 Yanjun Zhang1
Yanjun Zhang1 Liuwen Du1,2
Liuwen Du1,2 Ao Liu1,2Jianjun Chen1Li Wu1
Ao Liu1,2Jianjun Chen1Li Wu1 Weiming Hu1Wei Zhang3Kyunghee Kim4,5Sang-Choon Lee4
Weiming Hu1Wei Zhang3Kyunghee Kim4,5Sang-Choon Lee4 Tae-Jin Yang4*
Tae-Jin Yang4* Ying Wang5*
Ying Wang5*- 1Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China
- 2College of Life Science, University of Chinese Academy of Sciences, Beijing, China
- 3College of Life Sciences, Xinyang Normal University, Xinyang, China
- 4Department of Plant Science, College of Agriculture and Life Sciences, Plant Genomics and Breeding Institute, and Research Institute of Agriculture and Life Sciences, Seoul National University, Seoul, South Korea
- 5Key Laboratory of South China Agricultural Plant Molecular Analysis and Genetic Improvement, Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
Epimedium L. is a phylogenetically and economically important genus in the family Berberidaceae. We here sequenced the complete chloroplast (cp) genomes of four Epimedium species using Illumina sequencing technology via a combination of de novo and reference-guided assembly, which was also the first comprehensive cp genome analysis on Epimedium combining the cp genome sequence of E. koreanum previously reported. The five Epimedium cp genomes exhibited typical quadripartite and circular structure that was rather conserved in genomic structure and the synteny of gene order. However, these cp genomes presented obvious variations at the boundaries of the four regions because of the expansion and contraction of the inverted repeat (IR) region and the single-copy (SC) boundary regions. The trnQ-UUG duplication occurred in the five Epimedium cp genomes, which was not found in the other basal eudicotyledons. The rapidly evolving cp genome regions were detected among the five cp genomes, as well as the difference of simple sequence repeats (SSR) and repeat sequence were identified. Phylogenetic relationships among the five Epimedium species based on their cp genomes showed accordance with the updated system of the genus on the whole, but reminded that the evolutionary relationships and the divisions of the genus need further investigation applying more evidences. The availability of these cp genomes provided valuable genetic information for accurately identifying species, taxonomy and phylogenetic resolution and evolution of Epimedium, and assist in exploration and utilization of Epimedium plants.
Introduction
Epimedium comprising about 58 species, is a phylogenetically and economically important genus in the family Berberidaceae (Stearn, 2002; Ying et al., 2011). As the diversity center of Epimedium, China possesses about 48 species, and has used Epimedium plants as herb-medicine for more than 2000 years. Herb epimedii has been verified with activity in nourishing the kidney, reinforcing the Yang, regulating bone remodeling, curing cardiovascular diseases, possessing anti-cancer, and anti-aging benefits (Ma et al., 2011; Jiang et al., 2015). The kind and quantity of drugs and health products with herb epimedii as raw materials have been increasing in the last 20 years, which has led to substantial appreciation of prices of the medicinal materials. Furthermore, bearing attractive foliage and flowers, Epimedium plants were previously mainly introduced as perennial garden plant in Europe and America. At present, the horticultural values of Epimedium plants have been widely paid attention with great commercial prospects (Lubell and Brand, 2005; Ren et al., 2008; Avent, 2010).
Epimedium is taxonomically and phylogenetically regarded as one of the most challengingly difficult taxa in plants. The updated system of Epimedium classified the genus into two subgenera, four sections, and four series mainly based on geographical distribution, and leaf, and flower morphology (Stearn, 2002). However, molecular phylogenetic analyses based on internal transcribed spacer (ITS), trnK-matK, atpB-rbcL spacer sequences, and amplified fragment length polymorphisms (AFLPs) only consistently supported subg. Rhizophyllum and four sections of subg. Epimedium as five distinctive clades (Sun et al., 2005; Zhang et al., 2007, 2014; De Smet et al., 2012). The two subgenera were not well-supported, the relationships between five clades were unresolved except for sect. Epimedium as sister to sect. Macroceras, as well as the four series of sect. Diphyllon being poorly supported. As a genus of basal eudicots in North Temperate Zone, the five clades of Epimedium have their unique distribution regions, respectively, and with enormous gaps. It needs more effective molecular markers to investigate the relationships between the five clades and classification system of Epimedium, as well as the origin, evolution, migration, and dispersal of the genus in North Temperate Zone.
It has been intractable for the species identification of Epimedium, particularly for those of sect. Diphyllon, which baffled the effective exploration and utilization of the genus. Chinese sect. Diphyllon has highest species diversity level with about 47 species, and sympatric distribution, and hybridization made the interspecies relationship very complicated. Furthermore, many species, such as E. sagittatum, E. pubescens, and E. acuminatum, have abundant infra-species variations in morphology and medicinal ingredients. However, only AFLPs were heretofore and successfully applied to identify the species of sect. Diphyllon (Zhang et al., 2014). Internal primer binding sites (iPBS) were used to investigate the intra-species variations of E. sagittatum (Chen et al., 2015). For conservation, utilization, and domestication of Epimedium plants, more effective molecular markers are needed to identify Epimedium species and conduct the population genetics and breeding for the Epimedium genus.
The chloroplast (cp) is an important plastid that plays a key role in plant cell for photosynthesis and carbon fixation (Neuhaus and Emes, 2000). The cp genomes in angiosperms are circular DNA molecules ranging from 115 to 165 kb in length and consisting of two copies of a large inverted repeat (IR) region separated by a large-single-copy (LSC) region and a small-single-copy (SSC) region (Raubeson and Jansen, 2005; Wicke et al., 2011). The cp genomes could provide valuable information for taxonomy and phylogeny as a result of sequence divergence between plant species and individuals (Jansen et al., 2007; Moore et al., 2007; Parks et al., 2009; Huang et al., 2014; Jung et al., 2014). Owing to being haploid, maternal inheritance, and high conservation in gene content and genome structure, the cp genomes have been popular to study the evolutionary relationships at almost any taxonomic level in plants. With the advent of high-throughput sequencing technologies, it is now more practical and inexpensive to obtain cp genome sequences and promote cp-based phylogenetics to phylogenomics.
In this study, we sequenced the cp genomes of four Epimedium species using the next-generation sequencing platform, which is also the first comprehensive analysis on cp genomes for Epimedium combining the cp genome of E. koreanum previously reported (Lee et al., 2015). Our study aims were as follows: (1) to investigate global structural patterns of Epimedium cp genomes; (2) to screen sequence divergence hotspot regions in the five Epimedium cp genomes; (3) to examine variations of simple sequence repeats (SSRs) and repeat sequences among the five Epimedium cp genomes; (4) to reconstruct phylogenetic relationships among the five Epimedium species using their cp genome sequences. The results will provide abundant information for further species identification, taxonomy and phylogenetic resolution of Epimedium, and assist in exploration and utilization of Epimedium plants.
Materials and Methods
Sample Preparation, Sequencing, Assembly, and Validation
Fresh leaves of five Epimedium species, four from China, and one from Korea, were sampled. The samples of four Chinese species were used for complete cp genome sequencing, while that of E. koreanum from Korea was only used for PCR-based validating its cp genome sequence (KM207675) previously reported (Lee et al., 2015). The voucher herbarium specimens of four Chinese species were deposited at the Herbaria of Wuhan Botanical Garden, Chinese Academy of Sciences (HIB), and the sample of E. koreanum was deposited at Wuhan Botanical Garden, Chinese Academy of Sciences, Hallym University and Seoul National University (Table S1). Total genomic DNA per species was extracted from 100 mg fresh leaves using the DNeasy Plant MiniKit (Qiagen, CA, USA).
For the four Chinese Epimedium species, Purified DNA (5 mg) was sheared by nebulization with compressed nitrogen gas, yielding fragments of 300 bp in length, and fragmentation quality was checked on a Bioanalyzer 2100 (Agilent Technologies). Paired-end libraries were constructed following the manufacturer's protocol (Illumina, San Diego, California, USA). Genomic DNAs of four species were sequenced on a single lane on HiSeq2000 flow cell lanes (Illumina Inc.) by National Instrumentation Center for Environmental Management (NICEM; http://nature.snu.ac.kr/kr.php), Seoul, Korea.
For each of the four Chinese Epimedium species, cp genome reads were extracted by mapping all raw reads to the reference cp genome of Nandina domestica (DQ923117) with BWA (Li and Durbin, 2009). High quality reads were obtained using the CLC-quality trim tool with Phred scores of < 20 and assembled using the CLC genome assembler v4.06 (http://www.clcbio.com/products/clc-assembly-cell) with default parameters. Sequence gaps were filled by Gapcloser included in the SOAP package v1.12 (Li et al., 2010). All the contigs were aligned to the reference cp genome of Nandina domestica using MUMmer (Kurtz et al., 2004), and aligned contigs were ordered according to the reference cp genome. Based on the reference cp genome, the four junctions between LSC/IRs and SSC/IRs of the five sampled Epimedium species were validated with PCR-based conventional Sanger sequencing, respectively. To avoid assembly errors and obtain high quality complete cp genome sequences, validation of assembly was also carried out on 10 chloroplast genes (Table S2).
Genome Annotation and Analysis
Initial gene annotation of the five chloroplast genomes (including that of E. koreanum, KM207675) was performed with Dual Organellar GenoMe Annotator (DOGMA; Wyman et al., 2004). DOGMA annotations were manually corrected for the start and stop codons and intron/exon boundaries by comparison to homologous genes from other sequenced cp genomes in Ranales. The tRNA genes were also verified with ARAGORN (Laslett and Canback, 2004) and tRNAscan-SE (Lowe and Eddy, 1997; Schattner et al., 2005). The circular cp genome maps were drawn using the OrganellarGenome DRAW tool (ORDRAW; Lohse et al., 2007), with subsequent manual editing.
Cp genome comparison among the five Epimedium species was performed with the mVISTA program (Frazer et al., 2004). Genome, protein coding gene, intron, and spacer sequence divergences were evaluated using DnaSP 5.10 (Rozas et al., 2003) after aligned. The genome sequences were aligned using MAFFT v5 (Katoh and Toh, 2010) and adjusted manually where necessary. For the protein coding gene sequences, introns and spacers, every gene or fragment was edited using ClustalW multiple alignment option within the software BioEdit v7.0.9.0 (Hall, 2011).
Microsatellites (mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats) were detected using the Perl script MISA (Thiel et al., 2003) with thresholds of ten repeat units for mononucleotide SSRs, five repeat units for di- and trinucleotide SSRs, and three repeat units for tetra-, penta-, and hexanucleotide SSRs. Size and location of both direct (forward) and inverted (palindromic) repeats in the Epimedium cp genome were identified by running REPuter (Kurtz et al., 2001) according to the following criteria: cutoff n ≥30% bp and 90% sequence identities (Hamming distance of 3).
Phylogenetic Analysis
It was found that trnQ-UUG genes were duplicated in the LSC of the five Epimedium cp genomes, which was not found in other basal eudicotyledons. For investigating the evolution of trnQ-UUG gene of Epimedium, phylogenetic analyses was conducted based on the nucleotide sequence of the gene of Epimedium and other taxa of basal eudicots. The phylogenetic analyses were also performed for the five Epimedium species with Nandina domestica and Aconitum barbatum of Ranales as outgroups. The analyses were carried out based on the following three data sets: (1) the complete cp DNA sequences; (2) protein coding sequences; (3) the introns and spacers. The nucleotide sequence data of trnQ-UUG gene and cp genome, except those of the four Chinese Epimedium species, were obtained from NCBI, which the sequence data of trnQ-UUG gene were also obtained from the corresponding Genbank files of cp genome sequence data (Table S3).
Maximum parsimony (MP) analyses were conducted using PAUP v4b10 (Swofford, 2003). Heuristic search were performed with 1000 random addition sequences, 10 trees held at each step, tree-bisection-reconnection (TBR) branch swapping and MulTrees switched off. Branch support was assessed with 1000 bootstrap replicates with 10 random taxon additions each and TBR and MulTtrees ON. Maximum likelihood (ML) analyses were performed using RAxML-HPC BlackBox v.8.1.24 on the CIPRES Science Gateway website (Stamatakis et al., 2008; Miller et al., 2010). The best-fitting model was selected using ModelTest v.0.1.1 (Posada, 2008), and branch support was estimated with 1000 bootstrap replicates.
Results and Discussions
Genome Assembly and PCR-Based Validation
Using the Illumina HiSeq 2000 system, five Epimedium species were sequenced to produce 4,573,881–4,675,703 paired-end raw reads (101 bp in average reads length). After screening these paired-end reads through alignment with reference cp genomes of Nandina domestica, 84,589 to 236,730 cp genome reads were extracted with 50 × to 145 × coverage (Table 1). Four junction regions and 10 cp genes were validated by PCR-based sequencing in each of the five Epimedium cp genomes. The PCR-based sequencing on E. koreanum demonstrated identical with its original de novo assembly of complete cp genome sequence (KM20267; Lee et al., 2015). However, some initial gene annotations on the sequence were inaccurate, for example that only one trnQ-UUG was identified while two copies of trnQ-UUG were actually located in LSC. We hereon updated the annotation on the cp genome sequence of E. koreanum with Genbank accession number KU522471. The four Chinese Epimedium cp genome sequences were also deposited in GenBank (accession numbers, KU522469, KU522470, KU522472, and KU522473).
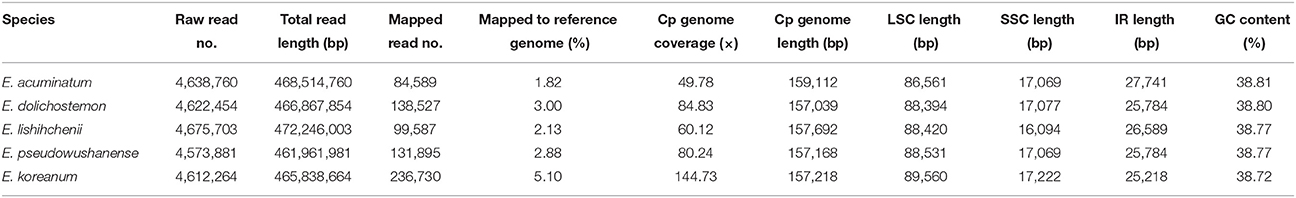
Table 1. Summary of the sequencing data for five Epimedium species.
Genome Features
The nucleotide sequences of the five Epimedium cp genomes ranged from 157,039 bp (E. acuminatum) to 159,112 bp (E. dolichostemon; Figure 1, Table 1). All the five cp genomes displayed the typical quadripartite structure of angiosperms, which consisted of a pair of IR regions (25,218–27,741 bp) separated by a LSC region (86,561–89,560 bp), and a SSC region (16,094–17,222 bp). The average GC content was ~38.77%, which is almost identical with each other among the five complete Epimedium cp genomes.
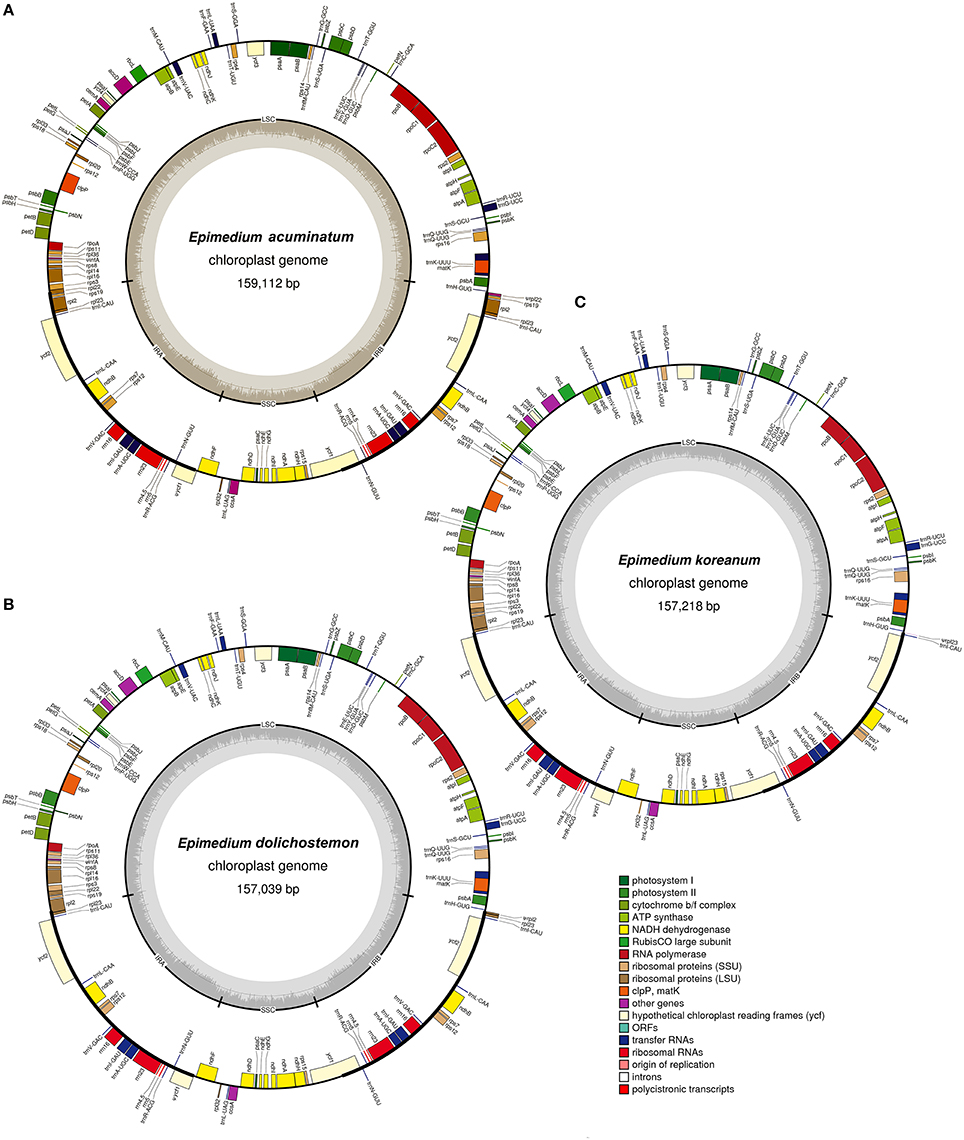
Figure 1. Gene maps of three Epimediumchloroplast genomes. (A) E. acuminatum, (B). E. dolichostemon, (C). E. koreanum. Genes shown outside the outer circle are transcribed clockwise, and those inside are transcribed counterclockwise. Genes belonging to different functional groups are color coded. The dashed area in the inner circle indicates GC content of the chloroplast genomes.
When duplicated genes in IR regions were counted only once, the five Epimedium cp genomes identically harbored 112 different genes arranged in the same order, including 78 protein-coding genes, 30 tRNA, and 4 rRNA. Twelve of the protein-coding genes and six of the tRNA genes contain introns, 15 of which contained a single intron, whereas, three have two introns (Table 2). Among 78 protein-coding genes, 75 genes had the standard AUG as the initiator codon, but rps14 and rps19 started with GUG while rpl2 and ndhD with ACG. An ACG codon may be restored to a canonical start codon (AUG) by RNA editing (Hoch et al., 1991; Takenaka et al., 2013), whereas, a GUG initiation codon has been reported in other cp genomes (Kuroda et al., 2007; Gao et al., 2009).
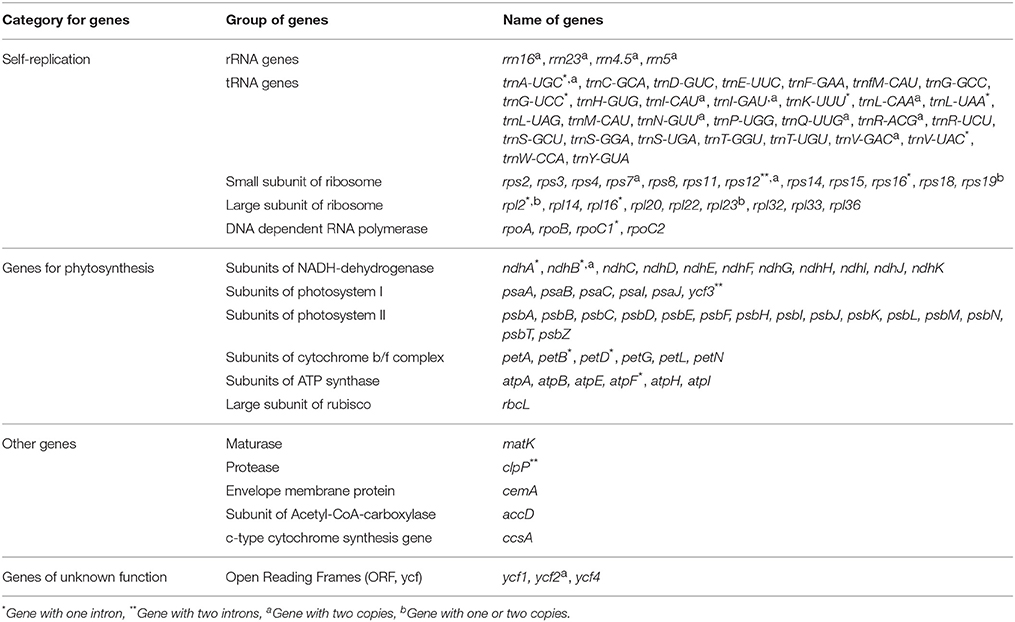
Table 2. List of genes encoded by five Epimedium chloroplast genome.
The trnQ-UUG genes were duplicated in the LSC of the five Epimedium cp genomes and coherently separated by 101 bp with the same orientation. The nucleotide sequence of each copy was identical among the five Epimedium species. The length of one copy was 72 bp and the other with 73 bp, and the two copies were with 19% sequence divergence. The trnQ-UUG duplication had been reported in the family Geraniaceae (Weng et al., 2013), but the gene duplication of Epimedium was firstly found in the basal eudicotyledons. Both MP and ML phylogenetic trees based on trnQ-UUG sequences of Epimedium, and other 11 basal eudicotyledons demonstrated that the two copies of the gene in Epimedium had most close relationship (Figure 2). This raised the possibility of independent duplications of trnQ-UUG in the genus Epimedium.
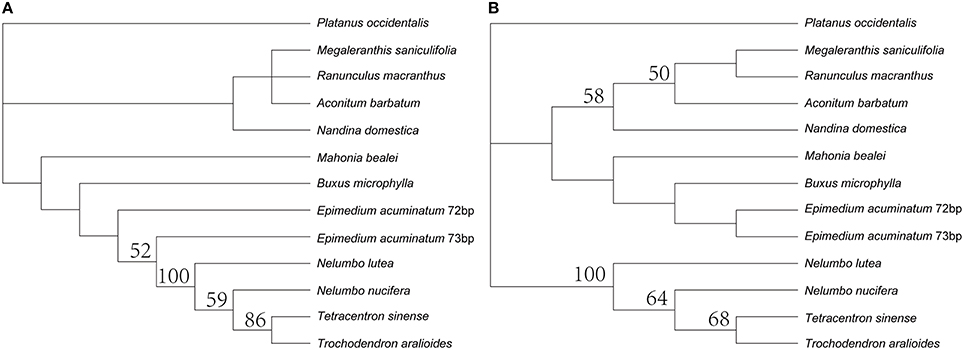
Figure 2. Phylogenetic trees constructed by trnQ-UUG sequences of Epimedium, and other 11 species of basal eudicotyledons with maximum parsimony (A) and maximum likelihood (B). Numbers above node are bootstrap support values (>50%).
The expansion and contraction of the IR region and the single-copy (SC) boundary regions was considered as a primarily mechanism causing the length variation of angiosperm cp genomes (Kim and Lee, 2004). Although overall genomic structure including gene number and gene order were well-conserved, the five Epimedium cp genomes exhibited obvious different at the IR/SC boundary regions (Figure 3). The gene ycf1 crossed the SSC/IRB region, and the pseudogene fragment ψycf1 was located at the IRA region with 2181–3056 bp. The gene rpl22 crossed the LSC/IRA region in E. acuminatum, and ψrpl22 with 296 bp was located at IRB region; rpl2 crossed the LSC/IRA region in E. dolichostemon, E. lishihchenii, and E. pseudowushanense, and ψrpl2 with 226 bp was located at IRB region; rpl23 crossed the LSC/IRA region in E. koreanum, and ψrpl23 with 33 bp was located at IRB region. At the junction of IRA/SSC region, the distance between ψycf1 and ndhF ranged from 47 to 282 bp. At the junction of IRB/LSC region, the distance between ψrpl22 and trnH in E. acuminatum was 76 bp, the distance between ψrpl2 and trnH was from 71 to 76 bp in E. dolichostemon, E. lishihchenii, and E. pseudowushanense, and the distance between ψrpl23 and trnH in E. koreanum was 92 bp. The variations at IR/SC boundary regions in the five Epimedium cp genomes led to their length variation of the four regions and whole genome sequences.
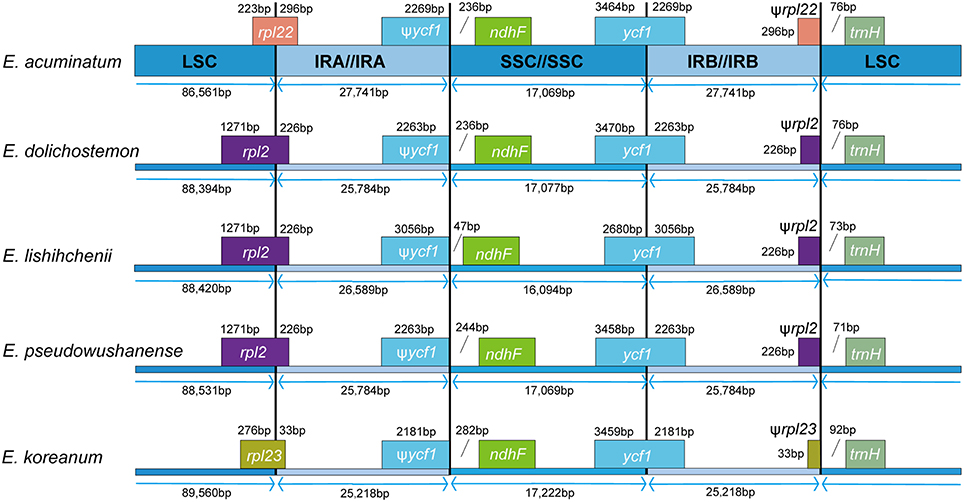
Figure 3. Comparisons of LSC, SSC, and IR region borders among the five Epimedium chloroplast genomes.
Divergence Hotspot Regions
For purposes of the subsequent phylogenetic analyses and plant identification, the complete cp genomes of the five Epimedium species were compared and plotted using the mVISTA program to elucidate the level of sequence divergence (Figure 4). The IRs had lower sequence divergence than that in the SC regions, which also occurred in most higher plants and possibly due to copy correction between IR sequences by gene conversion (Khakhlova and Bock, 2006). The whole genomes, protein-coding regions (pCDS), and non-coding regions (introns and spacers) exhibited divergence proportions of 3.97%, 1.10%, and 5.81%, respectively. For protein coding regions, rps16, psbK, rps132, rps14, and rps15 had over 3% sequence divergences (Table S4). The non-coding regions had higher variability proportions, and four of the regions at the junction of the IRB and LSC had divergence proportions of 100% because of difference in expansion and contraction of IRB (Table S5). Fifty-two non-coding regions had variability proportions ranging from 3.03 to 86.55%, among which 17 regions, such as ycf1/ndhF, trnC-GCA/petN, and trnQ-UUG/psbK, had over 10% variability proportions. These divergence hotspot regions of the five Epimedium cp genome sequences provided abundant information for developing molecular markers for phylogenetic analyses and plant identification of Epimedium species.
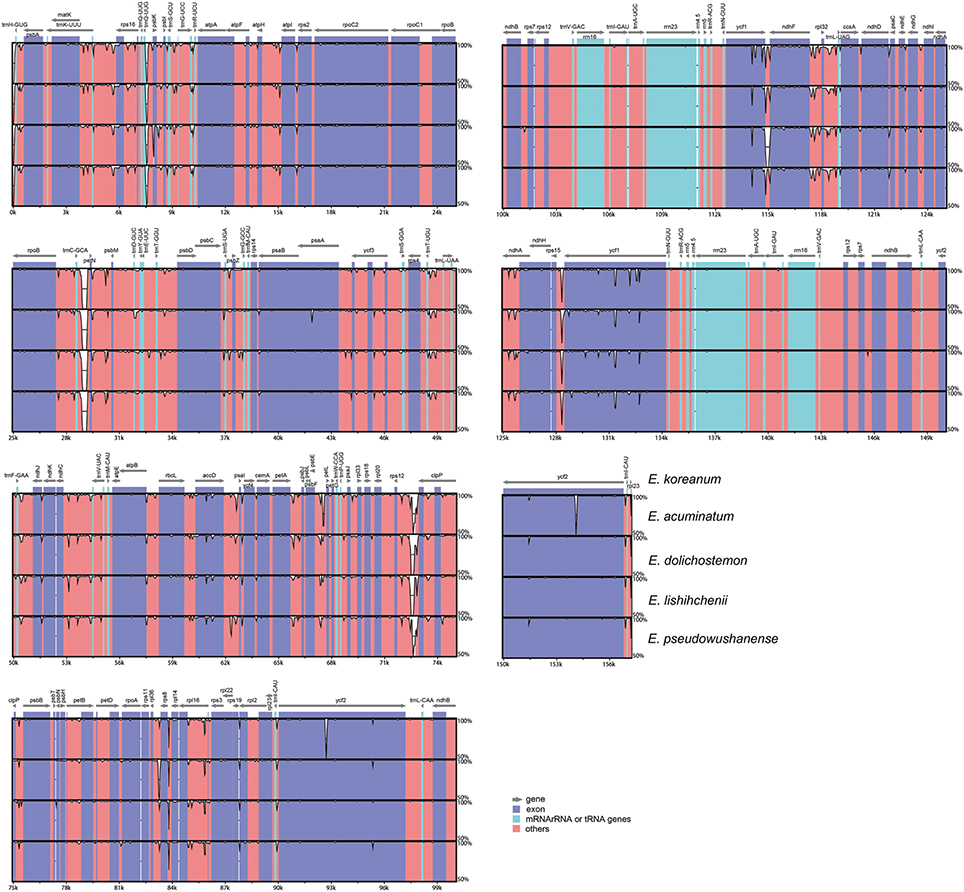
Figure 4. Sequence identity plots among the five Epimedium chloroplast genomes.
SSR Polymorphisms
SSRs in the cp genome present high diversity in copy numbers, and are important molecular markers for plant population genetics, and evolutionary, and ecological studies (Huang et al., 2014; Zhao et al., 2015). With MISA analysis, 116 SSRs with a length of at least 10 bp were detected in the five Epimedium cp genomes with 103 loci showing polymorphism (Table 3, Table S6, S7). Each Epimedium cp genome was found to contain 80 to 87 SSRs, of which 13 SSRs appeared same for the five cp genomes, and the numbers of polymorphic SSRs ranged from 67 to 74. Among the 116 SSRs, the mono-, di-, trin-, tetra-, penta-, and hexanucleotide SSRs were all detected, the mononucleotide SSRs were richest with a portion of 72.76%, and the mononucleotide A and T repeat units occupied the highest portion with 35.34% and 44.83%, respectively. These 116 SSR loci mainly located in intergenic spacer (IGS, 62.07%), following by pCDS (13.79%) and introns (23.28%). Only one SSR crossed the pCDS and IGS (psbI-psbI/trnS-GCU) in the cp genome of E. acuminatum. We observed that 16 SSRs located in 10 protein-coding genes [rpoc2, rpoB, psbC, psaA, psbF, ycf2 (×4), ycf1 (×4), rpl32, ndhE, ndhH] of the five Epimeidum cp genomes. Most of those SSR loci were located in LSC region, followed by SSC and IR regions. In general, the cp SSRs of the five Epimedium represented abundant variation, and undoubtedly useful for assays detecting polymorphisms at population-level as well as comparing more distantly phylogenetic relationships among Epimedium species.
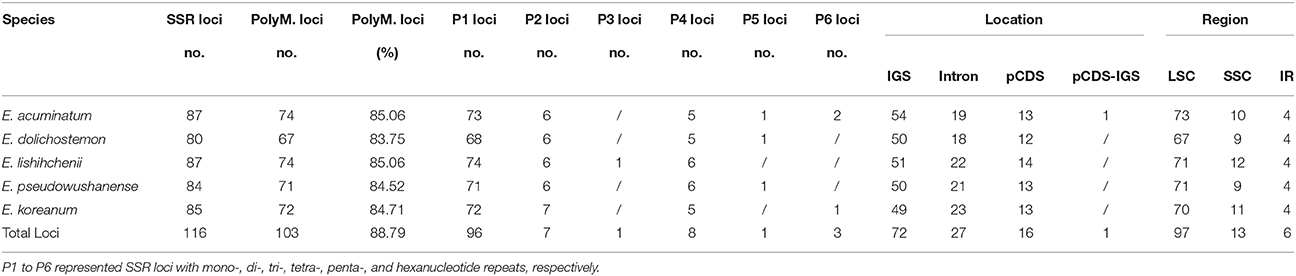
Table 3. Simple sequence repeats (SSRs) in the five Epimedium cp genomes.
Repetitive Sequences
With the criterion of copy size 30 bp or longer and sequence identity >90%, REPuter identified a total of 49 repeats in the five Epimedium cp genomes, including direct, and palindromic repeats (Figure 5, Table S8). Except for E. koreanum with 24 direct repeats and 25 palindromic repeats, the other four Epimedium species identically possessed 23 direct repeats, and 26 palindromic repeats. The lengths of repeats in the five Epimedium cp genomes ranged from 31 to 131 bp, and the copy lengths with 30–49 bp are most common (61.22%) while those with more than 100 bp were least (7.76%). Under the criterion with identical lengths located in homologous regions as shared repeats, we investigated the repeats shared among the five Epimedium cp genomes. There were 16 repeats shared by the five Epimedium cp genomes, 15 repeats shared by the four Epimedium species endemic to China, 13 repeats shared by E. koreanum and the three of four Chinese Epimedium species, and four repeats shared by two or three Chinese Epimedium species. E. koreanum had the most unique repeats (20), followed with E. acuminatum (16), while the other three Epimedium species had one to three unique repeats. The repeats of the five Epimedium cp genomes were mainly located in pCDS and IGS, while the minority was located in intron and rrn gene coding region (rCDS), or covered across IGS and one of pCDS, rCDS, or trn gene coding region (tCDS). Except for E. koreanum, the proportions of repeat locations were identical in the other four Epimedium species.
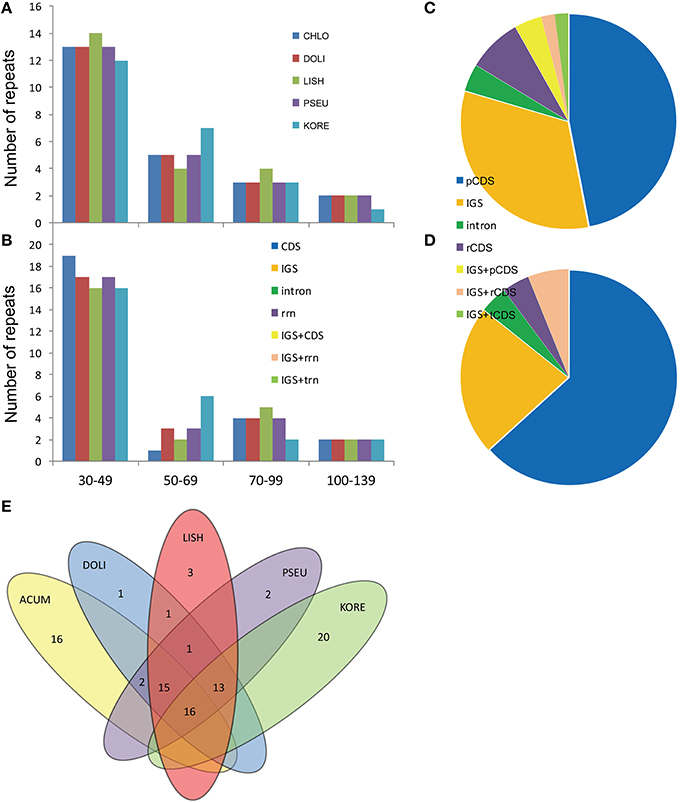
Figure 5. Analysis of repeated sequences in the five Epimedium chloroplast genomes. (A) Frequency of the direct repeats by length; (B). Frequency of the palindromic repeats; (C). Location of repeats in the four Epimedium cp genomes endemic to China; (D). Location of repeats in the cp genome of E. koreanum; (E). Summary of shared repeats among the five Epimedium chloroplast genomes. chlo, E. acuminatum; dewu, E. dolichostemon; lish, E. lishihchenii; pseu, E. pseudowushanense; kore, E. koreanum; IGS, intergenic spacer.
Contrasting to the major repeats of most angiosperm plant cp genomes located in noncoding regions (Uthaipaisanwong et al., 2012; Yao et al., 2015), the proportions of repeats located in coding regions (CDS) were higher than those in noncoding regions in Epimedium species. In E. koreanum cp genomes, the proportion of the repeats located in pCDS led to 63.27%, while the repeats located in IGS only accounted for 22.45%. Previous work suggested that repeat sequences have played an important role in sequence rearranging and variation in cp genomes through illegitimate recombination and slipped-strand mispairing (Bausher et al., 2006; Saski et al., 2007; Huang et al., 2014). Our research also showed that divergent regions of cp genomes were associated with various repeat sequences such as ycf1 gene and intergenic trnQ-UUG/psbK. These repeats may further serve as genetic markers for phylogenetic and population genetic studies on Epimedium species.
Phylogenetic Analysis
The cp genome sequences are addressed successfully for the phylogenetic studies of angiosperm (Jansen et al., 2007; Huang et al., 2014; Kim et al., 2015). In the present studies, three datasets (protein coding exons, introns and spacers, and whole complete cp genome sequences) from cp genomes of five Epimedium species and two outgroups were used to perform phylogenetic analysis. Among the three datasets, introns and spacers contained the highest parsimony informative characters (6.85%), followed by whole complete cp genome sequences (5.22%) and protein coding exons (5.03%). Using MP and ML analyses, phylogenetic trees were built based on the three datasets (Figure 6). The topologies based on both analyses were highly concordant in each dataset, as well as the dendrograms based on the noncoding sequences and whole complete cp genome sequences, and the phylogenetic trees of the three datasets were largely congruent with each other. For the five Epimedium species, E. koreanum is distributed in Northeast China, Japan, and Korea, and belongs to sect. Macroceras, while the other four species are native to Central and Southwest China, being attributed to sect. Diphyllon (Stearn, 2002). The resulting six phylogenetic trees identically exhibited that E. koreanum were firstly separated from the other four Epimedium species. For the four Epimedium species of sect. Diphyllon, E. dolichostemon has relatively small flowers and short spurs, being a member of ser. Brachycerae; the other three species has large flowers with petals bearing long spurs, of which E. acuminatum and E. lishihchenii possess petal without basal laminae, being attributed to ser. Dolichocerae, while E. pseudowushanense possesses petal with slight basal lamina, belonging to ser. Davidianae. In accordance with classical taxonomy of Epimedium (Stearn, 2002), phylogenetic trees based on noncoding regions and whole complete cp genome sequences all supported that E. dolichostemon was early divided from the other three species of sect. Diphyllon. However, the basal position of E. dolichostemon among four species of sec. Diphyllon was inconsistent with Stearn's (2002) and Ying's (2002) interpretation on floral evolution of the genus. Furthermore, all trees based on the three datasets identically supported that E. lishihchenii firstly clustered with E. pseudowushanense, not with E. acuminatum from the same series, which were coincident with the previous phylogenetic studies based on AFLPs (Zhang et al., 2014). These results showed that Stearn's (2002) taxonomic system of Epimedium is reasonable on the whole and the phylogenetic relationships within Chinese sect. Diphyllon are closely related with corolla characters, especially with petals. However, the evolutionary relationships and the divisions within the section need further investigation applying more evidences.
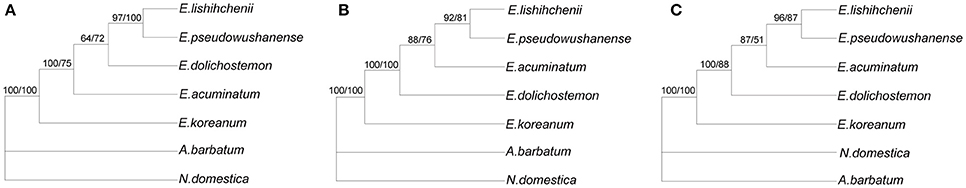
Figure 6. Phylogenetic relationships of the five Epimedium species constructed by CDS regions (A), noncoding regions (B), and whole cp genome sequences (C) with maximum parsimony (MP) and maximum likelihood. Numbers above node are bootstrap support values (>50%) with MP bootstrap values on the left and ML bootstrap on the right.
Author Contributions
YZ, YW, and TY conceived and designed the experiment, and writed the paper. JC, WZ, AL, KK, and SL collected the materials. KK, SL, YZ, and LD performed the experiments. KK and SL completed the sequence assembly. LD, YZ, LW, and WH conducted the comprehensive analyses on the cp genome sequences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research is supported by the National Natural Science Foundation of China (30900076) and Key Research Program of the Chinese Academy of Sciences (KSZD-EW-Z-004), and by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2015M3A9A5030733). We thank Jia Li and Ke Tao for their help on data analysis, and Lei Gao and Bo Wang for their valuable comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00306
References
Avent, T. (2010). An overview of Epimedium. Plantsman 9, 10–17. Available online at: http://www.cabdirect.org/abstracts/20103075307.html
Bausher, M. G., Singh, N. D., Lee, S. B., Jansen, R. K., and Daniell, H. (2006). The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 6:21. doi: 10.1186/1471-2229-6-21
Chen, J., Xu, Y., Wei, G., Liao, S., Zhang, Y., Huang, W., et al. (2015). Chemotypic and genetic diversity in Epimedium sagittatum from different geographical regions of China. Phytochemistry 116, 180–187. doi: 10.1016/j.phytochem.2015.04.005
De Smet, Y., Goetghebeur, P., Wanke, S., Asselman, P., and Samain, M. S. (2012). Additional evidence for recent divergence of Chinese Epimedium (Berberidaceae) derived from AFLP, chloroplast and nuclear data supplemented with characterisation of leaflet pubescence. Plant Ecol. Evol. 145, 73–87. doi: 10.5091/plecevo.2012.646
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, L., Yi, X., Yang, Y. X., Su, Y. J., and Wang, T. (2009). Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 9:130. doi: 10.1186/1471-2148-9-130
Hoch, B., Maier, R. M., Appel, K., Igloi, G. L., and Kössel, H. (1991). Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353:178–180. doi: 10.1038/353178a0
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Leebens-Mack, J., Müller, K. F., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jiang, J., Song, J., and Jia, X. B. (2015). Phytochemistry and ethnopharmacology of Epimedium L. species. Chin. Herbal Med. 7, 204–222. doi: 10.1016/S1674-6384(15)60043-0
Jung, J., Kim, K. H., Yang, K., Bang, K. H., and Yang, T. J. (2014). Practical application of DNA markers for high-throughput authentication of Panax ginseng and Panax quinquefolius from commercial ginseng products. J. Ginseng Res. 38, 123–129. doi: 10.1016/j.jgr.2013.11.017
Katoh, K., and Toh, H. (2010). Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900. doi: 10.1093/bioinformatics/btq224
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kim, K., Lee, S.-C., Lee, J., Yu, Y., Yang, K., Choi, B.-S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5:15655. doi: 10.1038/srep15655
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kuroda, H., Suzuki, H., Kusumegi, T., Hirose, T., Yukawa, Y., and Sugiura, M. (2007). Translation of psbC mRNAs starts from the downstream GUG, not the upstream AUG, and requires the extended Shine–Dalgarno sequence in tobacco chloroplasts. Plant Cell Physiol. 48, 1374–1378. doi: 10.1093/pcp/pcm097
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lee, J. H., Kim, K., Kim, N. R., Lee, S. C., Yang, T. J., and Kim, Y. D. (2015). The complete chloroplast genome of a medicinal plant Epimedium koreanum Nakai (Berberidaceae). Mitochondrial DNA. doi: 10.3109/19401736.2015.1089492. [Epub ahead of print].
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964. doi: 10.1093/nar/25.5.0955
Lubell, J. D., and Brand, M. H. (2005). Division size and timing influence propagation of four Species of Epimedium L. HortScience 40, 1444–1447.
Ma, H., He, X., Yang, Y., Li, M., Hao, D., and Jia, Z. (2011). The genus Epimedium: an ethnopharmacological and phytochemical review. J. Ethnopharmacol. 134, 519–541. doi: 10.1016/j.jep.2011.01.001
Miller, M., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of Gateway Computing Environments Workshop (GCE) (New Orleans, LA. IEEE), 1–8.
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Neuhaus, H., and Emes, M. (2000). Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 51, 111–140. doi: 10.1146/annurev.arplant.51.1.111
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants, ed R. J. Henry (Cambridge, MA: CABI Press), 45–68.
Ren, L., Dai, S. L., and Wang, Y. (2008). The germplasm resources of Epimedium in China and its application in landscape architecture. Wuhan Bot. Res. 26, 644–649.
Rozas, J., Sánchez-DelBarrio, J. C., Messeguer, X., and Rozas, R. (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497. doi: 10.1093/bioinformatics/btg359
Saski, C., Lee, S. B., Fjellheim, S., Guda, C., Jansen, R. K., Luo, H., et al. (2007). Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes. Theor. Appl. Genet. 115, 571–590. doi: 10.1007/s00122-007-0567-4
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stearn, W. T. (2002). The Genus Epimedium and Other Herbaceous Berberidaceae. Portland: Timber Press.
Sun, Y., Fung, K. P., Leung, P. C., and Shaw, P. C. (2005). A phylogenetic analysis of Epimedium (Berberidaceae) based on nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol. 35, 287–291. doi: 10.1016/j.ympev.2004.12.014
Swofford, D. L. (2003). PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods). Version 4b10. Sunderland, Massachusetts: Sinauer.
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B., and Brennicke, A. (2013). RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Uthaipaisanwong, P., Chanprasert, J., Shearman, J., Sangsrakru, D., Yoocha, T., Jomchai, N., et al. (2012). Characterization of the chloroplast genome sequence of oil palm (Elaeis guineensis Jacq.). Gene 500, 172–180. doi: 10.1016/j.gene.2012.03.061
Weng, M. L., Blazier, J. C., Govindu, M., and Jansen, R. K. (2013). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. doi: 10.1093/molbev/mst257
Wicke, S., Schneeweiss, G. M., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yao, X., Tang, P., Li, Z., Li, D., Liu, Y., and Huang, H. (2015). The first complete chloroplast genome sequences in Actinidiaceae: genome structure and comparative analysis. PLoS ONE 10:e0129347. doi: 10.1371/journal.pone.0129347
Ying, T. S. (2002). Petal evolution and distribution patterns of Epimedium L. (Berberidaceae). Acta Phytotax. Sin. 40, 481–489.
Ying, T. S., Boufford, D. E., and Brach, A. R. (2011). “Epimedium L.,”in Flora of China, eds Z. Y. Wu, P. H. Raven, and D. Y. Hong (Beijing; St. Louis, MO: Science Press, Missouri Botanical Garden Press), 787–799.
Zhang, M. L., Uhink, C. H., and Kadereit, J. W. (2007). Phylogeny and biogeography of Epimedium/Vancouveria (Berberidaceae): Western North American-East Asian disjunctions, the origin of European mountain plant taxa, and East Asian species diversity. Syst. Bot. 32, 81–92. doi: 10.1600/036364407780360265
Zhang, Y., Yang, L., Chen, J., Sun, W., and Wang, Y. (2014). Taxonomic and phylogenetic analysis of Epimedium L. based on amplified fragment length polymorphisms. Sci. Hortic. 170, 284–292. doi: 10.1016/j.scienta.2014.02.025
Keywords: Epimedium, chloroplast genome, genome structure, phylogenetic relationships, taxonomic identification
Citation: Zhang Y, Du L, Liu A, Chen J, Wu L, Hu W, Zhang W, Kim K, Lee S-C, Yang T-J and Wang Y (2016) The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Received: 17 January 2016; Accepted: 26 February 2016;
Published: 15 March 2016.
Edited by:
Daniel Pinero, Universidad Nacional Autónoma de México, MexicoReviewed by:
Caiguo Zhang, University of Colorado, USASithichoke Tangphatsornruang, National Center for Genetic Engineering and Biotechnology, Thailand
Copyright © 2016 Zhang, Du, Liu, Chen, Wu, Hu, Zhang, Kim, Lee, Yang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tae-Jin Yang, tjyang@snu.ac.kr;
Ying Wang, yingwang@scib.ac.cn