 Yonghong Zhang1Lanlan Zheng1Yan Zheng1Chao Zhou2Ping Huang1Xiao Xiao1Yongheng Zhao1Xincai Hao1
Yonghong Zhang1Lanlan Zheng1Yan Zheng1Chao Zhou2Ping Huang1Xiao Xiao1Yongheng Zhao1Xincai Hao1 Zhubing Hu3Qinhua Chen4Hongliang Li5
Zhubing Hu3Qinhua Chen4Hongliang Li5 Xuanbin Wang5Kenji Fukushima6
Xuanbin Wang5Kenji Fukushima6 Guodong Wang7
Guodong Wang7 Chen Li1*
Chen Li1*- 1Laboratory of Medicinal Plant, Institute of Basic Medical Sciences, School of Basic Medicine, Biomedical Research Institute, Hubei Key Laboratory of Wudang Local Chinese Medicine Research, Hubei University of Medicine, Shiyan, China
- 2Key Laboratory of Three Gorges Regional Plant Genetics and Germplasm Enhancement (CTGU)/Biotechnology Research Center, China Three Gorges University, Yichang, China
- 3Center for Multi-Omics Research Key Laboratory of Plant Stress Biology, State Key Laboratory of Cotton Biology, School of Life Sciences, Henan University, Kaifeng, China
- 4Affiliated Dongfeng Hospital, Hubei University of Medicine, Shiyan, China
- 5Laboratory of Chinese Herbal Pharmacology, Oncology Center, Renmin Hospital, Biomedical Research Institute, Hubei University of Medicine, Shiyan, China
- 6Institute for Molecular Plant Physiology and Biophysics, University of Würzburg, Würzburg, Germany
- 7Key Laboratory of Medicinal Resources and Natural Pharmaceutical Chemistry, Ministry of Education, National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest China, College of Life Sciences, Shaanxi Normal University, Xi’an, China
Polygonum cuspidatum (Japanese knotweed, also known as Huzhang in Chinese), a plant that produces bioactive components such as stilbenes and quinones, has long been recognized as important in traditional Chinese herbal medicine. To better understand the biological features of this plant and to gain genetic insight into the biosynthesis of its natural products, we assembled a draft genome of P. cuspidatum using Illumina sequencing technology. The draft genome is ca. 2.56 Gb long, with 71.54% of the genome annotated as transposable elements. Integrated gene prediction suggested that the P. cuspidatum genome encodes 55,075 functional genes, including 6,776 gene families that are conserved in the five eudicot species examined and 2,386 that are unique to P. cuspidatum. Among the functional genes identified, 4,753 are predicted to encode transcription factors. We traced the gene duplication history of P. cuspidatum and determined that it has undergone two whole-genome duplication events about 65 and 6.6 million years ago. Roots are considered the primary medicinal tissue, and transcriptome analysis identified 2,173 genes that were expressed at higher levels in roots compared to aboveground tissues. Detailed phylogenetic analysis demonstrated expansion of the gene family encoding stilbene synthase and chalcone synthase enzymes in the phenylpropanoid metabolic pathway, which is associated with the biosynthesis of resveratrol, a pharmacologically important stilbene. Analysis of the draft genome identified 7 abscisic acid and water deficit stress-induced protein-coding genes and 14 cysteine-rich transmembrane module genes predicted to be involved in stress responses. The draft de novo genome assembly produced in this study represents a valuable resource for the molecular characterization of medicinal compounds in P. cuspidatum, the improvement of this important medicinal plant, and the exploration of its abiotic stress resistance.
Introduction
Polygonum cuspidatum Sieb. et Zucc., commonly known as Huzhang in Chinese and Japanese knotweed in English, is a medicinal plant that is widely distributed in eastern Asia whose roots have served for centuries as an important traditional Chinese medicine for dispelling wind-evil, damp elimination, analgesic therapy, relieving coughs, and reducing sputum (Peng et al., 2013; Hong et al., 2016). P. cuspidatum belongs to the Polygonaceae family of eudicots, which includes many other key medicinal plants, such as Rheum palmatum (Chinese rhubarb), Fagopyrum cymosum (tall buckwheat), and Polygonum multiflorum, as well as the pseudocereal crop Fagopyrum tataricum (Tartary buckwheat). In contrast to its medicinal uses, P. cuspidatum is regarded as an invasive plant in Europe and North America due to its aggressive growth, allelopathic effects, and extremely strong abiotic stress tolerance (Murrell et al., 2011; Rouifed et al., 2012; Dommanget et al., 2016). This species shows strong adaptability and tolerance to a wide range of stress conditions, such as dense shade, high temperatures, cold, drought, waterlogging, burning, heavy metals, various soil types, and extreme pH, salt, and high sulfur dioxide conditions (Clements and Ditommaso, 2012; Michalet et al., 2017). However, few molecular genetic studies have been conducted on this pharmaceutically and economically important herbaceous plant.
Modern pharmacological studies have indicated that extracts from P. cuspidatum have anti-inflammatory, antioxidant, and hepatoprotective properties and could be to treat cancer and other diseases (Zhang et al., 2013; Olaru et al., 2015; Sohn et al., 2015; Abu-Amero et al., 2016; Liu and Huang, 2016; Geng et al., 2018). Extensive photochemistry investigations have led to the isolation and identification of more than 67 compounds from the roots and leaves of P. cuspidatum, providing chemical evidence for its pharmacological effect (Vastano et al., 2000; Yi et al., 2007; Shan et al., 2008). The major compounds in P. cuspidatum are stilbenes and quinones (Vastano et al., 2000; Shan et al., 2008). The most abundant stilbenes in P. cuspidatum are polydatin and resveratrol, which have been identified in several other plants, including grapevine (Vitis vinifera) (Langcake and Pryce, 1976), peanuts (Arachis hypogaea) (Sanders et al., 2000), blueberries (Vaccinium spp.) (Lyons et al., 2003), cocoa (Theobroma cacao) (Counet et al., 2006), and sorghum (Sorghum bicolor) (Burdette et al., 2010; Vanamala et al., 2017). Resveratrol functions in plant disease resistance by activating constitutive and inducible defense responses (Chong et al., 2009). Resveratrol, especially in the form of trans-resveratrol, has many pharmacological uses in treating inflammation, HIV, and cardiovascular-related diseases (Hao et al., 2012; Ouyang et al., 2014; Hong et al., 2016). Although some recent clinical studies have cast doubt on its pharmacological activities, resveratrol has been widely utilized in the nutraceutical and cosmetics industries for decades (Donnez et al., 2009; Lu et al., 2016). The largest portion of resveratrol in the global market is currently produced from the root extracts of field-grown P. cuspidatum (Mei et al., 2015).
The anthraquinone emodin and its derivative, physcion, are major quinones in P. cuspidatum and have potential antimicrobe and anticancer applications (Jayasuriya et al., 1992; Teich et al., 2004; Su et al., 2005; Olsen et al., 2007; Yang et al., 2007; Shan et al., 2008; Narender et al., 2013; Chen et al., 2015; Xiong et al., 2015; Han et al., 2016; Hong et al., 2016; Li et al., 2016; Liu et al., 2016; Pan et al., 2016; Gao et al., 2017). Physcion significantly inhibits cancer cell proliferation and tumor growth in mice by specifically decreasing the activity of 6-phosphogluconate dehydrogenase, thus affecting the oxidative pentose phosphate pathway (Lin et al., 2015).
Resveratrol is produced by a well-characterized biosynthetic pathway that includes four major enzymes: phenylalanine ammonia lyase (PAL), cinnamic acid 4-hydroxylase (C4H), 4-coumarate CoA ligase (4CL), and stilbene synthase (STS) (Ferrer et al., 1999; Hao et al., 2012; Jiang et al., 2018; Zhao et al., 2018). PAL, C4H, and 4CL are members of the common phenylpropanoid pathway in plants, which synthesizes phenolic compounds. By contrast, STS is a type III polyketide synthase (PKS), which catalyzes the condensation of resveratrol in the final step of the pathway (Hao et al., 2012). Plant type III PKSs have been widely studied, including chalcone synthase (CHS), which plays an important role in plant metabolite biosynthesis (Yu et al., 2012). STS shares high amino acid sequence identity (>70%) with CHS; these enzymes comprise the STS/CHS superfamily (Melchior and Kindl, 1990; Tropf et al., 1995; Ferrer et al., 1999). STS and CHS catalyze the same iterative condensation to yield tetraketide intermediates in the same manner (Austin and Noel, 2003; Funa et al., 2006). Although STS is thought to have arisen from CHS several times during the course of evolution (Tropf et al., 1994), no genome-wide investigation of CHS/STS family genes in P. cuspidatum has been conducted thus far. Although CHS and STS share the same substrate in flowering plants, CHS is responsible for the biosynthesis of chalcones, which serve as starting molecules for flavonoid compounds (Schröder et al., 1988; Wang et al., 2017a). While CHS is widely present in many plants, STS is only found in plants that produce resveratrol (Kiselev, 2011).
Several genes that are likely involved in resveratrol biosynthesis have been identified (Ferrer et al., 1999; Hao et al., 2012; Jiang et al., 2018; Zhao et al., 2018), but little is known about the genetic basis of the anthraquinone biosynthetic pathway. Our current understanding on anthraquinone biosynthesis were obtained from the family Rubiaceae and especially in the genera Rubia and related species, such as Rubia cordifolia (Wijnsma and Verpoorte, 1986; Han et al., 2001; Han et al., 2002; Wurglics and Schubert-Zsilavecz, 2006; Perassolo et al., 2011; Huang et al., 2014). These species were known to produce substantial amount of anthraquinone derivatives. It has been reported that the mevalonic acid (MVA) and the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathways produce dimethylallyldiphosphate, a precursor of the anthraquinone backbone, while the shikimate pathway produces another backbone, 1,4-dihydroxy-2-napthoyl-CoA, via isochorismate (Han et al., 2001; Hao et al., 2012; Peng et al., 2013). In P. cuspidatum, MVA, MEP, and shikimate pathways are shown to be involved in the biosynthesis of anthraquinones such as emodin and physcion (Hao et al., 2012; Peng et al., 2013). Nevertheless, the anthraquinone biosynthetic pathway in P. cuspidatum is largely elusive.
With the rapid development of next-generation sequencing technology, whole-genome information has been obtained for many medicinal plants. Herbgenomics is a new field of study that investigates the genetics and regulatory mechanisms of traditional Chinese herbal medicines via genomics, which clarifies the mechanisms of action of traditional Chinese medicines and facilitates molecular breeding from the perspective of the genome (Chen and Song, 2016; Xin et al., 2018). Herbgenomics mainly involves analysis of medicinal plant genomes, transcriptomes, proteomes, metabolomes, and so on. To date, the genomes of Ganoderma lucidum (Chen et al., 2012), Salvia miltiorrhiza (Wenping et al., 2011; Ma et al., 2012; Xu et al., 2016), Dendrobium officinale (Yan et al., 2015), Andrographis paniculata (Sun et al., 2019), Macleaya cordata (Liu et al., 2017), Panax ginseng (Xu et al., 2017), Panax notoginseng (Zhang et al., 2017a), Chrysanthemum nankingense (Song et al., 2018), and other important Chinese herbs have been described.
Dissecting the metabolic pathways of useful natural products from a genomics perspective will provide fundamental resources for the large-scale production and generation of novel chemicals via synthetic biology (Esvelt and Wang, 2013; Baltes and Voytas, 2015). Given that P. cuspidatum is a popular traditional Chinese medicine with widespread applications, the in vivo distributions of major components and their underlying metabolic pathways should be investigated. With the help of next-generation sequencing, genes involved in certain metabolic pathways in many medicinal plants could be identified. These approaches will not only shed light on how natural products are synthesized and how their production is regulated, but they could also be utilized for the genetic improvement of medicinal plants.
Due to their similar appearance, it is challenging to distinguish dry roots of P. cuspidatum from those of closely related species, such as Fallopia multiflora and R. palmatum. Traditional authentication methods such as chromatographic fingerprint analysis and spectroscopy have limited utility (Xie et al., 2006; Yap et al., 2007). DNA marker-based technology could also be used to explore intra-species genetic variation (Hon et al., 2003), which would provide important information about the relationship between genetic diversity and environmental interactions in P. cuspidatum. Therefore, the availability of whole-genome resources would facilitate the development of DNA marker-based technology in P. cuspidatum.
The lack of genomic information represents a major obstacle to exploring the biological features of P. cuspidatum. To address this problem, in this study, we sequenced and assembled a draft genome of P. cuspidatum. We annotated genes in the genome and predicted whole-genome duplication (WGD) events. Our study paves the way for the genetic analysis of P. cuspidatum. The draft genome produced in this study should facilitate the improvement of P. cuspidatum, provide a better understanding of the metabolic pathways of its natural products, give insight on its remarkable stress tolerance, and facilitate the development of methods for the biocontrol of this plant for weed management.
Materials and Methods
Plant Material and Sequencing
P. cuspidatum plants were cultivated in a field in Hubei Province, China (25°20ʹ5.496ʹʹ N, 114?57ʹ52.459ʹʹE). The fresh leaves of 1-year-old plants were collected for DNA extraction. To construct sequencing libraries, 5 μg of DNA was used. Libraries were constructed using an Illumina TruSeq DNA Preparation Kit following the manufacturer’s recommendations. The four sequencing libraries, with insert sizes of 550 bp, 2 to 3 kb, 5 to 7 kb, and 10 to 15 kb, were sequenced on the Illumina HiSeq platform (150-bp paired-end reads, PE150). In addition, 250-bp paired-end reads were generated on the Illumina HiSeq 2500 platform.
Genome Assembly
A genome survey was performed using 67 Gb of clean Illumina sequencing data (PE150) using Jellyfish software following the instructions from the GenomeScope website (http://qb.cshl.edu/genomescope/) (Marçais and Kingsford, 2011). The genome survey indicated that this species has very high heterogeneity. Two software packages were used for genome assembly: SOAPdenovo, which is highly effective for short-read assembly, and platanus, which is thought to perform well using genomes with high heterozygosity (Luo et al., 2012). For SOAPdenovo-guided assembly, 150-bp paired-end sequencing reads were assembled into contigs, and sequencing reads with a large insertion size were used to construct scaffolds. Reads with a length of PE250 were used to fill gaps in the scaffolds with GapFiller software (Nadalin et al., 2012; Kajitani et al., 2014). For platanus-guided assembly, all sequencing reads with different insertion sizes and lengths (PE150 and PE250) were used for contig and scaffold assembly, followed by gap filling. This software was run with different Kmers, and the best assemblies from different Kmers were determined based on contig and scaffold length, gap content, and genome completeness. Due to possible sequence redundancy from the platanus assembly, Redundans software was used to filter the scaffolds (Pryszcz and Gabaldón, 2016). For scaffolds <1 kb long, BLAST analysis was performed by aligning these sequences against sequences longer than 1 kb (E-value < 1e-5, identity >70%, and match length >60%).
Transposable Element Prediction
Due to the relatively low conformance of repeat sequences among species, repeat sequence databases are often constructed to predict the repeats for a specific species. By integrating four software programs, including LTR FINDER (Xu and Wang, 2007), mite-hunter (Han and Wessler, 2010), RepeatScout (Price et al., 2005), and PILER-DF (Edgar and Myers, 2005), a repeat sequence database was constructed for P. cuspidatum based on the principle of structural prediction and de novo prediction. Sequences in this database were classified using PASTEClassifier (Hoede et al., 2014) and merged with the Repbase database to form the final repeat sequence database. RepeatMasker (version 4.0.6) was then used to predict the repeat sequences in the genome based on the newly constructed database (Tarailo-Graovac and Chen, 2009).
Gene Prediction and Annotation
Three approaches were used to predict genes in the P. cuspidatum genome: de novo prediction, homolog-based prediction, and transcriptome-based prediction. For de novo prediction, Augustus software (version 2.4) was utilized with a transposable element (TE)-masked genome (Stanke et al., 2006). For homolog-based approach, the GeMoMa (version 1.3.1) was used to predict genes with gene models from the other four species (three dicot species and one monocot species) (Keilwagen et al., 2016), including the dicot species Tartary buckwheat (http://www.mbkbase.org/Pinku1/), Arabidopsis thaliana (https://www.arabidopsis.org/) and grape (http://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis/), and the monocot species rice (Oryza sativa) (http://rice.plantbiology.msu.edu/). For transcriptome-based prediction, the HISAT (version 2.0.4) and StringTie (version 1.2.3) programs were used for transcript assembly (Kim et al., 2015; Pertea et al., 2015). In addition, TransDecoder (version 2.0, https://github.com/TransDecoder/TransDecoder/) was used to predict gene models. All gene models obtained using these approaches were integrated with EVM software (Haas et al., 2008). To predict the putative functions of genes, all gene models were aligned against the GenBank Non-Redundant, TrEMBL, Pfam, SwissProt, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases.
Gene Family Identification and Analysis
To identify gene families based on those in other plants, OrthoMCL analysis was performed (Li et al., 2003). Protein data were downloaded from four other dicot species, including Tartary buckwheat (http://www.mbkbase.org/Pinku1/), Arabidopsis (https://www.arabidopsis.org/), grape (http://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis/), and tomato (Solanum lycopersicum) (ftp://ftp.solgenomics.net/tomato_genome/annotation/ITAG3.2_release/). All data were subjected to BLASTp analysis (E-value < 1e-5), and the resulting data were grouped into gene families with OrthoMCL. To predict the expansion and contraction of the gene families and to infer lineage-specific gene gains and losses, the five genomes were analyzed using OrthoFinder v.2.2.6 (Emms and Kelly, 2015) and CAFE v.4.2.1 (De Bie et al., 2006) with default parameters. A dated species tree was downloaded from the TimeTree database (Kumar et al., 2017) and used as a guide tree. The birth-death parameter (lambda) was estimated using orthogroups in which no more than 100 genes were derived from a single genome.
Identification of Species-Specific Genes
To explore how many genes were unique to the P. cuspidatum genome, gene models were downloaded from 79 plant species, and the protein data for P. cuspidatum were compared with data from the 79 species by BLASTp (E-value < 1e-5). Only genes with no homologs in the 79 other species were retained; these genes were defined as species-specific genes.
RNA-Seq Data Analysis
The roots and mixed samples of aboveground tissues of 1-year-old plants were collected in three biological replicates, immediately frozen in liquid nitrogen, and stored at −80°C before an RNA extraction. The RNA-Seq libraries were constructed using an Illumina HiSeq Library Preparation Kit (Li et al., 2018). After validating the libraries, they were sequenced on the Illumina HiSeq platform. Trimmomatic software was used to filter out sequencing adapters and low-quality bases (Bolger et al., 2014). The clean data were mapped to the P. cuspidatum genome using HISAT, and gene expression levels were estimated using StringTie software (Kim et al., 2015; Pertea et al., 2015).
Phylogenetic Analysis
To identify homologous genes in different species, BLASTp analysis was performed using Arabidopsis query sequences (E-value < 1e-20). The results were manually checked based on gene annotation information. Partial genes with incomplete assembly or representing pseudogenes were excluded from further analysis. The protein sequences were aligned with ClustalX software (Chenna et al., 2003) and subjected to phylogenetic analysis with MEGA X software (Kumar et al., 2018). A phylogenetic tree was constructed using the neighbor-joining method with 500 bootstrap replicates.
Validation of RNA-Seq Data by Quantitative Real-Time RT-PCR (qRT-PCR)
qRT-PCR was performed as described previously with minor modification (Jiao et al., 2019; Zhang et al., 2019). Briefly, total RNA was isolated from roots and a mixture of aboveground tissues using Tranzol (Transgene) based on the manufacturer’s protocol (Li et al., 2019). cDNA was prepared using a PrimeScript RT Reagent Kit (RR047A, Takara). Relative expression levels were determined by qRT-PCR using the ABI 7500 Real-Time PCR system (Life Technologies). Three biological replicates and three qRT-PCR technical replicates were performed for each sample. The primer sequences used for qRT-PCR are listed in Supplementary Table S1.
Extraction and HPLC Analysis of Resveratrol and Emodin
After harvest, aboveground tissues and root tissues were collected and immediately dried in an oven at 105°C for 15 min, followed by 80°C until they were completely dry. Powdered dry samples were used for HPLC analysis using a SHIMADZU LC-20AT liquid chromatograph with a model SIL-10AF autosampler injector. The HPLC was equipped with a SPD-20A programmable multiwavelength ultraviolet (UV) detector. An InertSustain C18 column (250 mm × 4.6 mm, 5 µm) with a suitable guard column (C18, 7.5 mm × 4.6 mm, 5 µm) was used. The mobile phase consisted of deionized water and acetonitrile using the following gradient program: 28% for 0–7 min, 72% for 7–9 min, and 28% for 9–20 min, with a flow rate of 1.2 ml/min and column temperature of 25°C.
Results and Discussion
Genome Sequencing, Assembly, and Scaffolding
Since genome information for P. cuspidatum was not available, we sequenced 102 Gb of Illumina data to estimate the size and heterozygosity of the P. cuspidatum genome via genome survey analysis (Figure 1, Supplementary Table S2). The P. cuspidatum genome size is ca. 2.6 Gb. The Kmer distribution (Figure 1) indicates that the P. cuspidatum genome contains a large proportion of repeat sequences and high heterozygosity. The genome heterozygosity was estimated to be ca. 1.6%, while the GC content was predicted to be 37.1% (Supplementary Table S2). These results indicate that the P. cuspidatum genome is highly complex, making de novo assembly quite challenging.
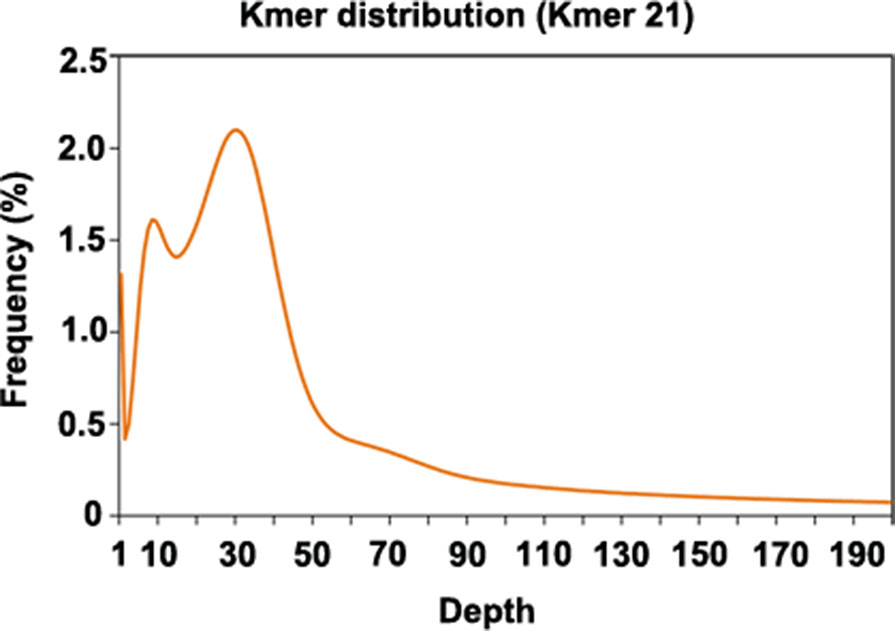
Figure 1 Kmer distribution of the genome survey result. The x-axis shows the depth of each Kmer, and the y-axis shows the frequency of each Kmer. In this analysis, all Kmers were 21 bp long.
Based on the genome survey, we constructed four libraries with different insertion lengths for sequencing, including insertion sizes of 550 bp, 2–3 kb, 5–7 kb, and 10–15 kb. The libraries were sequenced on the Illumina platform (150-bp paired-end reads, PE150). We also sequenced PE250 data for genome assembly. In total, we generated 377 Gb of short-read data (Supplementary Table S3). To conduct de novo assembly of P. cuspidatum, we used two different software programs, SOAPdenovo (Luo et al., 2012) and platanus (Kajitani et al., 2014). The assemblies from platanus were more complete than those generated using SOAPdenovo, which is consistent with the observation that platanus performs better for highly heterozygous genomes (Kajitani et al., 2014). Following contig and scaffold assembly, we performed gap filling and removed redundant reads. The final assembled genome was 2.56 Gb long, including 948,118 scaffolds (Table 1). We performed BUSCO analysis (Supplementary Table S4) using a plant genome dataset (Simão et al., 2015), finding that our assembly covered 76.1% of P. cuspidatum genes. These results indicate that our assembly should be referred to as a draft genome sequence, with a large number of scaffolds and assembly gaps. Nevertheless, this study represents the first attempt to assemble the P. cuspidatum genome, providing a valuable resource for the community.

Table 1 Summary of the genome assembly and annotation in P. cuspidatum.
TE and Functional Gene Annotations
Based on the genome assembly described above, we annotated the TEs in the P. cuspidatum genome. Based on this analysis, 71.54% of the sequences in the P. cuspidatum genome are TEs (2.0 Gb). Of these sequences, 55% were classified as class I TEs, including 30.68% gypsy-type long terminal repeats (LTRs), 9.88% copia LTRs, and 10.67% large retrotransposon derivatives (LARD) (Supplementary Table S5), whereas 4.59% of the sequences were classified as class II repeats. The proportions of these categories of TE sequences in the P. cuspidatum genome are in agreement with the finding that class I TEs comprise the largest proportion of all TEs in other major plant species (Quesneville et al., 2005; Flutre et al., 2011).
After masking these annotated TE sequences, we performed genome-wide gene prediction analysis of P. cuspidatum. To ensure accurate gene prediction, we conducted comparative transcriptome analysis between tissues sampled from roots and aboveground plant parts using a combination of de novo gene prediction, homology-based prediction, and transcriptome-based prediction. The results from the three approaches were integrated to constitute the final gene dataset (Supplementary Table S6). In total, 55,075 genes were predicted in the draft genome. Detailed characterization of the genes suggested that each gene possesses an average of four exons with an average length of 223 bp.
We aligned all predicted proteins against several databases to perform functional prediction, finding that 98.3% (54,155/55,075) of the genes could be annotated in at least one database (Supplementary Table S7). According to analysis of homologous genes in the GenBank Non-Redundant (nr) database, Beta vulgaris contains the highest proportion (12.39%) of these genes among species examined, indicating that P. cuspidatum and B. vulgaris share many homologous genes (Figure 2A). In addition, 29,674 genes could be assigned to at least one GO term in the categories: cellular component, molecular function, and biological process (Figure 2B). In the cellular component category, the most highly enriched GO terms were cell, membrane, organelle, and cell part. Two large groups of genes in the molecular function category were assigned to the GO terms “catalytic activity” and “binding,” indicating that P. cuspidatum contains versatile regulators of metabolite production. Consistently, most genes in the biological process category were assigned to GO terms “cellular process” and “metabolic process.”
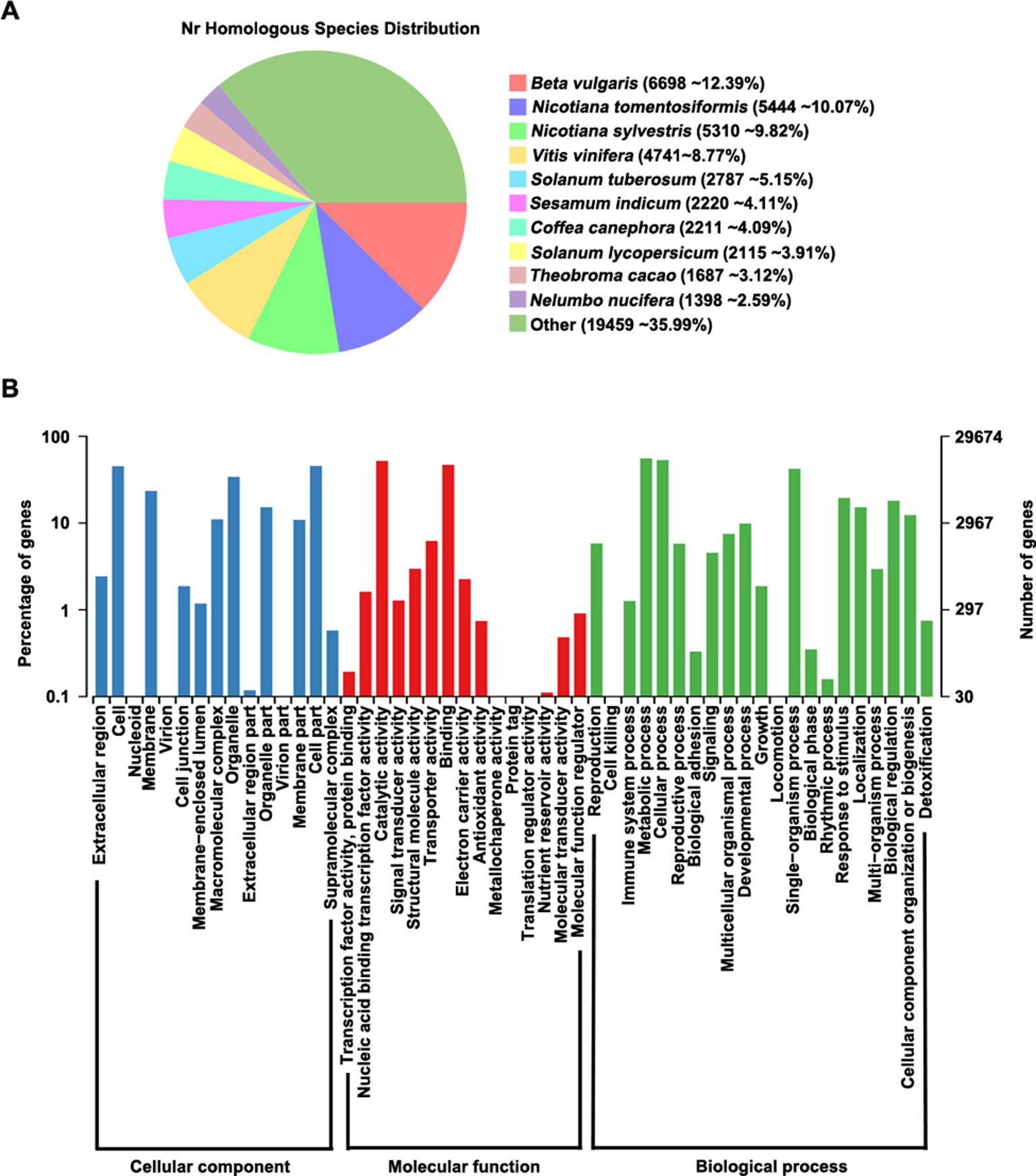
Figure 2 Gene annotation in P. cuspidatum. (A) Sequence alignment of all genes against the nr database. The proportions of genes with the closest homologs in different species are shown. (B) GO annotation of genes in P. cuspidatum. The GO terms were categorized into three different groups, including cellular component, molecular function, and biological process.
Transcription factors (TFs) play important roles in regulating secondary metabolism, development, and environmental responses in plants. Through a comparison with Arabidopsis, we identified 4,753 unigenes as putative TF genes (Supplementary Table S8). Of the annotated TFs, 2,075 (44%) were expressed [fragments per kilobase of transcript per million mapped reads (FPKM) > 0.3] in at least one tissue, including 52 TF genes specifically expressed in roots (FPKM of roots >2 and FPKM of aboveground tissues <0.3) and 132 specifically expressed in aboveground tissues (FPKM of aboveground tissue >2 and FPKM of roots <0.3) (Supplementary Table S8).
Classification of Gene Families and Characterization of Species-Specific Genes
To uncover functional gene families in P. cuspidatum, we compared all genes with those of four other species, including F. tataricum, S. lycopersicum, Arabidopsis, and V. vinifera. Among these, F. tataricum (Tartary buckwheat) is the most closely related species to P. cuspidatum, which also belongs to the Polygonaceae family. The reference genome of Tartary buckwheat was recently released (Zhang et al., 2017b). S. lycopersicum and Arabidopsis are representative eudicots and are widely studied model plants. V. vinifera, a well-known resveratrol-producing plant (Langcake and Pryce, 1976), is an excellent plant for genome duplication analysis since it experienced no duplication events after the gamma hexaploidization (Martin et al., 2010). OrthoMCL analysis (Li et al., 2003) categorized the P. cuspidatum genes into 16,661 families/clusters, 6,776 of which were shared by all five species (Figure 3A). By contrast, 2,386 families were unique in P. cuspidatum, which is larger than the number of unique gene families in any of the other species. We conducted detailed analysis of genes in these families, including 39,491 genes in P. cuspidatum. A total of 6,776 genes in each species were clustered as single-copy orthologs. Notably, P. cuspidatum had the largest number of multicopy orthologs among the five species. However, 6,817 P. cuspidatum genes were not clustered with genes from any of the other species (Figure 3B). Collectively, these results indicate that P. cuspidatum harbors many species-specific genes. Finally, we performed expansion and contraction analysis of the gene families in these five species. Overall, the P. cuspidatum genome contains many contracted gene families, whereas we detected the expansion of only a few families. This pattern differs from that of other species. Among the species examined, P. cuspidatum had the lowest ratio of contracted versus expanded gene families, with 1,220 contracted families and 6,449 expanded families (Figure 4A). Compared to the four other species, even including the closely related species F. tataricum, P. cuspidatum has experienced many more gene family expansions than contractions (6,449 expansions/1,220 contractions; Figure 4A), indicating that increasing numbers of gene gains and retentions have been occurring in the P. cuspidatum genome.
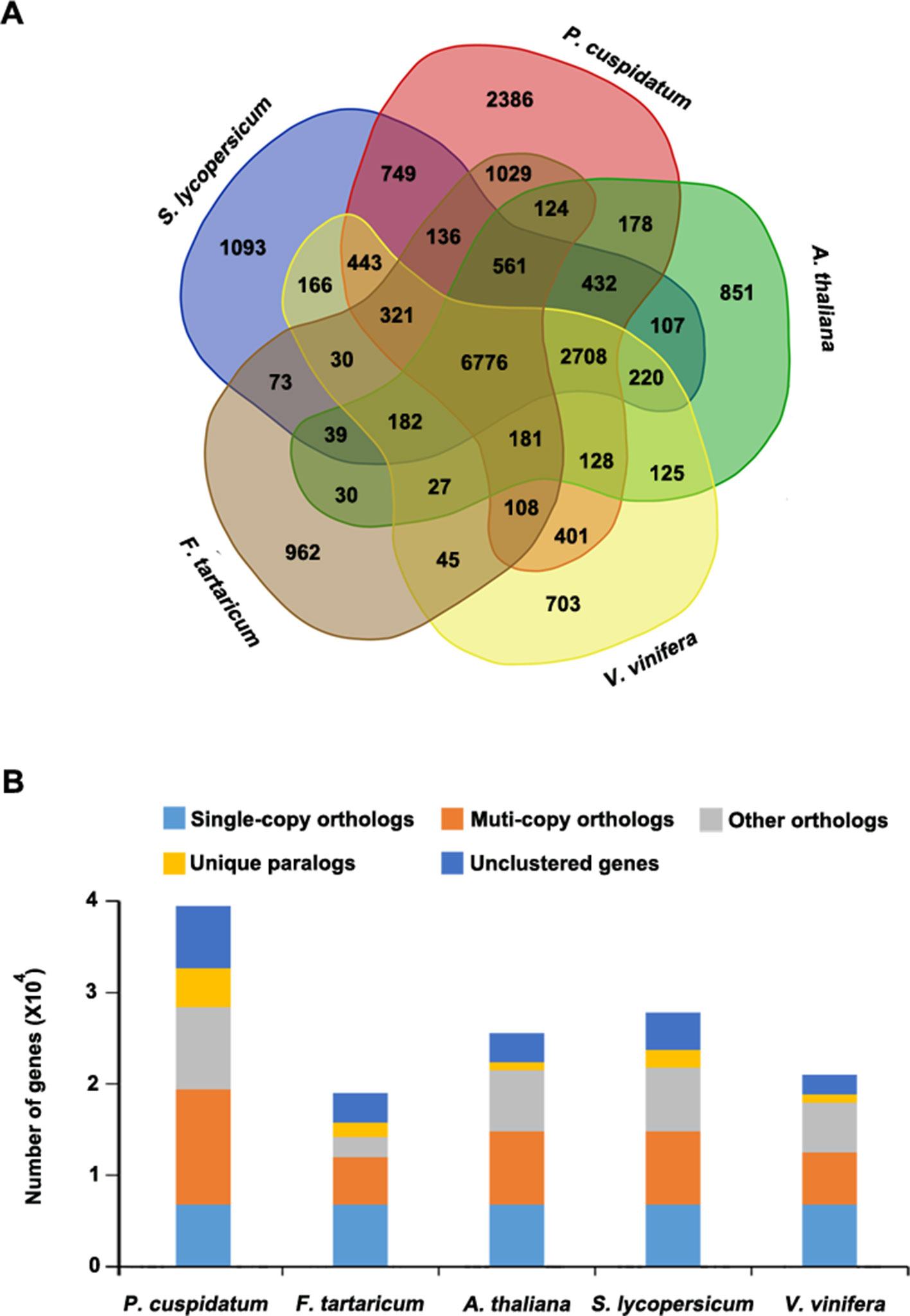
Figure 3 Classification of gene families. (A) Venn diagram showing the number of gene families in five plant species. Each number represents the number of gene families in each species or those shared by different species. The analysis was carried out using OrthoMCL software. (B) Summary of the number of genes in different groups. The genes were parsed from OrthoMCL clustering analysis.
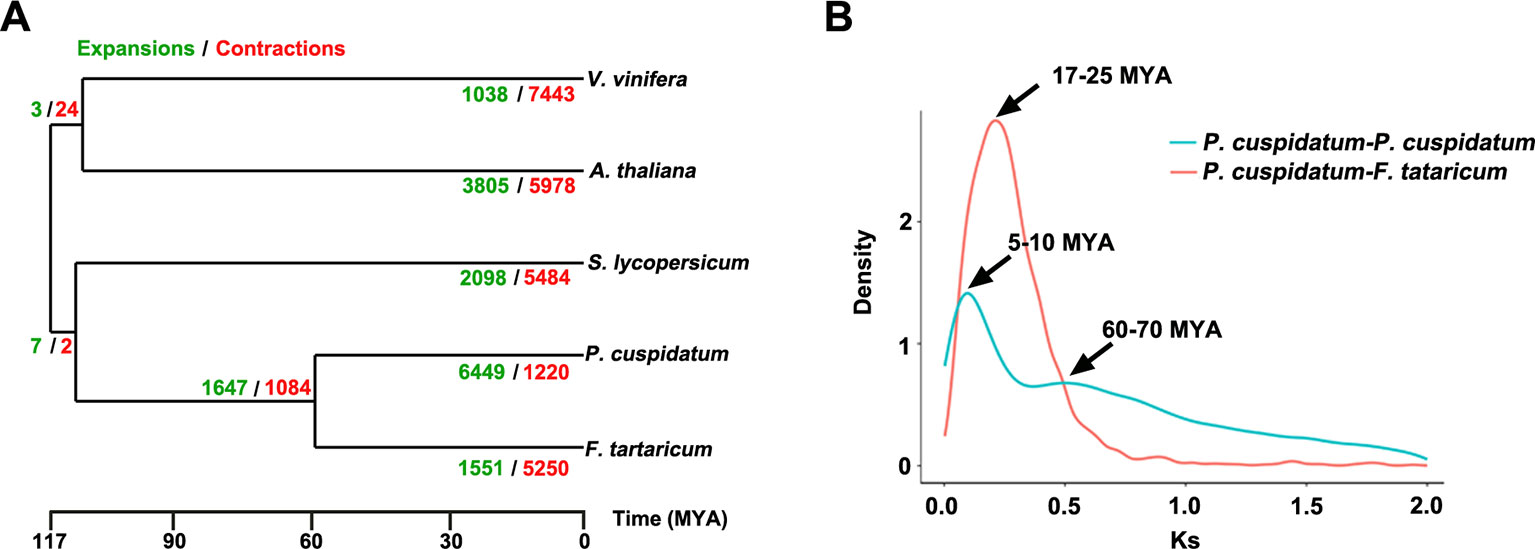
Figure 4 Dynamic evolution of gene families. (A) Gene family expansion and contraction in five plants species. The gene families that have undergone expansion and contraction are shown in green and red, respectively. The numbers separated by slashes (expansions/contractions) indicate the number of gene families. The scale bar indicates the divergence time [million years ago (MYA)]. (B) Ks distribution of paralogous gene pairs in P. cuspidatum and F. tataricum. In this analysis, two peaks were identified, which are thought to represent two WGD events. The x-axis shows Ks values, and the y-axis shows the density of distribution.
To further explore the species-specific genes in P. cuspidatum, we downloaded 79 publically available genomes (Goodstein et al., 2012) from various species, including 5 algae species and 74 plants, including bryophytes, ferns, gymnosperms, basal angiosperms, monocots, and dicots. We separately aligned all of the P. cuspidatum genes to each of the 79 genomes, finding that 1,159 genes had no homologs in any of the species (Supplementary Table S9; E-value < 1e-5). By integrating our RNA-Seq data, we determined that 509 genes were upregulated in roots compared to aboveground tissues (Supplementary Table S10), suggesting that these species-specific genes might play distinct roles in the corresponding tissues. Of these species-specific genes, 824 (71%) were annotated as genes of unknown function. Interestingly, we identified two gene families reported to be involved in stress responses, including seven conserved abscisic acid and water-deficit stress (ABA/WDS)-induced genes (EVM0009225, EVM0028174, EVM0029800, EVM0030471, EVM0042890, EVM0043230, and EVM0051394) that play vital roles in responses to abiotic stresses (such as water-deficit, salt, and cold stress) during senescence and fruit development (Çakir et al., 2003; Liu et al., 2019; Qu et al., 2019), as well as 14 cysteine-rich transmembrane module (TM) stress tolerance genes (EVM0004810, EVM0007286, EVM0010978, EVM0012820, EVM0019558, EVM0023411, EVM0025148, EVM0025816, EVM0026927, EVM0038117, EVM0038295, EVM0042574, EVM0044873, and EVM0047701) (Bhosale et al., 2018; Xiao et al., 2018). These results are consistent with the observation that P. cuspidatum has high levels of abiotic stress tolerance and that many stress factors induce the accumulation of resveratrol (Hasan and Bae, 2017), providing a molecular understanding of the interaction between P. cuspidatum and its growth environment. Our characterization of species-specific genes provides a valuable resource for further investigating these biological processes.
WGD Analysis
The genomes of flowering plants have undergone multiple WGD events with profound effects on genome organization, gene duplication, and fractionation (Zhang et al., 2017c). Whether the P. cuspidatum genome has undergone duplication events has not been previously addressed. Our assembly of the draft genome sequence of P. cuspidatum provides the opportunity to trace genome duplication events compared to other flowering plants. By performing all-versus-all sequence alignment, we identified 28,786 homologous gene pairs in P. cuspidatum, which allowed us to calculate the synonymous mutation rates (Ks) between single-copy collinear paralogous genes of P. cuspidatum and the closely related species F. tataricum at the whole-genome level. The analysis of Ks values revealed two peaks of distribution in P. cuspidatum (Figure 4B), suggesting that two WGD events have occurred approximately 6.6 million years ago (MYA) and 65 MYA. A comparison with F. tataricum (Zhang et al., 2017c) suggested that these plants shared a common WGD event at ca. 65 MYA. To further investigate the divergence time between P. cuspidatum and F. tataricum, we calculated the Ks values for single-copy genes via OrthoMCL analysis (Figure 4B). We identified a Ks peak at ∼21 MYA, which represents a speciation event between P. cuspidatum and F. tataricum. Collectively, these results indicate that P. cuspidatum had a WGD at 65 MYA, supporting the notion that many plant species shared a common WGD event at 65 MYA, which has facilitated plant survival during this period (Fawcett et al., 2009). After its divergence from a common ancestor with F. tataricum 21 MYA, P. cuspidatum experienced lineage-specific WGD at 6.6 MYA. This WGD analysis lays the foundation for genome evolution studies of P. cuspidatum in the future.
Characterization of Genes That Are Preferentially Expressed in Roots
Although crude P. cuspidatum root tissue serves as an effective agent in traditional Chinese medicine and a major source of bioactive metabolite derivatives (for instance, resveratrol and emodin), little is known about the regulation of the biosynthetic pathways of these compounds. We therefore investigated genes that are preferentially expressed in P. cuspidatum roots on a genome-wide scale by conducting comparative transcriptome analysis between roots and aboveground tissues based on genome assembly and gene annotation information.
RNA-Seq revealed 21,415 genes that were expressed in roots, 23,100 that were expressed in aboveground tissues (FPKM > 0.1), and 20,492 that were expressed in both tissues. Of these, 923 genes were expressed only in roots and 2,608 genes were expressed only in aboveground tissues (Supplementary Table S11). Finally, 24,023 genes were differentially expressed between tissues, including 2,173 genes that were upregulated in roots and 3,773 that were upregulated in aboveground tissues (Figure 5A, Supplementary Table S11).
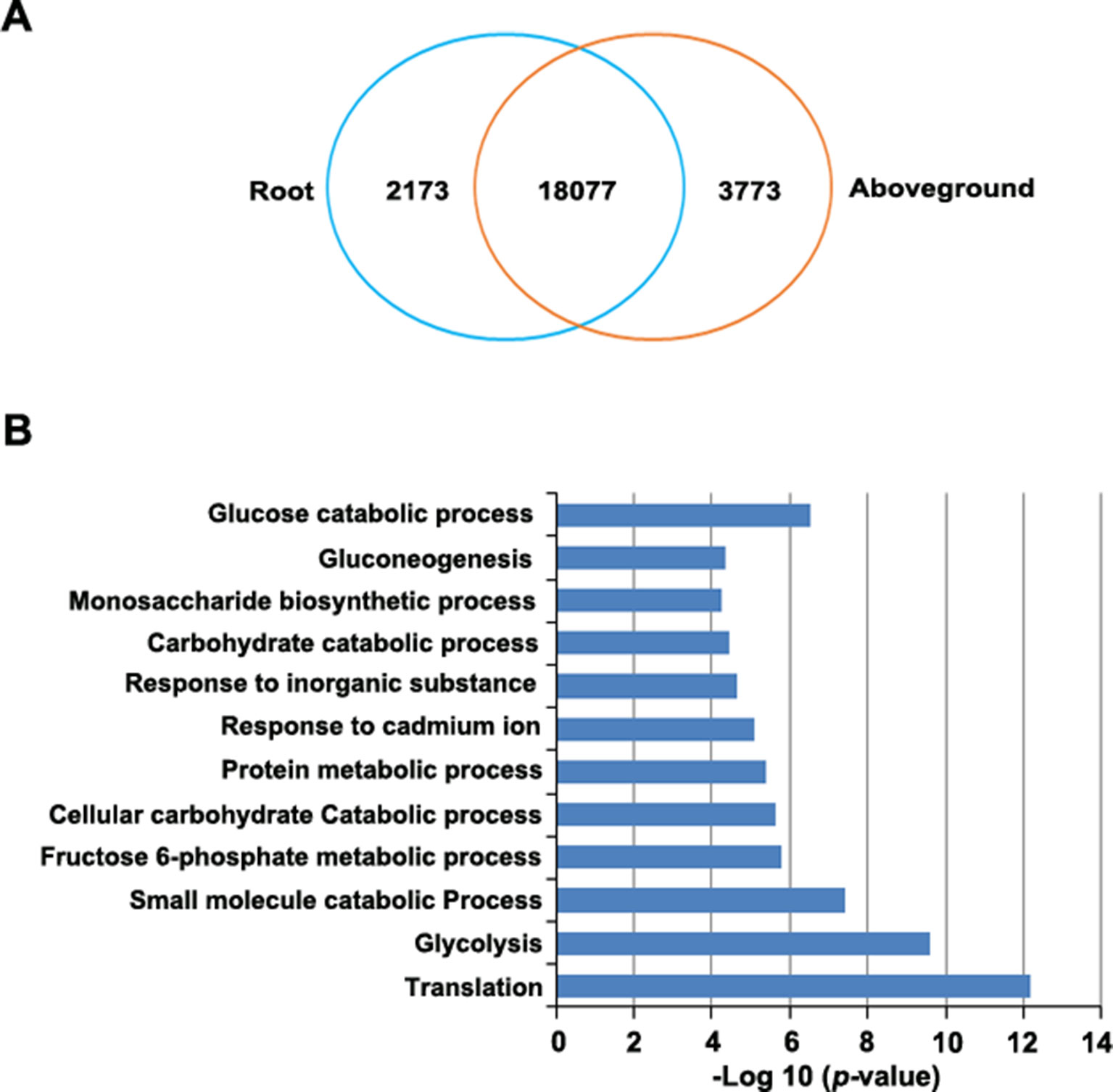
Figure 5 Identification of differentially expressed genes in roots versus aboveground tissues. (A) Venn diagram of upregulated genes in roots and aboveground tissues. The number in the overlapping region represents the number of genes that were expressed (FPKM > 0.1) in at least one sample but did not appear to be differentially expressed. The other numbers represent the number of genes that were upregulated in each sample. (B), GO enrichment analysis of genes that were upregulated in root samples. The most highly enriched GO terms in different biological process categories are shown.
To predict the functional roles of differentially expressed genes (DEGs), we performed GO enrichment analysis. DEGs that were upregulated in roots were highly enriched in several important biological processes, such as in glycolysis and gluconeogenesis and in the monosaccharide biosynthetic process, the fructose 6-phosphate metabolic process, and glucose catabolic process (Figure 5B, Supplementary Table S12). This observation is not unexpected since various phytochemicals, such as resveratrol and anthraquinone, are important for P. cuspidatum roots (Zhang et al., 2013). By contrast, most upregulated genes in aboveground tissues were enriched in GO terms associated with various fundamental pathways, such as photosynthesis (Supplementary Table S12).
Since TFs play important roles in regulating basic biological processes, we investigated TF genes that are specifically expressed in roots to determine whether they function in the regulation of root development in P. cuspidatum. Several important TF families were identified (Supplementary Table S8), such as WRKYs (4), which play roles in abiotic and/or biotic stress responses; MYBs (5), which are important regulators of metabolite biosynthesis; lateral organ boundary domain (LBDs) (5), which are involved in lateral root development; NAM, ATAF, and CUC (NACs) (3) and APETALA2/ethylene responsive factors (AP2/ERFs) (7), which regulate the formation of ground tissues in roots; basic helix–loop–helix (bHLH) (3), most of which are involved in root hair development and auxin response factors (ARFs) (1), which modulate auxin responses during root formation. These results provide a basis for further functional analysis of genes that contribute to the formation of root architecture and the production of bioactive metabolite derivatives in P. cuspidatum roots.
Genes Involved in the Resveratrol Biosynthetic Pathway
Although proposed resveratrol biosynthetic pathways have been described for several plant species (Lu et al., 2016), and resveratrol is known to be produced via a metabolic pathway that largely overlaps with the phenylpropanoid pathway (Figure 6A), genetic information about the enzymes in P. cuspidatum on a whole-genome scale is still lacking. To address this issue, we identified and investigated the expression of the four known major players in the resveratrol biosynthetic pathway, including PAL, C4H, 4CL, and STS. PAL, C4H, and 4CL are members of the common phenylpropanoid pathway in plants, which synthesizes phenolic compounds, whereas STS, a member of the type III polyketide synthase STS/CHS superfamily, catalyzes the condensation of resveratrol in the final step of this pathway (Hao et al., 2012). We identified six PAL, four C4H, four 4CL, and nine STS/CHS genes in P. cuspidatum (Figure 6A; Supplementary Table S13). Since we could not identify the functions of novel genes in P. cuspidatum through complementation experiments, we performed phylogenetic analysis to compare STS/CHS genes with those from S. lycopersicum, Arabidopsis, V. vinifera, and the closely related species F. tataricum to elucidate their potential molecular characteristics (Figure 6B). All STS/CHS genes in P. cuspidatum had homologous genes in the four other species (Figure 6). Interestingly, a clade of CHS genes in P. cuspidatum appears to have been duplicated compared to those in F. tataricum. Of these genes, one gene was upregulated in roots, whereas the others were expressed at very high levels in both roots and other tissues (Figure 6B). The expansion of STS/CHS genes might be related to the importance of resveratrol and flavonoid metabolism in P. cuspidatum roots.
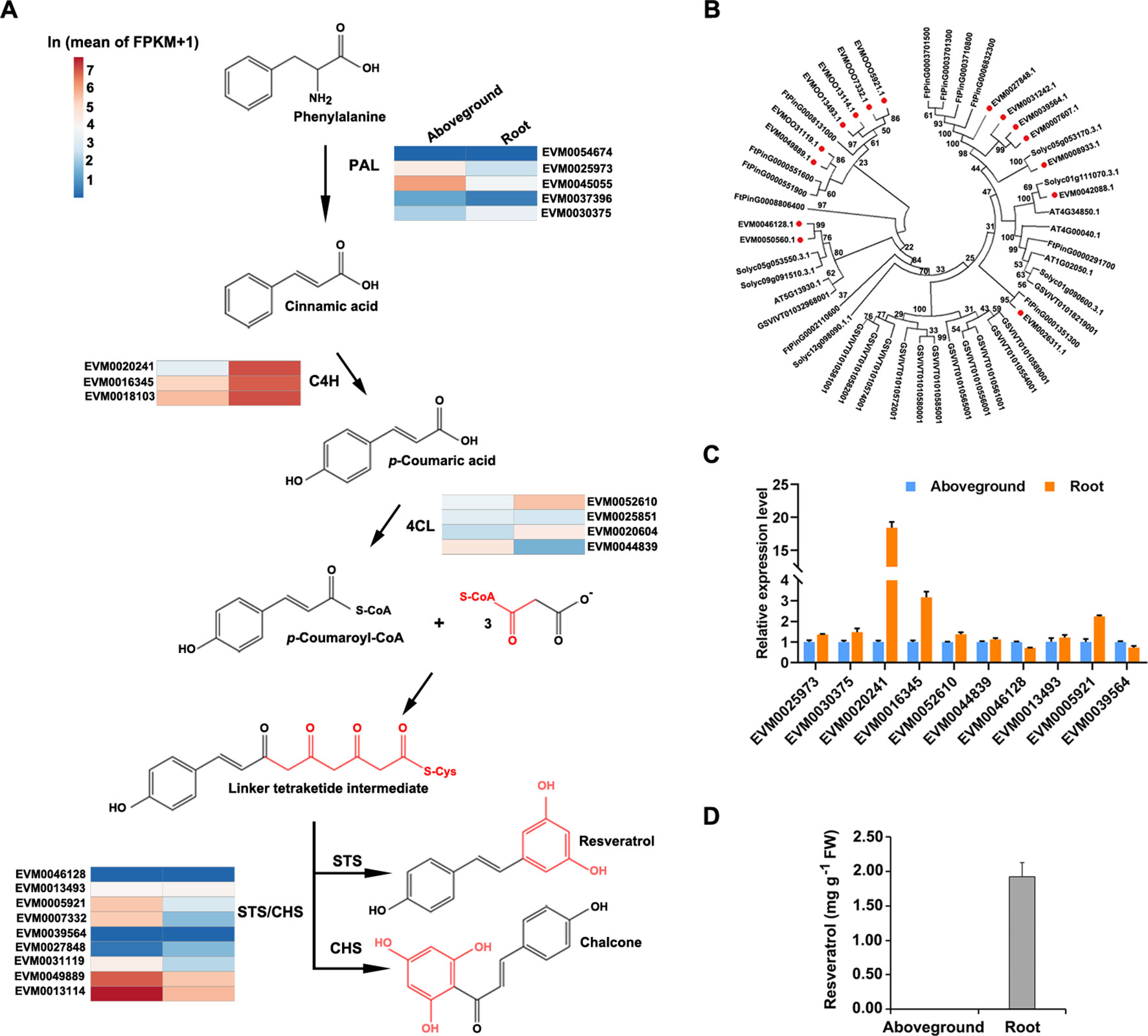
Figure 6 Phylogenetic and expression analysis of gene families involved in the resveratrol biosynthetic pathway in five species. (A), Proposed pathway for resveratrol biosynthesis derived from the phenylpropanoid pathway in P. cuspidatum. PAL, phenylalanine ammonia lyase; C4H, cinnamic acid 4-hydroxylase; 4CL, 4-coumarate CoA ligase; STS, stilbene synthase; CHS, chalcone synthase. The colors represent the expression levels of each gene in ln(FPKM) in aboveground and root tissues. (B), Phylogenetic tree of the STS/CHS gene family. Genes in P. cuspidatum are indicated by red dots. The tree was constructed using the neighbor-joining method with 500 bootstrap replicates. (C), Expression analysis of the representative resveratrol biosynthetic genes, as determined by qRT-PCR. (D), Resveratrol levels in roots and aboveground tissues. Each bar represents the mean value (n = 3). Error bars represent the SD.
To experimentally validate the expression patterns of genes identified by transcriptome sequencing, we subjected the genes involved in resveratrol biosynthesis to qRT-PCR analysis. Several resveratrol biosynthesis genes were upregulated in roots compared to aboveground tissues, such as C4H (EVM0020241), 4CL (EVM0052610), and STS/CHS (EVM0039564) (Figure 6C), which is consistent with the transcriptome data. These results suggest that resveratrol is primarily synthesized in root tissue. To confirm this notion, we measured the levels of resveratrol and the anthraquinone emodin in various tissues by HPLC and determined that resveratrol is mainly present in root tissue (Figure 6D; Supplementary Figure S1D). Even though some biosynthetic genes were also expressed in aboveground tissues, we could only detect resveratrol and emodin in the root tissue. This discrepancy between the activity of biosynthetic genes and their product might be caused by a common explanation that gene expression at transcriptional level sometimes doesn’t properly reflect the level of its products since there are also post-transcriptional and post-translational regulations. Alternatively, even if they might be produced at both root and aboveground tissues, resveratrol and emodin might accumulate predominately in the root tissue through unknown transportation mechanisms. These findings suggest that the resveratrol metabolic pathway is activated in P. cuspidatum roots.
Genes Involved in the Anthraquinone Biosynthetic Pathway
Anthraquinones such as emodin and physcion are another group of major pharmaceutically active compounds in P. cuspidatum (Hong et al., 2016). The MVA, MEP, and shikimate pathways are involved in anthraquinone biosynthesis in plants (Wijnsma and Verpoorte, 1986; Han et al., 2001; Hao et al., 2012; Zhang et al., 2013). Although the detailed metabolic pathways are currently unknown, we identified 14, 14, and 16 unigenes that were assigned to these three metabolic pathways, respectively (Supplementary Table S13). We examined the expression levels of all genes in these pathways based on the transcriptome data (Supplementary Figure S1), which indicated that MVA pathway genes (e.g., EVM0022304) involved in anthraquinone biosynthesis are actively expressed in P. cuspidatum roots.
Concluding Remarks
Due to the lack of genomic information about P. cuspidatum, little is known about this plant at the molecular level. The lack of genomic information has hindered the development of molecular markers to identify different varieties of P. cuspidatum, as well as investigations of the functions of key chemical components in P. cuspidatum. Here, we assembled a draft genome of P. cuspidatum based on Illumina short-read sequencing data. It is challenging to perform de novo assembly of large genomes with a high degree of heterozygosity. Since we determined that the P. cuspidatum genome is highly complex, with a high proportion of repeats and high heterogeneity, considerable effort will be needed to obtain a high-quality genome assembly of this plant in the future. Even though the draft genome sequence produced in this study contains many assembly gaps and perhaps assembly errors in highly complex regions, this study represents the first attempt to assemble such a sequence. The genome sequence obtained here should accelerate genomic and molecular studies of P. cuspidatum.
In this study, we identified 55,075 predicted genes in P. cuspidatum by integrating three approaches. These genes were categorized into gene families, including 1,159 that were identified as species-specific genes. By integrating our RNA-Seq data, we identified genes that are preferentially expressed in roots, which were predicted to be related to the unique biology of roots. For example, the STS/CHS gene family appears to have expanded and is preferentially expressed in roots, which might contribute to resveratrol biosynthesis. The results of this study provide a reference for future detailed analysis of the metabolic pathways in P. cuspidatum and could facilitate the utilization of P. cuspidatum as an important medical herb.
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in BIG Data Center (Wang et al., 2017b; Members, 2019), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA001939, CRA001941 that are publicly accessible at https://bigd.big.ac.cn/gsa.
Author Contributions
CL, YZhang, and GW designed the study. LZ, YZhang, YZheng, YZhao, PH, XX, XH, QC, HL, CZ, ZH and XW performed the research. CL, LZ, KF, YZhang, and GW wrote the paper. All the authors analyzed the data, discussed the results, and made comments on the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (31701294, 31801210, and 31771556), the Cultivating Project for Young Scholar at Hubei University of Medicine (2017QDJZR26, 2016QDJZR11, and 2016QDJZR14), the Natural Science Foundation of Hubei Provincial Department of Education (Q20182104), the Fundamental Research Funds for the Central Universities (GK201702016), Hubei Provincial Natural Science Foundation of China (2017CFB674), the Foundation of Health Commission of Hubei Province (ZY2019Q004), Open Research Fund of Key Laboratory of Medicinal Resources and Natural Pharmaceutical Chemistry Ministry of Education (2019005), Fund for Key Laboratory Construction of Hubei Province (2018BFC360, WLSP201905), Hubei Provincial Outstanding Young and Middle-aged Science and Technology Innovation Team Project (Grant No. T201813), the Scientific and Technological Project of Shiyan City of Hubei Province (18Y06) and the Hubei Provincial Technology Innovation Project (2017ACA176).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Dr. Qinghua Zhang for his assistance with experiments and helpful suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01274/full#supplementary-material
References
Abu-Amero, K. K., Kondkar, A. A., Chalam, K. V. (2016). Resveratrol and ophthalmic diseases. Nutrients 8, 200–200. doi: 10.3390/nu8040200
Austin, M. B., Noel, J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110. doi: 10.1039/b100917f
Baltes, N. J., Voytas, D. F. (2015). Enabling plant synthetic biology through genome engineering. Trends Biotechnol. 33, 120–131. doi: 10.1016/j.tibtech.2014.11.008
Bhosale, R., Boudolf, V., Cuevas, F., Lu, R., Eekhout, T., Hu, Z. B., et al. (2018). A spatiotemporal DNA endoploidy map of the Arabidopsis root reveals roles for the endocycle in root development and stress adaptation. Plant Cell 30, 2330–2351. doi: 10.1105/tpc.17.00983
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burdette, A., Garner, P. L., Mayer, E. P., Hargrove, J. L., Hartle, D. K., Greenspan, P. (2010). Anti-inflammatory activity of select sorghum (Sorghum bicolor) brans. J. Med. Food 13, 879–887. doi: 10.1089/jmf.2009.0147
Çakir, B., Agasse, A., Gaillard, C., Saumonneau, A., Delrot, S., Atanassova, R. (2003). A grape ASR protein involved in sugar and abscisic acid signaling. Plant Cell 15, 2165–2180. doi: 10.1105/tpc.013854
Chen, S., Song, J. (2016). Herbgenomics. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J. Chin. Mater. Med. 41, 3881–3889. doi: 10.4268/cjcmm20162101
Chen, S., Xu, J., Liu, C., Zhu, Y., Nelson, D. R., Zhou, S., et al. (2012). Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 3, 913. doi: 10.1038/ncomms1923
Chen, X., Gao, H., Han, Y., Ye, J., Xie, J., Wang, C. (2015). Physcion induces mitochondria-driven apoptosis in colorectal cancer cells via downregulating EMMPRIN. Eur. J. Pharmacol. 764, 124–133. doi: 10.1016/j.ejphar.2015.07.008
Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G., et al. (2003). Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500. doi: 10.1093/nar/gkg500
Chong, J., Poutaraud, A., Hugueney, P. (2009). Metabolism and roles of stilbenes in plants. Plant Sci. 177, 143–155. doi: 10.1016/j.plantsci.2009.05.012
Clements, D. R., Ditommaso, A. (2012). Predicting weed invasion in Canada under climate change: evaluating evolutionary potential. Can. J. Plant Sci. 92, 1013–1020. doi: 10.4141/cjps2011-280
Counet, C., Callemien, D., Collin, S. (2006). Chocolate and cocoa: New sources of trans-resveratrol and trans-piceid. Food Chem. 98, 649–657. doi: 10.1016/j.foodchem.2005.06.030
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Dommanget, F., Cavaillé, P., Evette, A., Martin, F. (2016). “Asian knotweeds-an example of a raising threat?”. Introduced tree species in European forests: opportunities and challenges. European Forest Institute, 202–211. doi: 10.1007/978-3-7091-8846-0
Donnez, D., Jeandet, P., Clement, C., Courot, E. (2009). Bioproduction of resveratrol and stilbene derivatives by plant cells and microorganisms. Trends Biotechnol. 27, 706–713. doi: 10.1016/j.tibtech.2009.09.005
Edgar, R. C., Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158. doi: 10.1093/bioinformatics/bti1003
Emms, D. M., Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Esvelt, K. M., Wang, H. H. (2013). Genome-scale engineering for systems and synthetic biology. Mol. Syst. Biol. 9, 641. doi: 10.1038/msb.2012.66
Fawcett, J. A., Maere, S., Van De Peer, Y. (2009). Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proc. Natl. Acad. Sci. 106, 5737–5742. doi: 10.1073/pnas.0900906106
Ferrer, J.-L., Jez, J. M., Bowman, M. E., Dixon, R. A., Noel, J. P. (1999). Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Mol. Biol. 6, 775. doi: 10.1038/11553
Flutre, T., Duprat, E., Feuillet, C., Quesneville, H. (2011). Considering transposable element diversification in de novo annotation approaches. PloS One 6, e16526. doi: 10.1371/journal.pone.0016526
Funa, N., Ozawa, H., Hirata, A., Horinouchi, S. (2006). Phenolic lipid synthesis by type III polyketide synthases is essential for cyst formation in Azotobacter vinelandii. Proc. Natl. Acad. Sci. 103, 6356–6361. doi: 10.1073/pnas.0511227103
Gao, F., Liu, W., Guo, Q., Bai, Y., Yang, H., Chen, H. (2017). Physcion blocks cell cycle and induces apoptosis in human B cell precursor acute lymphoblastic leukemia cells by downregulating HOXA5. Biomed. Pharmacother. 94, 850–857. doi: 10.1016/j.biopha.2017.07.149
Geng, Q., Wei, Q., Wang, S., Qi, H., Zhu, Q., Liu, X., et al. (2018). Physcion 8-O-β-glucopyranoside extracted from Polygonum cuspidatum exhibits anti-proliferative and anti-inflammatory effects on MH7A rheumatoid arthritis-derived fibroblast-like synoviocytes through the TGF-β/MAPK pathway. Int. J. Mol. Med. 42, 745–754. doi: 10.3892/ijmm.2018.3649
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Han, Y.-S., Heijden, R. V. D., Lefeber, A. W. M., Erkelens, C., Verpoorte, R. (2002). Biosynthesis of anthraquinones in cell cultures of Cinchona “Robusta” proceeds via the methylerythritol 4-phosphate pathway. Phytochemistry 59, 45–55. doi: 10.1016/S0031-9422(01)00296-5
Han, Y.-S., Van Der Heijden, R., Verpoorte, R. (2001). Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tissue Organ Cult. 67, 201–220. doi: 10.1023/A:1012758922713
Han, Y.-T., Chen, X.-H., Gao, H., Ye, J.-L., Wang, C.-B. (2016). Physcion inhibits the metastatic potential of human colorectal cancer SW620 cells in vitro by suppressing the transcription factor SOX2. Acta Pharmacol. Sin. 37, 264. doi: 10.1038/aps.2015.115
Han, Y., Wessler, S. R. (2010). MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199–e199. doi: 10.1093/nar/gkq862
Hao, D., Ma, P., Mu, J., Chen, S., Xiao, P., Peng, Y., et al. (2012). De novo characterization of the root transcriptome of a traditional Chinese medicinal plant Polygonum cuspidatum. Sci. China Life Sci. 55, 452–466. doi: 10.1007/s11427-012-4319-6
Hasan, M., Bae, H. (2017) An overview of stress-induced resveratrol synthesis in grapes: perspectives for resveratrol-enriched grape products. Molecules 22, 294. doi: 10.3390/molecules22020294
Hoede, C., Arnoux, S., Moisset, M., Chaumier, T., Inizan, O., Jamilloux, V., et al. (2014). PASTEC: an automatic transposable element classification tool. Plos One 9, e91929. doi: 10.1371/journal.pone.0091929
Hon, C., Chow, Y., Zeng, F., Leung, F. (2003). Genetic authentication of ginseng and other traditional Chinese medicine. Acta Pharmacol. Sin. 24, 841–846.
Hong, M., Tan, H. Y., Li, S., Cheung, F., Wang, N., Nagamatsu, T., et al. (2016). Cancer stem cells: the potential targets of Chinese medicines and their active compounds. Int. J. Mol. Sci. 17 (6), 893. doi: 10.3390/ijms17060893
Huang, L.-F., Wang, Z.-H., Chen, S.-L. (2014). Hypericin: chemical synthesis and biosynthesis. Chin. J. Nat. Med. 12, 81–88. doi: 10.1016/S1875-5364(14)60014-5
Jayasuriya, H., Koonchanok, N. M., Geahlen, R. L., Mclaughlin, J. L., Chang, C.-J. (1992). Emodin, a protein tyrosine kinase inhibitor from Polygonum cuspidatum. J. Nat. Prod. 55, 696–698. doi: 10.1021/np50083a026
Jiang, J., Xi, H., Dai, Z., Lecourieux, F., Yuan, L., Liu, X., et al. (2018). VvWRKY8 represses stilbene synthase gene through direct interaction with VvMYB14 to control resveratrol biosynthesis in grapevine. J. Exp. Bot. 70(2), 715-729. doi: 10.1093/jxb/ery401
Jiao, K., Li, X., Guo, Y., Guan, Y., Guo, W., Luo, D., et al. (2019). Regulation of compound leaf development in mungbean (Vigna radiata L.) by cup-shaped cotyledon/no apical meristem (CUC/NAM) gene. Planta 249, 765–774. doi: 10.1007/s00425-018-3038-z
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., Hartung, F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89–e89. doi: 10.1093/nar/gkw092
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357. doi: 10.1038/nmeth.3317
Kiselev, K. V. (2011). Perspectives for production and application of resveratrol. Appl. Microbiol. Biotechnol. 90, 417–425. doi: 10.1007/s00253-011-3184-8
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., Suleski, M., Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34(7), 1812–1819. doi: 10.1093/molbev/msx116
Langcake, P., Pryce, R. (1976). The production of resveratrol by Vitis vinifera and other members of the Vitaceae as a response to infection or injury. Physiol. Plant Pathol. 9, 77–86. doi: 10.1016/0048-4059(76)90077-1
Li, L., Hou, M. J., Cao, L., Xia, Y., Shen, Z. G., Hu, Z. B. (2018). Glutathione S-transferases modulate Cu tolerance in Oryza sativa. Environ. Exp. Bot. 155, 313–320. doi: 10.1016/j.envexpbot.2018.07.007
Li, L., Stoeckert, C. J., Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, X., Liu, W., Zhuang, L., Zhu, Y., Wang, F., Chen, T., et al. (2019). Bigger organs and elephant ear-like leaf1 control organ size and floral organ internal asymmetry in pea. J. Exp. Bot. 70, 179–191. doi: 10.1093/jxb/ery352
Li, Y., Tian, S., Yang, X., Wang, X., Guo, Y., Ni, H. (2016). Transcriptomic analysis reveals distinct resistant response by physcion and chrysophanol against cucumber powdery mildew. PeerJ 4, e1991. doi: 10.7717/peerj.1991
Lin, R., Elf, S., Shan, C., Kang, H.-B., Ji, Q., Zhou, L., et al. (2015). 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumor growth by inhibiting LKB1-AMPK signaling. Nat. Cell Biol. 17, 1484.
Liu, C., Huang, Y. (2016). Chinese herbal medicine on cardiovascular diseases and the mechanisms of action. Front. Pharmacol. 7, 469–469. doi: 10.3389/fphar.2016.00469
Liu, H., Guo, S., Lu, M., Zhang, Y., Li, J., Wang, W., et al. (2019). Biosynthesis of DHGA 12 and its roles in Arabidopsis seedling establishment. Nat. Commun. 10, 1768. doi: 10.1038/s41467-019-09467-5
Liu, W., He, J., Yang, Y., Guo, Q., Gao, F. (2016). Upregulating miR-146a by physcion reverses multidrug resistance in human chronic myelogenous leukemia K562/ADM cells. Am. J. Cancer Res. 6, 2547.
Liu, X., Liu, Y., Huang, P., Ma, Y., Qing, Z., Tang, Q., et al. (2017). The genome of medicinal plant Macleaya cordata provides new insights into benzylisoquinoline alkaloids metabolism. Mol. Plant 10, 975–989. doi: 10.1016/j.molp.2017.05.007
Lu, Y., Shao, D., Shi, J., Huang, Q., Yang, H., Jin, M. (2016). Strategies for enhancing resveratrol production and the expression of pathway enzymes. Appl. Microbiol. Biotechnol. 100, 7407–7421. doi: 10.1007/s00253-016-7723-1
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1(1), 18. doi: 10.1186/2047-217X-1-18
Lyons, M. M., Yu, C., Toma, R., Cho, S. Y., Reiboldt, W., Lee, J., et al. (2003). Resveratrol in raw and baked blueberries and bilberries. J. Agric. Food Chem. 51, 5867–5870. doi: 10.1021/jf034150f
Ma, Y., Yuan, L., Wu, B., Li, X. E., Chen, S., Lu, S. (2012). Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J. Exp. Bot. 63, 2809–2823. doi: 10.1093/jxb/err466
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Martin, D. M., Aubourg, S., Schouwey, M. B., Daviet, L., Schalk, M., Toub, O., et al. (2010). Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 10, 226. doi: 10.1186/1471-2229-10-226
Mei, Y. Z., Liu, R. X., Wang, D. P., Wang, X., Dai, C. C. (2015). Biocatalysis and biotransformation of resveratrol in microorganisms. Biotechnol. Lett. 37, 9–18. doi: 10.1007/s10529-014-1651-x
Melchior, F., Kindl, H. (1990). Grapevine stilbene synthase cDNA only slightly differing from chalcone synthase cDNA is expressed in Escherichia coli into a catalytically active enzyme. FEBS Lett. 268, 17–20. doi: 10.1016/0014-5793(90)80961-H
Members, B.I.G.D.C. (2019). Database resources of the BIG Data Center in 2019. Nucleic Acids Res. 47, D8–D14.
Michalet, S., Rouifed, S., Pellassa-Simon, T., Fusade-Boyer, M., Meiffren, G., Nazaret, S., et al. (2017). Tolerance of Japanese knotweed s. l. to soil artificial polymetallic pollution: early metabolic responses and performance during vegetative multiplication. Environ. Sci. Pollut. Res. 24, 20897–20907. doi: 10.1007/s11356-017-9716-8
Murrell, C., Gerber, E., Krebs, C., Parepa, M., Schaffner, U., Bossdorf, O. (2011). Invasive knotweed affects native plants through allelopathy. Am. J. Bot. 98, 38–43. doi: 10.3732/ajb.1000135
Nadalin, F., Vezzi, F., Policriti, A. (2012). GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13, S8. doi: 10.1186/1471-2105-13-S14-S8
Narender, T., Sukanya, P., Sharma, K., Bathula, S. R. (2013). Apoptosis and DNA intercalating activities of novel emodin derivatives. RSC Adv. 3, 6123–6131. doi: 10.1039/c3ra23149f
Olaru, O. T., Venables, L., Van De Venter, M., Nitulescu, G. M., Margina, D., Spandidos, D. A., et al. (2015). Anticancer potential of selected Fallopia Adans species. Oncol. Lett. 10, 1323–1332. doi: 10.3892/ol.2015.3453
Olsen, B. B., Bjørling-Poulsen, M., Guerra, B. (2007). Emodin negatively affects the phosphoinositide 3-kinase/AKT signalling pathway: a study on its mechanism of action. Int. J. Biochem. Cell Biol. 39, 227–237. doi: 10.1016/j.biocel.2006.08.006
Ouyang, L., Luo, Y., Tian, M., Zhang, S. Y., Lu, R., Wang, J. H., et al. (2014). Plant natural products: from traditional compounds to new emerging drugs in cancer therapy. Cell Prolif. 47, 506–515. doi: 10.1111/cpr.12143
Pan, X., Wang, H., Tong, D., Wang, C., Sun, L., Zhao, C., et al. (2016). Physcion induces apoptosis in hepatocellular carcinoma by modulating miR-370. Am. J. Cancer Res. 6, 2919.
Peng, W., Qin, R., Li, X., Zhou, H. (2013). Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb. et Zucc.: a review. J. Ethnopharmacol. 148, 729–745. doi: 10.1016/j.jep.2013.05.007
Perassolo, M., Quevedo, C. V., Busto, V. D., Giulietti, A. M., Talou, J. R. (2011). Role of reactive oxygen species and proline cycle in anthraquinone accumulation in Rubia tinctorum cell suspension cultures subjected to methyl jasmonate elicitation. Plant Physiol. Biochem. 49, 758–763. doi: 10.1016/j.plaphy.2011.03.015
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290. doi: 10.1038/nbt.3122
Price, A. L., Jones, N. C., Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Pryszcz, L. P., Gabaldón, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113–e113. doi: 10.1093/nar/gkw294
Qu, X., Cao, B., Kang, J., Wang, X., Han, X., Jiang, W., et al. (2019). Fine-tuning stomatal movement through small signaling peptides. Front. Plant Sci. 10, 1–6. doi: 10.3389/fpls.2019.00069
Quesneville, H., Bergman, C. M., Andrieu, O., Autard, D., Nouaud, D., Ashburner, M., et al. (2005). Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 1, e22. doi: 10.1371/journal.pcbi.0010022
Rouifed, S., Byczek, C., Laffray, D., Piola, F. (2012). Invasive knotweeds are highly tolerant to salt stress. Environ. Manage. 50, 1027–1034. doi: 10.1007/s00267-012-9934-2
Sanders, T. H., Mcmichael, R. W., Hendrix, K. W. (2000). Occurrence of resveratrol in edible peanuts. J. Agric. Food Chem. 48, 1243–1246. doi: 10.1021/jf990737b
Schröder, G., Brown, J. W., Schr der, J. (1988). Molecular analysis of resveratrol synthase: cDNA, genomic clones and relationship with chalcone synthase. Eur. J. Biochem. 172, 161–169. doi: 10.1111/j.1432-1033.1988.tb13868.x
Shan, B., Cai, Y.-Z., Brooks, J. D., Corke, H. (2008). Antibacterial properties of Polygonum cuspidatum roots and their major bioactive constituents. Food Chem.109, 530–537. doi: 10.1016/j.foodchem.2007.12.064
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sohn, E., Kim, J., Kim, C. S., Jo, K., Kim, J. S. (2015). Extract of rhizoma Polygonum cuspidatum reduces early renal podocyte injury in streptozotocin-induced diabetic rats and its active compound emodin inhibits methylglyoxal-mediated glycation of proteins. Mol. Med. Rep. 12, 5837–5845. doi: 10.3892/mmr.2015.4214
Song, C., Liu, Y., Song, A., Dong, G., Zhao, H., Sun, W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant 11, 1482–1491. doi: 10.1016/j.molp.2018.10.003
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Su, Y.-T., Chang, H.-L., Shyue, S.-K., Hsu, S.-L. (2005). Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem. Pharmacol. 70, 229–241. doi: 10.1016/j.bcp.2005.04.026
Sun, W., Leng, L., Yin, Q., Xu, M., Huang, M., Xu, Z., et al. (2019). The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J. 97, 841–857. doi: 10.1111/tpj.14162
Tarailo-Graovac, M., Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 25, 4.10.11–14.10.14. doi: 10.1002/0471250953.bi0410s25
Teich, L., Daub, K. S., Krügel, V., Nissler, L., Gebhardt, R., Eger, K. (2004). Synthesis and biological evaluation of new derivatives of emodin. Bioorg. Med. Chem. 12, 5961–5971. doi: 10.1016/j.bmc.2004.08.024
Tropf, S., Kärcher, B., Schröder, G., Schröder, J. (1995). Reaction mechanisms of homodimeric plant polyketide synthases (stilbene and chalcone synthase): a single active site for the condensing reaction is sufficient for synthesis of stilbenes, chalcones, and 6′-deoxychalcones. J. Biol. Chem. 270, 7922–7928. doi: 10.1074/jbc.270.14.7922
Tropf, S., Lanz, T., Rensing, S., Schröder, J., Schröder, G. (1994). Evidence that stilbene synthases have developed from chalcone synthases several times in the course of evolution. J. Mol. Evol. 38, 610–618. doi: 10.1007/BF00175881
Vanamala, J. K., Massey, A. R., Pinnamaneni, S. R., Reddivari, L., Reardon, K. F. (2017). Grain and sweet sorghum (Sorghum bicolor L. Moench) serves as a novel source of bioactive compounds for human health. Crit. Rev. Food Sci. Nutr., 58(17) 2867–2881. doi: 10.1080/10408398.2017.1344186
Vastano, B. C., Chen, Y., Zhu, N., Ho, C.-T., Zhou, Z., Rosen, R. T. (2000). Isolation and identification of stilbenes in two varieties of Polygonum cuspidatum. J. Agric. Food Chem. 48, 253–256. doi: 10.1021/jf9909196
Wang, C., Zhi, S., Liu, C., Xu, F., Zhao, A., Wang, X., et al. (2017a). Isolation and characterization of a novel chalcone synthase gene family from mulberry. Plant Physiol. Biochem. 115, 107–118. doi: 10.1016/j.plaphy.2017.03.014
Wang, Y., Song, F., Zhu, J., Zhang, S., Yang, Y., Chen, T., et al. (2017b). GSA: genome sequence archive. Genom. Proteom. Bioinf. 15, 14–18. doi: 10.1016/j.gpb.2017.01.001
Wenping, H., Yuan, Z., Jie, S., Lijun, Z., Zhezhi, W. (2011). De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98, 272–279. doi: 10.1016/j.ygeno.2011.03.012
Wijnsma, R., Verpoorte, R. (1986). “Anthraquinones in the Rubiaceae,” in Fortschritte der Chemie organischer Naturstoffe/Progress in the chemistry of organic natural products (Springer), 49, 79–149. doi: 10.1007/978-3-7091-8846-0_2
Wurglics, M., Schubert-Zsilavecz, M. (2006). Hypericum perforatum: a “modern” herbal antidepressant. Clin. Pharmacokinet. 45, 449–468. doi: 10.2165/00003088-200645050-00002
Xiao, T. W., Mi, M. M., Wang, C. Y., Qian, M., Chen, Y. H., Zheng, L. Q., et al. (2018). A methionine-R-sulfoxide reductase, OsMSRB5, is required for rice defense against copper toxicity. Environ. Exp. Bot. 153, 45–53. doi: 10.1016/j.envexpbot.2018.04.006
Xie, P., Chen, S., Liang, Y.-Z., Wang, X., Tian, R., Upton, R. (2006). Chromatographic fingerprint analysis—a rational approach for quality assessment of traditional Chinese herbal medicine. J. Chromatogr. A 1112, 171–180. doi: 10.1016/j.chroma.2005.12.091
Xin, T., Zhang, Y., Pu, X., Gao, R., Xu, Z., Song, J. (2018). Trends in herbgenomics. Sci. China Life Sci., 62(3) 1–21. doi: 10.1007/s11427-018-9352-7
Xiong, Y., Ren, L., Wang, Z., Hu, Z., Zhou, Y. (2015). Anti-proliferative effect of physcion on human gastric cell line via inducing ROS-dependent apoptosis. Cell Biochem. Biophys. 73, 537–543. doi: 10.1007/s12013-015-0674-9
Xu, H., Song, J., Luo, H., Zhang, Y., Li, Q., Zhu, Y., et al. (2016). Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant 9, 949–952. doi: 10.1016/j.molp.2016.03.010
Xu, J., Chu, Y., Liao, B., Xiao, S., Yin, Q., Bai, R., et al. (2017). Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience 6, gix093. doi: 10.1093/gigascience/gix093
Xu, Z., Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yan, L., Wang, X., Liu, H., Tian, Y., Lian, J., Yang, R., et al. (2015). The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 8, 922–934. doi: 10.1016/j.molp.2014.12.011
Yang, X., Yang, L., Wang, S., Yu, D., Ni, H. (2007). Synergistic interaction of physcion and chrysophanol on plant powdery mildew. Pest Manag. Sci. 63, 511–515. doi: 10.1002/ps.1362
Yap, K. Y.-L., Chan, S. Y., Lim, C. S. (2007). Authentication of traditional Chinese medicine using infrared spectroscopy: distinguishing between ginseng and its morphological fakes. J. Biomed. Sci. 14, 265–273. doi: 10.1007/s11373-006-9133-3
Yi, T., Zhang, H., Cai, Z. (2007). Analysis of rhizoma polygoni cuspidati by HPLC and HPLC-ESI/MS. Phytochem. Anal. 18, 387–392. doi: 10.1002/pca.993
Yu, D., Xu, F., Zeng, J., Zhan, J. (2012). Type III polyketide synthases in natural product biosynthesis. IUBMB Life 64, 285–295. doi: 10.1002/iub.1005
Zhang, D., Li, W., Xia, E.-H., Zhang, Q.-J., Liu, Y., Zhang, Y., et al. (2017a). The medicinal herb Panax notoginseng genome provides insights into ginsenoside biosynthesis and genome evolution. Mol. Plant 10, 903–907. doi: 10.1016/j.molp.2017.02.011
Zhang, H., Li, C., Kwok, S.-T., Zhang, Q.-W., Chan, S.-W. (2013). A review of the pharmacological effects of the dried root of Polygonum cuspidatum (Hu Zhang) and its constituents. Evid. Based Complement Altern. Med.: eCAM 2013, 208349–208349. doi: 10.1155/2013/208349
Zhang, L., Li, X., Ma, B., Gao, Q., Du, H., Han, Y., et al. (2017b). The Tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol. Plant 10, 1224–1237. doi: 10.1016/j.molp.2017.08.013
Zhang, L., Li, X., Ma, B., Gao, Q., Du, H., Han, Y., et al. (2017c). The Tartary buckwheat genome provides insights into Rutin biosynthesis and abiotic stress tolerance. Mol. Plant 10, 1224–1237. doi: 10.1016/j.molp.2017.08.013
Zhang, L., Shi, X., Zhang, Y., Wang, J., Yang, J., Ishida, T., et al. (2019). CLE9 peptide-induced stomatal closure is mediated by abscisic acid, hydrogen peroxide, and nitric oxide in Arabidopsis thaliana. Plant Cell Environ. 42, 1033–1044. doi: 10.1111/pce.13475
Keywords: Polygonum cuspidatum, genome assembly, resveratrol biosynthesis, whole-genome duplication, medicinal plant, stress tolerance
Citation: Zhang Y, Zheng L, Zheng Y, Zhou C, Huang P, Xiao X, Zhao Y, Hao X, Hu Z, Chen Q, Li H, Wang X, Fukushima K, Wang G and Li C (2019) Assembly and Annotation of a Draft Genome of the Medicinal Plant Polygonum cuspidatum. Front. Plant Sci. 10:1274. doi: 10.3389/fpls.2019.01274
Received: 03 January 2019; Accepted: 12 September 2019;
Published: 18 October 2019.
Edited by:
Xiaoquan Qi, Chinese Academy of Sciences, ChinaReviewed by:
Wei Sun, China Academy of Chinese Medical Sciences, ChinaYansheng Zhang, Chinese Academy of Sciences, China
Copyright © 2019 Zhang, Zheng, Zheng, Zhou, Huang, Xiao, Zhao, Hao, Hu, Chen, Li, Wang, Fukushima, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guodong Wang, guodong_wang@snnu.edu.cn; Chen Li, lee.childe@gmail.com