10.1016/j.cardiores.2007.03.004
Liu, Y., Lei, S., Gao, X., Mao, X., Wang, T., Wong, G. T., et al. (2012). PKCbeta inhibition with ruboxistaurin reduces oxidative stress and attenuates left ventricular hypertrophy and dysfunction in rats with streptozotocin-induced diabetes. Clin. Sci. 122, 161–173. doi: 10.1042/CS20110176
Love, D. C., and Hanover, J. A. (2005). The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci. Stke 2005:re13. doi: 10.1126/stke.3122005re13
Lu, H., Bogdanovic, E., Yu, Z., Cho, C., Liu, L., Ho, K., et al. (2018). Combined hyperglycemia- and hyperinsulinemia-induced insulin resistance in adipocytes is associated with dual signaling defects mediated by PKC-zeta. Endocrinology 159, 1658–1677. doi: 10.1210/en.2017-00312
Maamoun, H., Zachariah, M., McVey, J. H., Green, F. R., and Agouni, A. (2017). Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 127, 46–59. doi: 10.1016/j.bcp.2016.12.009
Maessen, D. E., Stehouwer, C. D., and Schalkwijk, C. G. (2015). The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 128, 839–861. doi: 10.1042/CS20140683
Mayer, M. (2000). Association of serum bilirubin concentration with risk of coronary artery disease. Clin. Chem. 46, 1723–1727.
McCoubrey, W. K., Huang, T. J., and Maines, M. D. (1997). Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur. J. Biochem. 247, 725–732. doi: 10.1111/j.1432-1033.1997.00725.x
Michiels, C. (2003). Endothelial cell functions. J. Cell Physiol. 196, 430–443. doi: 10.1002/jcp.10333
Mishra, M., and Ndisang, J. F. (2014). A critical and comprehensive insight on heme oxygenase and related products including carbon monoxide, bilirubin, biliverdin and ferritin in type-1 and type-2 diabetes. Curr. Pharm. Des. 20, 1370–1391. doi: 10.2174/13816128113199990559
Morgan, M. J., Kim, Y. S., and Liu, Z. G. (2008). TNF alpha and reactive oxygen species in necrotic cell death. Cell Res. 18, 343–349. doi: 10.1038/cr.2008.31
Morse, D., Pischke, S. E., Zhou, Z., Davis, R. J., Flavell, R. A., Loop, T., et al. (2003). Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 278, 36993–36998. doi: 10.1074/jbc.M302942200
Munoz-Sanchez, J., and Chanez-Cardenas, M. E. (2014). A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014:604981. doi: 10.1155/2014/604981
Mustafa, A. K., Gadalla, M. M., and Snyder, S. H. (2009). Signaling by gasotransmitters. Sci. Signal. 2:re2. doi: 10.1126/scisignal.268re2
Ndisang, J. F., and Jadhav, A. (2009). Heme oxygenase system enhances insulin sensitivity and glucose metabolism in streptozotocin-induced diabetes. Am. J. Physiol. Endocrinol. Metab. 296, E829–E841. doi: 10.1152/ajpendo.90783.2008
Negi, G., Kumar, A., and Sharma, S. S. (2011). Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kappaB and Nrf2 cascades. J. Pineal Res. 50, 124–131. doi: 10.1111/j.1600-079X.2010.00821.x
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., et al. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404, 787–790. doi: 10.1038/35008121
Nitti, M., Piras, S., Marinari, U. M., Moretta, L., Pronzato, M. A., and Furfaro, A. L. (2017). HO-1 induction in cancer progression: a matter of cell adaptation. Antioxidants 6:E29. doi: 10.3390/antiox6020029
Nowis, D., Legat, M., Grzela, T., Niderla, J., Wilczek, E., Wilczynski, G. M., et al. (2006). Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 25, 3365–3374. doi: 10.1038/sj.onc.1209378
Osterby, R., and Nyberg, G. (1987). New vessel formation in the renal corpuscles in advanced diabetic glomerulopathy. J. Diabet. Complications 1, 122–127. doi: 10.1016/S0891-6632(87)80069-7
Otterbein, L. E., Bach, F. H., Alam, J., Soares, M., Tao Lu, H., Wysk, M., et al. (2000). Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 6, 422–428. doi: 10.1038/74680
Owen, C., Lees, E. K., Grant, L., Zimmer, D. J., Mody, N., Bence, K. K., et al. (2013). Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 56, 2286–2296. doi: 10.1007/s00125-013-2992-z
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. doi: 10.1126/science.1103160
Paridaens, A., Laukens, D., Vandewynckel, Y. P., Coulon, S., Van Vlierberghe, H., Geerts, A., et al. (2014). Endoplasmic reticulum stress and angiogenesis: is there an interaction between them? Liver Int. 34, e10–e18. doi: 10.1111/liv.12457
Perisic, O., Wilson, M. I., Karathanassis, D., Bravo, J., Pacold, M. E., Ellson, C. D., et al. (2004). The role of phosphoinositides and phosphorylation in regulation of NADPH oxidase. Adv. Enzyme Regul. 44, 279–298. doi: 10.1016/j.advenzreg.2003.11.003
Pieper, G. M., Langenstroer, P., and Siebeneich, W. (1997). Diabetic-induced endothelial dysfunction in rat aorta: role of hydroxyl radicals. Cardiovasc. Res. 34, 145–156. doi: 10.1016/S0008-6363(96)00237-4
Pitocco, D., Zaccardi, F., Di Stasio, E., Romitelli, F., Santini, S. A., Zuppi, C., et al. (2010). Oxidative stress, nitric oxide, and diabetes. Rev. Diabet. Stud. 7, 15–25. doi: 10.1900/RDS.2010.7.15
Poss, K. D., and Tonegawa, S. (1997a). Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. U.S.A. 94, 10919–10924.
Poss, K. D., and Tonegawa, S. (1997b). Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10925–10930.
Rajapakse, A. G., Ming, X. F., Carvas, J. M., and Yang, Z. (2009a). O-linked beta-N-acetylglucosamine during hyperglycemia exerts both anti-inflammatory and pro-oxidative properties in the endothelial system. Oxid. Med. Cell. Longev. 2, 172–175.
Rajapakse, A. G., Ming, X. F., Carvas, J. M., and Yang, Z. (2009b). The hexosamine biosynthesis inhibitor azaserine prevents endothelial inflammation and dysfunction under hyperglycemic condition through antioxidant effects. Am. J. Physiol. Heart Circ. Physiol. 296, H815–H822. doi: 10.1152/ajpheart.00756.2008
Rochette, L., Zeller, M., Cottin, Y., and Vergely, C. (2014). Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 1840, 2709–2729. doi: 10.1016/j.bbagen.2014.05.017
Rochette, L., Zeller, M., Cottin, Y., and Vergely, C. (2018). Redox functions of heme oxygenase-1 and biliverdin reductase in diabetes. Trends Endocrinol. Metab. 29, 74–85. doi: 10.1016/j.tem.2017.11.005
Rolo, A. P., and Palmeira, C. M. (2006). Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 212, 167–178. doi: 10.1016/j.taap.2006.01.003
Ryter, S. W., Alam, J., and Choi, A. M. (2006). Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650. doi: 10.1152/physrev.00011.2005
Salazar, G. (2018). NADPH oxidases and mitochondria in vascular senescence. Int. J. Mol. Sci. 19:E1327. doi: 10.3390/ijms19051327
Schmeichel, A. M., Schmelzer, J. D., and Low, P. A. (2003). Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 52, 165–171. doi: 10.2337/diabetes.52.1.165
Schulz, E., Gori, T., and Munzel, T. (2011). Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 34, 665–673. doi: 10.1038/hr.2011.39
Searles, C. D. (2006). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 291, C803–C816. doi: 10.1152/ajpcell.00457.2005
Shan, Y., Lambrecht, R. W., Donohue, S. E., and Bonkovsky, H. L. (2006). Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. 20, 2651–2653. doi: 10.1096/fj.06-6346fje
Sheikh-Ali, M., Sultan, S., Alamir, A. R., Haas, M. J., and Mooradian, A. D. (2010). Hyperglycemia-induced endoplasmic reticulum stress in endothelial cells. Nutrition 26, 1146–1150. doi: 10.1016/j.nut.2009.08.019
Shen, G. X. (2010). Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Can. J. Physiol. Pharmacol. 88, 241–248. doi: 10.1139/Y10-018
Shen, Y. H., Zhang, L., Utama, B., Wang, J., Gan, Y., Wang, X., et al. (2006). Human cytomegalovirus inhibits Akt-mediated eNOS activation through upregulating PTEN (phosphatase and tensin homolog deleted on chromosome 10). Cardiovasc. Res. 69, 502–511. doi: 10.1016/j.cardiores.2005.10.007
Simons, M. (2005). Angiogenesis, arteriogenesis, and diabetes: paradigm reassessed? J. Am. Coll. Cardiol. 46, 835–837. doi: 10.1016/j.jacc.2005.06.008
Son, S. M. (2012). Reactive oxygen and nitrogen species in pathogenesis of vascular complications of diabetes. Diabetes Metab. J. 36, 190–198. doi: 10.4093/dmj.2012.36.3.190
Srisook, K., Han, S. S., Choi, H. S., Li, M. H., Ueda, H., Kim, C., et al. (2006). CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem. Pharmacol. 71, 307–318. doi: 10.1016/j.bcp.2005.10.042
Stapor, P. C., Sweat, R. S., Dashti, D. C., Betancourt, A. M., and Murfee, W. L. (2014). Pericyte dynamics during angiogenesis: new insights from new identities. J. Vasc. Res. 51, 163–174. doi: 10.1159/000362276
Stocker, R., and Perrella, M. A. (2006). Heme oxygenase-1: a novel drug target for atherosclerotic diseases? Circulation 114, 2178–2189. doi: 10.1161/CIRCULATIONAHA.105.598698
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046. doi: 10.1126/science.3029864
Taha, H., Skrzypek, K., Guevara, I., Nigisch, A., Mustafa, S., Grochot-Przeczek, A., et al. (2010). Role of heme oxygenase-1 in human endothelial cells: lesson from the promoter allelic variants. Arterioscler. Thromb. Vasc. Biol. 30, 1634–1641. doi: 10.1161/ATVBAHA.110.207316
Tamarat, R., Silvestre, J. S., Huijberts, M., Benessiano, J., Ebrahimian, T. G., Duriez, M., et al. (2003). Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 8555–8560. doi: 10.1073/pnas.1236929100
Tepper, O. M., Capla, J. M., Galiano, R. D., Ceradini, D. J., Callaghan, M. J., Kleinman, M. E., et al. (2005). Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood 105, 1068–1077. doi: 10.1182/blood-2004-03-1051
Tepper, O. M., Galiano, R. D., Capla, J. M., Kalka, C., Gagne, P. J., Jacobowitz, G. R., et al. (2002). Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 106, 2781–2786. doi: 10.1161/01.CIR.0000039526.42991.93
Thannickal, V. J., and Fanburg, B. L. (2000). Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005
Tiwari, S., and Ndisang, J. F. (2014). The heme oxygenase system and type-1 diabetes. Curr. Pharm. Des. 20, 1328–1337. doi: 10.2174/13816128113199990552
Tojo, T., Ushio-Fukai, M., Yamaoka-Tojo, M., Ikeda, S., Patrushev, N., and Alexander, R. W. (2005). Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 111, 2347–2355. doi: 10.1161/01.CIR.0000164261.62586.14
Triggle, C. R., and Ding, H. (2010). A review of endothelial dysfunction in diabetes: a focus on the contribution of a dysfunctional eNOS. J. Am. Soc. Hypertens. 4, 102–115. doi: 10.1016/j.jash.2010.02.004
Tu, B. P., and Weissman, J. S. (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164, 341–346. doi: 10.1083/jcb.200311055
van der Vlies, D., Makkinje, M., Jansens, A., Braakman, I., Verkleij, A. J., Wirtz, K. W., et al. (2003). Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid. Redox Signal. 5, 381–387. doi: 10.1089/152308603768295113
Venugopal, S. K., Devaraj, S., Yang, T., and Jialal, I. (2002). Alpha-tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-alpha. Diabetes 51, 3049–3054. doi: 10.2337/diabetes.51.10.3049
Volpe, C. M. O., Villar-Delfino, P. H., Dos Anjos, P. M. F., and Nogueira-Machado, J. A. (2018). Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 9:119. doi: 10.1038/s41419-017-0135-z
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, G., Hamid, T., Keith, R. J., Zhou, G., Partridge, C. R., Xiang, X., et al. (2010). Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 121, 1912–1925. doi: 10.1161/CIRCULATIONAHA.109.905471
Wang, W. W., Smith, D. L., and Zucker, S. D. (2004). Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 40, 424–433. doi: 10.1002/hep.20334
Werstuck, G. H., Khan, M. I., Femia, G., Kim, A. J., Tedesco, V., Trigatti, B., et al. (2006). Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes 55, 93–101. doi: 10.2337/diabetes.55.01.06.db05-0633
Werstuck, G. H., Lentz, S. R., Dayal, S., Hossain, G. S., Sood, S. K., Shi, Y. Y., et al. (2001). Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 107, 1263–1273. doi: 10.1172/JCI11596
Wilkinson-Berka, J. L. (2004). Vasoactive factors and diabetic retinopathy: vascular endothelial growth factor, cyclooxygenase-2 and nitric oxide. Curr. Pharm. Des. 10, 3331–3348. doi: 10.2174/1381612043383142
Wohaieb, S. A., and Godin, D. V. (1987). Alterations in free radical tissue-defense mechanisms in streptozocin-induced diabetes in rat. Effects of insulin treatment. Diabetes 36, 1014–1018. doi: 10.2337/diab.36.9.1014
Xu, B., Ji, Y., Yao, K., Cao, Y. X., and Ferro, A. (2005). Inhibition of human endothelial cell nitric oxide synthesis by advanced glycation end-products but not glucose: relevance to diabetes. Clin. Sci. 109, 439–446. doi: 10.1042/CS20050183
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yachie, A., Niida, Y., Wada, T., Igarashi, N., Kaneda, H., Toma, T., et al. (1999). Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 103, 129–135. doi: 10.1172/JCI4165
Yamashita, T., Yamamoto, E., Kataoka, K., Nakamura, T., Matsuba, S., Tokutomi, Y., et al. (2007). Apoptosis signal-regulating kinase-1 is involved in vascular endothelial and cardiac remodeling caused by nitric oxide deficiency. Hypertension 50, 519–524. doi: 10.1161/HYPERTENSIONAHA.107.092049
Yang, G., Li, Y., Wu, W., Liu, B., Ni, L., Wang, Z., et al. (2015). Anti-oxidant effect of heme oxygenase-1 on cigarette smoke-induced vascular injury. Mol. Med. Rep. 12, 2481–2486. doi: 10.3892/mmr.2015.3722
Younce, C. W., Wang, K., and Kolattukudy, P. E. (2010). Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1 production and induction of a novel zinc-finger protein MCPIP. Cardiovasc. Res. 87, 665–674. doi: 10.1093/cvr/cvq102
Zeng, B., Lin, G., Ren, X., Zhang, Y., and Chen, H. (2010). Over-expression of HO-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. J. Biomed. Sci. 17:80. doi: 10.1186/1423-0127-17-80
Zhao, H., Azuma, J., Kalish, F., Wong, R. J., and Stevenson, D. K. (2011). Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol. Reprod. 85, 1005–1012. doi: 10.1095/biolreprod.111.093039
Zhao, L., Bartnikas, T., Chu, X., Klein, J., Yun, C., Srinivasan, S., et al. (2018). Hyperglycemia promotes microvillus membrane expression of DMT1 in intestinal epithelial cells in a PKCalpha-dependent manner. FASEB J. doi: 10.1096/fj.201801855R [Epub ahead of print].
Zhou, J., Lhotak, S., Hilditch, B. A., and Austin, R. C. (2005). Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 111, 1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1
Keywords: heme oxygenase-1 (Ho-1), endothelial dysfunction, angiogenesis, oxidative stress, ER stress, diabetes, hyperglycemia
Citation: Maamoun H, Benameur T, Pintus G, Munusamy S and Agouni A (2019) Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 10:70. doi: 10.3389/fphys.2019.00070
Received: 19 June 2018; Accepted: 21 January 2019;
Published: 11 February 2019.
Edited by:
Bo Akerstrom, Lund University, SwedenReviewed by:
Cynthia J. Meininger, Texas A&M University, United StatesEric J. Belin De Chantemele, Augusta University, United States
Copyright © 2019 Maamoun, Benameur, Pintus, Munusamy and Agouni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdelali Agouni, aagouni@qu.edu.qa