 Muzi Li
Muzi Li Yuwei Xiao
Yuwei Xiao Steve Mount
Steve Mount Zhongchi Liu
Zhongchi Liu- Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, United States
Rosaceae, a large plant family of more than 3,000 species, consists of many economically important fruit and ornamental crops, including peach, apple, strawberry, raspberry, cherry, and rose. These horticultural crops are not only important economic drivers in many regions of the world, but also major sources of human nutrition. Additionally, due to the diversity of fruit types in Rosaceae, this plant family offers excellent opportunities for investigations into fleshy fruit diversity, evolution, and development. With the development of high-throughput sequencing technologies and computational tools, an increasing number of high-quality genomes and transcriptomes of Rosaceae species have become available and will greatly facilitate Rosaceae research and breeding. This review summarizes major genomic resources and genome research progress in Rosaceae, highlights important databases, and suggests areas for further improvement. The availability of these big data resources will greatly accelerate research progress and enhance the agricultural productivity of Rosaceae.
Introduction
Rosaceae is a large angiosperm family consisting of three subfamilies—Rosoideae, Amygdaloideae, and Dryadoideae—and ~3,000 species (Xiang et al., 2017). The Rosaceae family of plants is diverse in architecture, including herbs, shrubs, and trees, and has a large number of hybrids and ploidy levels. Most importantly, Rosaceae fruits and ornamentals, such as apple, pear, peach, plum, cherry, almond, strawberry, raspberry, flowering cherry, and rose, are of tremendous economic and agronomic value. Rosaceae fruits are also surprisingly diverse in morphology and fruit type, including fleshy pome, drupe, and achenetum as well as dry fruits (Xiang et al., 2017; Liu Z. et al., 2020). Therefore, the Rosaceae family is also an ideal family for investigations of fruit diversity, domestication, and evolution.
Second- and third-generation sequencing technologies have allowed genome sequencing and genome-wide analyses to revolutionize plant research. The increasing number of sequenced plant genomes and higher quality genomes make molecular research, genome editing, and marker-assisted breeding possible in species previously recalcitrant to molecular genetic research. Further, the establishment of various online databases provides easy access and interaction with the genomic data. These databases help organize genomic resources, facilitate data sharing, and enable genome comparison across different species. In this review, we summarize the latest genome assemblies and annotations of major Rosaceae species, giving examples of findings enabled by genome sequencing. In addition, we present databases useful for the study of Rosaceae species.
Genome Sequencing and Assemblies of Rosaceae Species
Since 2016, there has been a rapid increase in the number of new Rosaceae genomes, from three new genomes in 2016 to 16 new genomes in 2020 (Figure 1). This trend will likely accelerate as research groups are moving into pan-genome sequencing. Figure 2 shows the nuclear phylogeny of Rosaceae and illustrates genera with different fruit types. Table 1 summarizes the status of genome sequencing in a selective number of economically important Rosaceae lineages. A more comprehensive summary of Rosaceae genomes and transcriptomes is provided in Supplementary Table 1, in which a total of 72 Rosaceae genomes or transcriptome assemblies are included. In addition, Supplementary Table 1 provides specific information on species name, variety name, ploidy level, and genome assembly as well as annotation versions, references, available websites, associated transcriptomes, and accession numbers for accessing these resources. In the following sections, we discuss and highlight some of the important Rosaceae genome studies.
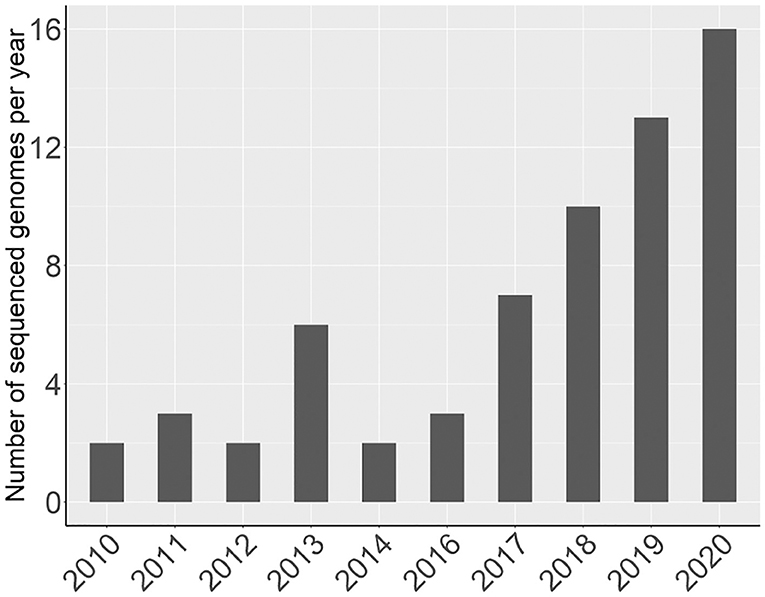
Figure 1. A graph showing the number of newly sequenced Rosaceae genomes per year.
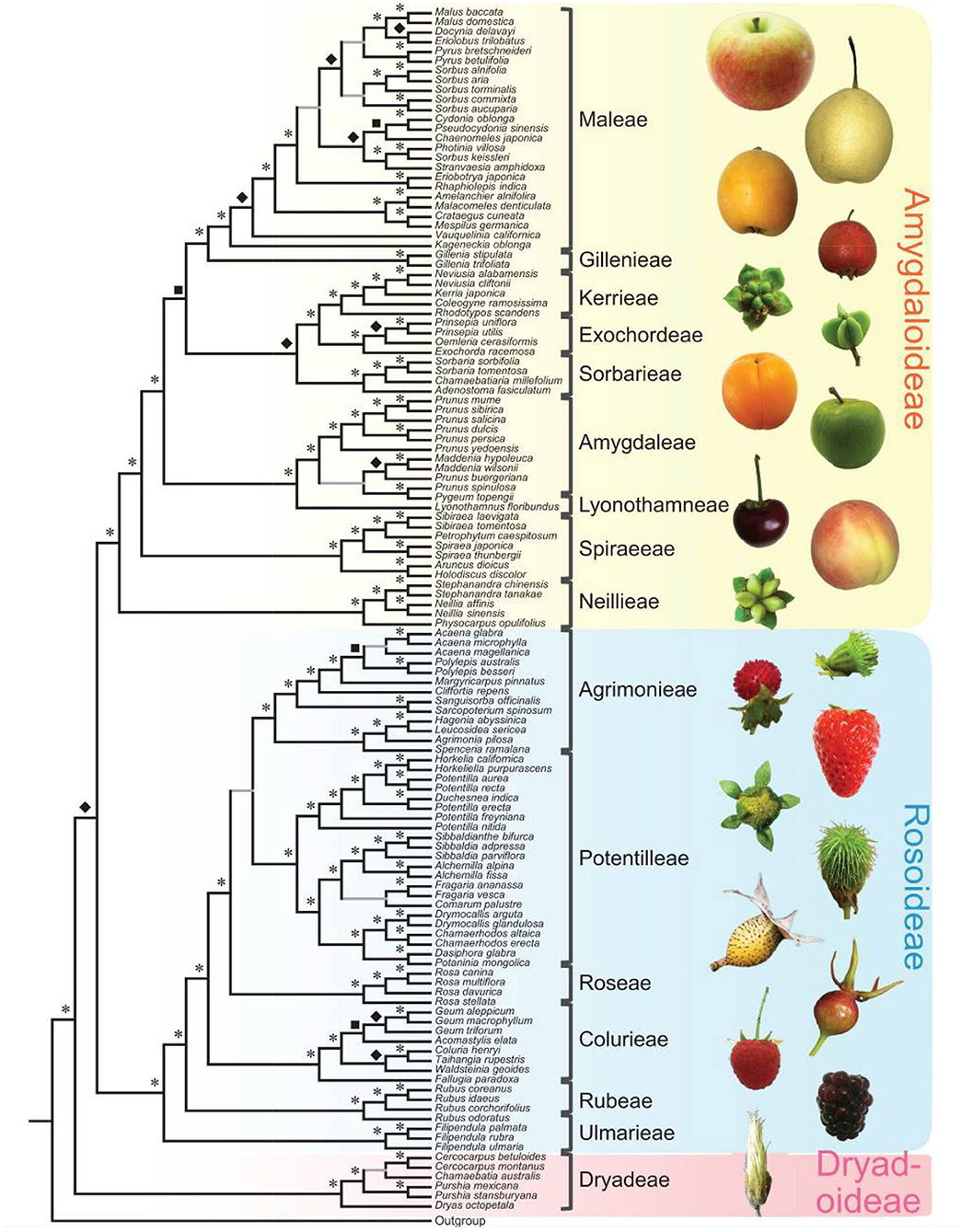
Figure 2. Rosaceae phylogeny and Rosaceae fruit types. At left is the nuclear phylogeny established in Xiang et al. (2017). Asterisks, diamonds, and squares indicate 100, 90, and 80 supports, respectively. Plant photographs on the right show the diversity of Rosaceae fruits. The figure is from Xiang et al. (2017) under the terms of Creative Commons Attribution License (CC BY 4.0).
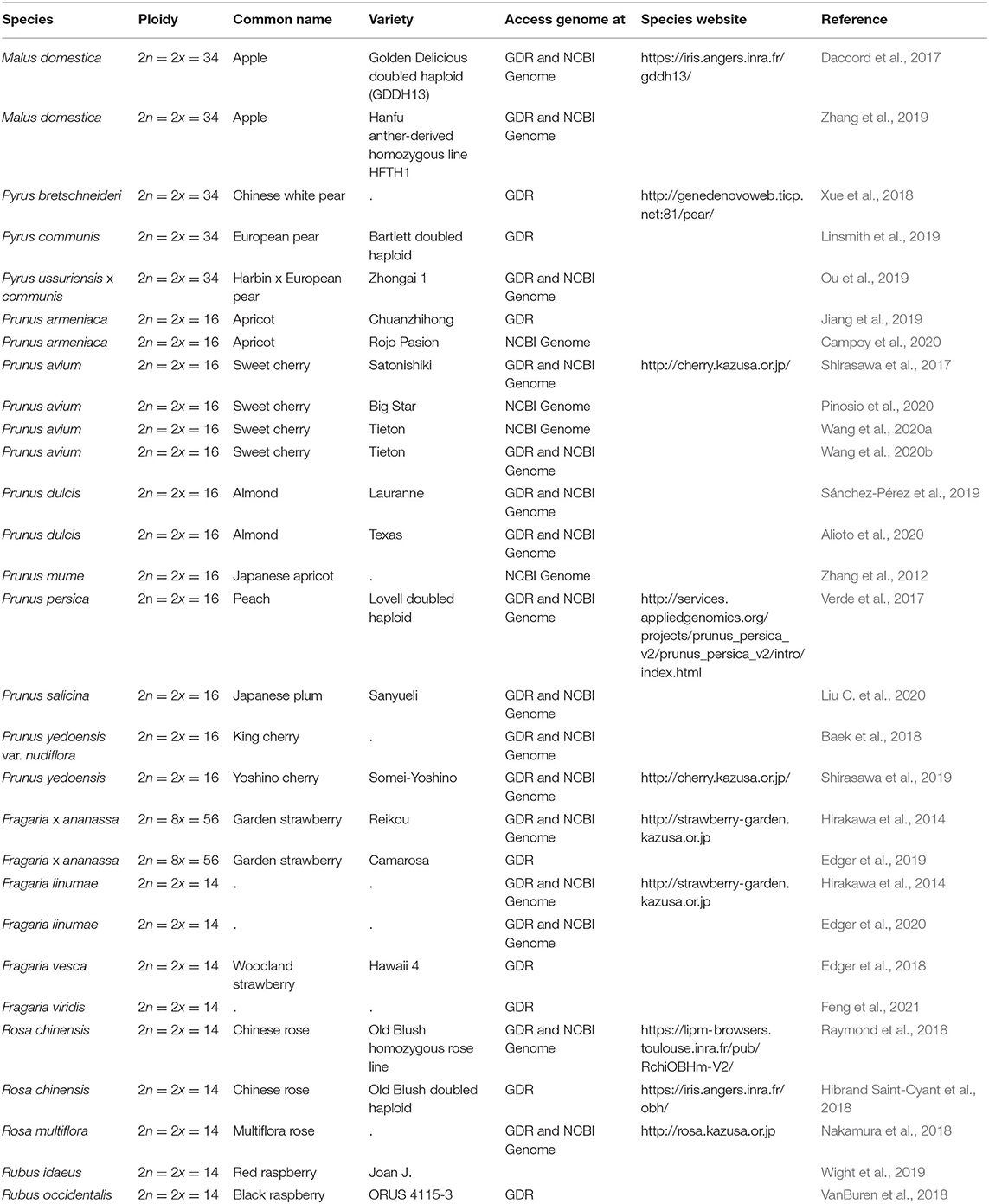
Table 1. List of selective Rosaceae species and corresponding genome resources.
Ornamentals
Two high-quality genomes of Chinese rose (Rosa chinensis cv. “Old Blush”) were generated from double haploid or homozygous lines (Hibrand Saint-Oyant et al., 2018; Raymond et al., 2018). The genome assembly by Raymond et al. (2018) consists of 82 contigs with an N50-value of 24 Mb, 36,377 protein-coding genes, and 3,971 long non-coding RNAs (lncRNAs), and the genome by Hibrand Saint-Oyant et al. (2018) is composed of 564 contigs (N50: 3.4 Mb), 39,669 predicted protein-coding genes, and 4,812 non-coding genes. The rose genomes show extensive synteny with the genome of diploid strawberry Fragaria vesca and provide valuable resources for identifying the molecular basis of key ornamental traits. For example, the “double flower” rose is more attractive due to large numbers of petals. Taking advantage of the sequenced genome, a GWAS study was conducted with 96 cultivated roses, which led to the identification of a transposon insertion in the intron of an APETALA2(AP2)/TOE homolog (Hibrand Saint-Oyant et al., 2018). Hence, the mis-regulated AP2/TOE appears to have resulted in reduced expression of AGAMOUS, leading to the double-flower phenotype.
Another worldwide ornamental tree is the flowering cherry native to Korea, Japan, and China. Due to a long history of cultivation, hybridization, and selection, there is confusion concerning the names and origins of many varieties. For example, the relationship between the King cherry (Prunus yedoensis var. nudiflora), a Korean cherry tree originating on Jeju Island, and the Yoshino cherry (Prunus x yedoensis), a popular hybrid cherry tree enjoyed in Japan and other regions of the world (Figures 3A,B), was unknown. A draft genome of King cherry was constructed, and genome-wide variome analysis using the King cherry assembly as a reference revealed that the King and Yoshino cherry trees can be clearly distinguished genetically (Baek et al., 2018).

Figure 3. Genomic studies to address questions about genetic relationship in cherry tree and fruit size in apple. (A) Yoshino cherry tree (P. x yedoensis) in the Washington, D.C., tidal basin. (B) A King cherry (P. yedoensis var. nudiflora Koehne) has superior flower, fruit, and tree shape. (C) A Golden Delicious apple and medium and small size apples from two isogenic lines, GDD13 and GDDH18, derived from the same haploid. The dramatic fruit size difference between GDDH13 and GDDH18 results from epigenetic differences. Photos in (B) and (C) are from Baek et al. (2018) and Daccord et al. (2017), respectively (both licensed under Creative Commons Attribution License 4.0).
Pome Fruits
Pear and apple share a recent whole-genome duplication event that occurred prior to their divergence and may underlie their pome fruit type (Xiang et al., 2017; Li et al., 2019). Pome fruits are characterized by their hypanthium-derived fruit flesh and agronomic importance world-wide. Multiple species and varieties of apple have been sequenced, including Malus domestica Golden Delicious (Daccord et al., 2017), Malus domestica Hanfu (Zhang et al., 2019), and Malus baccata (Chen et al., 2019) (Supplementary Table 1). Similarly, multiple species of pear, such as Chinese white pear Pyrus bretschneideri (Wu et al., 2013; Xue et al., 2018), European pear Pyrus communis “Bartlett” (Chagné et al., 2014; Linsmith et al., 2019), Pyrus ussuriensis x communis (Ou et al., 2019), and a wild Birchleaf pear (Pyrus betulifolia-Shanxi Duli, Pbe-SD) (Dong et al., 2020), have been sequenced (Supplementary Table 1). The ability to generate a double haploid line of “Golden Delicious” (GDDH13) provides an advantage in genome assembly (Daccord et al., 2017). Sequence analysis shows a major burst of different transposable elements (TEs) around 21 million years ago in the precursor of modern apple. The authors propose that the TE bursts may have possibly contributed to the divergence of apple from pear (Daccord et al., 2017). In addition, the higher quality genome allows the exploration of epigenomes and epigenetic effects on agronomic traits, such as fruit size (Figure 3C). GDDH13 and GDDH18 are isogenic lines obtained from the same haploid, but they develop different sized fruit. Whole genome bisulfide sequencing identified 22 genes with differentially methylated regions in their promoters at two developmental stages, three of which, SPL13 (MD16G0108400), ACS8 (MD15G0127800), and CYP71A25 (MD14G0147300), possess increased methylation in GDDH18 and could potentially contribute to the smaller fruit size of GDDH18 (Daccord et al., 2017).
Drupe, Stone Fruit, and Sweet Almond
Prunus develops drupe fruit, typically botanic fruit with ovary wall-derived fruit flesh (Figure 2). They include many agronomically important species, including peach, apricot, sweet cherry, and plum. They are also called stone fruit because their seeds are enclosed by large and hard (stony) endocarps. Almond (Prunus dulcis) is a closely related species with a highly syntenic genome to these Prunus species (Dirlewanger et al., 2004), but it offers high-value seeds instead of fleshy fruits. The genome resemblance among the Prunus species explains why they can form inter-specific hybrids. Although wild almonds accumulate a bitter and toxic amygdalin in their seeds, domesticated almonds develop sweet kernels/seeds. The genetic basis of this important domestication trait was recently revealed when the almond genomes were sequenced by two research groups using almond cultivars, Lauranne and Texas, respectively (Sánchez-Pérez et al., 2019; Alioto et al., 2020). The two genomes have similar contig N50-values (Lauranne: 82.26 kb, Texas: 103 kb) and protein-coding genes (Lauranne: 27,817, Texas: 27,969). Subsequent mapping identified a point mutation (Leu to Phe) in the bHLH2 gene that normally controls the expression of two P450 monooxygenase genes CYP79D16 and CYP71AN24 required for amygdalin biosynthesis (Sánchez-Pérez et al., 2019). The mutant bHLH2 fails to form a functional dimer, resulting in loss of P450 gene expression and, hence, sweet kernels. Alioto et al. (2020) compare the genomes between Prunus dulcis cv. Texas and peach, sweet cherry, and Japanese apricot and observed highly methylated TE insertions surrounding the CYP71AN24 gene, whose reduced expression was correlated with the sweet kernel trait in the almond cultivar Texas. Therefore, natural mutations and transposable elements contribute to the diversification of Prunus species and domestications of almond.
Following the publication of the peach (Prunus persica) genome and subsequent improvement (Verde et al., 2013, 2017), the pan-genome of peach (P. persica) is a much-welcomed next step (Cao et al., 2020). A pan-genome consists of the entire set of genes and genetic variations within a species, and the portion of the pan-genome common to all cultivars in the species forms the core genome. A pan-genome identifies genetic variations among cultivars, provides valuable resources, and supports evolutionary studies. In this study, 100 P. persica accessions were sequenced, giving rise to 27,796 genes in the pan-genome. Furthermore, the genomes of four wild peaches (Prunus mira, Prunus kansuensis, Prunus davidiana, Prunus ferganensis) were assembled de novo, and the core genome shared by peach and its four wild relatives consists of 15,216 gene families. The analysis reveals dramatic variation in gene content between congeneric species and suggests that P. mira is the primitive ancestor of the cultivated peach.
New Technologies for Improving Genome Assembly and Annotation
The rapid development of sequencing and related technologies, such as PacBio single-molecule real-time (SMRT) sequencing, Oxford Nanopore sequencing, Hi-C, and BioNano optical mapping over the past 10 years have greatly facilitated genome assembly and annotation. PacBio and Nanopore both belong to the third-generation (single-molecule and real-time) sequencing technology. Their long-read DNA-seq helps overcome challenges of genome assembly caused by repetitive regions (Rhoads and Au, 2015; Lu et al., 2016; Jiao and Schneeberger, 2017) and facilitates splicing isoform prediction and genome annotation (Rhoads and Au, 2015). Hi-C and BioNano optical mapping are two scaffolding technologies that help to construct chromosome-level scaffolds from contigs by providing long-range genomic information (Korbel and Lee, 2013; Tang et al., 2015; Jiao and Schneeberger, 2017). Many important crop species' genomes have benefitted from several rounds of genome assembly and annotation whenever a new technology was applied.
For heterozygous diploid species, most genomes were assembled into one pseudo-haploid sequence, ignoring sequence or structural differences between the two parental chromosomes. To generate homozygous lines, traditional methods involve breeding or creating double haploids; however, this can be extremely time-consuming or technically challenging. A recent advancement involves single-cell sequencing of haploid gametes, which enables separation of whole genome sequencing reads into haplotype-specific read sets. Using this method, two haploid genomes of a diploid apricot tree (Prunus armeniaca cultivar “Rojo Pasion”) were assembled based on whole genome sequencing of 445 pollen grains (Campoy et al., 2020). This is a much-needed advancement applicable to other Rosaceae species.
Several Updates in Apple Genome Assembly and Annotation
The progressive improvement of apple genome assemblies nicely illustrates the application of newer technologies. The first genome of apple (Malus domestica cv. “Golden Delicious”) was published in 2010 using traditional Sanger sequencing and 454 next-generation sequencing (Velasco et al., 2010). Six years later, an improved apple genome of “Golden Delicious” was assembled based on a combination of Illumina short reads and PacBio long reads (Li et al., 2016). Accordingly, the contig N50 of the apple genome was 111,619 bp, almost seven times the previous N50 (16,171 bp). In 2017, another de novo genome assembly of double haploid “Golden Delicious” (GDDH13) was published (Daccord et al., 2017). In addition to the Illumina and PacBio data, a BioNano optical mapping was used in scaffolding. As a result, the scaffold N50 was increased to 5,558 kb. In 2019, Illumina, PacBio, BioNano, and Hi-C technologies were integrated to construct a high-quality genome assembly of “Hanfu” (HFTH1) apple, a Malus domestica cultivar grown in northern China (Zhang et al., 2019). The scaffold N50 was increased to 6,988 kb. Compared with the HFTH1 genome, the three published “Golden Delicious” genomes shared 11,502 deletions and 6,590 insertions with an average length of 508 bp and 519 bp, respectively (Velasco et al., 2010; Li et al., 2016; Daccord et al., 2017). The average density of shared SNPs with the “Golden Delicious” genomes is 2.15/kb. The HFTH1 genome was utilized to completely fill 488 gaps in the GDDH13 genome; the average length of the filled gaps is 78,864 bp (Zhang et al., 2019). It would be useful if the gap-filled GDDH13 genome could be made publicly available. Because of the genetic variations between “Hanfu” and “Golden Delicious,” it is beneficial to use the genome assembly of the cultivar most closely related to the cultivars under one's study as a reference.
In addition to genome assembly, high-quality genome annotations are essential to enhance the utility of the genome. In the first “Golden Delicious” genome published in 2010, the genome annotation was based on the gene prediction programs and ESTs from Genbank (Korf et al., 2001; Birney et al., 2004; Majoros et al., 2004; Solovyev et al., 2006; Velasco et al., 2010; Sayers et al., 2020). In 2014, an improved apple reference transcriptome was constructed using RNA-Seq data generated from “Golden Delicious” fruits at 14 time points during development (Bai et al., 2014). In 2016, the de novo “Golden Delicious” genome assembly was supplemented by annotations based on RNA-Seq data from three distinct tissues (leaves, flowers, and stems) as well as ab initio and protein homology-based predictions (Li et al., 2016). To annotate the latest “Golden Delicious” GDDH13 genome, mRNA was extracted and sequenced from more tissues, including leaves, roots, fruits, apex, stems, and flowers (Daccord et al., 2017). The GDDH13 genome annotation has the lowest number of protein-coding genes at 42,140 (Daccord et al., 2017) compared with 53,922 (Li et al., 2016) and 63,141 (Velasco et al., 2010). However, GDDH13 possesses the highest BUSCO completeness at 94.9% (Daccord et al., 2017) compared with 51.5% (Li et al., 2016) and 86.7% (Velasco et al., 2010).
Several Updates in Strawberry Genome Assembly and Annotation
As with apples, the diploid woodland strawberry (Fragaria vesca ssp. vesca “Hawaii4”) genome assembly and annotation went through several rounds of updates. The first woodland strawberry genome became available at the end of 2010, and its genome annotation (v1.1) was generated by GeneMark-ES+ (Lomsadze et al., 2005), which integrated ab initio gene prediction and EST evidence (Shulaev et al., 2011). In 2015, a new annotation (v1.1.a2) was created that combined different evidence, such as de novo and genome-guided transcriptome assembly from RNA-Seq reads, ab initio gene models, and plant protein sequences from UniProt (Darwish et al., 2015). More than 2000 new genes were added in the v1.1.a2 annotation. In 2014, dense linkage maps were leveraged to construct an improved woodland strawberry genome assembly (v2.0.a1) (Tennessen et al., 2014). In 2017, based on PacBio long reads and Illumina short reads from F. vesca fruit receptacles as well as prior short-read RNA-Seq data, a new annotation (v2.0.a2) was generated (Li et al., 2018). Although the total number of protein-coding genes decreased slightly, 13,168 protein-coding genes were updated in their gene structures, alternatively spliced (AS) isoforms were identified for 7,370 genes, and the BUSCO completeness score was increased to 95.7% from the prior version (88.9%).
At the end of 2017, a high-quality woodland strawberry genome (v4.0.a1) was assembled using PacBio long reads, Illumina short reads, and BioNano optical mapping (Edger et al., 2018). This version uses a different gene-naming system, moving from the geneXXXXX to FvH4XgXXXXX format. Li Y. et al. (2019) include a supplementary table in their publication that correlates the F. vesca gene names between the old and new naming systems. In addition, a new annotation (v4.0.a2) was created based on comprehensive short- and long-read RNA-Seq data (Li Y. et al., 2019), adding 5,419 new protein-coding genes, improving the BUSCO completeness score to 98.1% from the prior 91.1%, and adding AS isoforms detected for about 30% of the genes.
In 2013, the first draft octoploid garden strawberry genome (Fragaria x ananassa cv. “Reikou”) was reported (Hirakawa et al., 2014). Homoeologous sequences of the allo-octoploid strawberry are integrated into a haploid genome named FANhybrid_r1.2 with an N50 of 5.14 kb. Gene prediction was done ab initio using Augustus. In the same study, the genomes of several Fragaria species were sequenced and assembled, including F. orientalis, F. iinume, F. nipponica, and F. bucharica, and F. bucharica (USDA accession CFRA522) was originally misidentified as Fragaria nubicola (Tennessen et al., 2014). In 2019, a near-complete chromosome-scale assembly of the Fragaria x ananassa cv. “Camarosa” was constructed with a contig N50 of about 79.97 kb, taking advantage of Illumina, 10X Genomics, and PacBio long reads (Edger et al., 2019). This chromosome-scaled genome consists of A, B, C, and D subgenomes, and the genome annotation (v1.0.a1) utilized RNA-Seq data from diverse tissue types (108,087 protein-coding genes) (Edger et al., 2019). In the same year, a garden strawberry reference transcriptome was constructed using PacBio sequencing (Yuan et al., 2019). The PacBio data in this study, together with other publicly available Illumina RNA-Seq data were recently utilized to improve the annotation of the Fragaria x ananassa cv. “Camarosa” genome (v1.0.a1) (Liu et al., 2021). Compared with Fragaria x ananassa v1.0.a1, the new annotation v1.0.a2 had a slight increase in the number of protein-coding genes (108,447). Importantly, the new annotation (v1.0.a2) for Fragaria x ananassa cv. “Camarosa” includes AS isoforms for 11,044 genes and adds 5′ and 3′ UTR information to a large proportion of the protein-coding genes (v1.0.a1: 38.93%, v1.0.a2: 73.61%).
The complete genome sequencing of the Fragaria x ananassa cultivar “Camarosa” allowed the identification of diploid progenitors, which has long been a mystery and recently a topic of intense debate. Based on the tree-searching algorithm (PhyDS), Edger et al. (2019) propose four diploid species (F. vesca, F. iinumae, F. viridis, and F. nipponica) as the four progenitors of the octoploid and suggest the hexaploid F. moschata as an intermediate species (Edger et al., 2019). However, Liston et al. (2020) reanalyzed the four subgenomes in a phylogenomic context and found support for F. vesca and F. iinumae but disputed F. viridis, F. nipponica, and F. moschata as progenitors (Liston et al., 2020). In response, a new chromosome-scale genome of F. iinumae was subsequently assembled, and a reanalysis using PhyDS supports their original proposal regarding the four diploid species as the progenitors (Edger et al., 2020). A third group recently sequenced and assembled the genomes of three wild diploid species, F. nilgerrensis, F. nubicola, and F. viridis (Feng et al., 2021). Combining these three genomes with the previously sequenced F. vesca and F. iinumae genomes, the group utilized sppIDer (Langdon et al., 2018) to map short-read sequencing data of F. x ananassa to a composite reference genome, and the result supports that F. vesca and F. iinumae, but not others, are the progenitor species of the cultivated garden strawberry (Feng et al., 2021).
Non-coding RNA in Rosaceae Genomes
Non-coding RNAs (ncRNAs) are RNAs that do not encode proteins. They are important components of the genomes and play roles in plant development and stress responses (Liu et al., 2017). However, ncRNA prediction is not always included in the annotation of published genomes. Computational tools, such as tRNAscan-SE (Chan and Lowe, 2019) and RNAmmer (Lagesen et al., 2007) are used to predict tRNA and rRNAs, respectively. Infernal (Nawrocki and Eddy, 2013) and Rfam (Kalvari et al., 2018) are often used to identify different types of ncRNAs. Besides the commonly used tools mentioned, additional approaches can be applied to detect ncRNAs, especially small RNAs. To predict miRNAs in the apricot (Prunus armeniaca cv. “Chuanzhihong”) genome, miRNA sequences derived from miRbase (Kozomara et al., 2019) were mapped against the genome, and the resulting miRNA candidates were further verified by RNAfold analysis (Lorenz et al., 2011; Jiang et al., 2019). In Rosa chinensis cv. “Old Blush,” an RNA library from pooled tissues was sequenced and analyzed for miRNA identification; tRNA and rRNA-like sequences were removed first, and miRNA precursors were then annotated using an established pipeline (Formey et al., 2014; Raymond et al., 2018). Previously, small RNA libraries derived from diverse tissues were sequenced to detect miRNAs and PhasiRNAs in wild diploid strawberry using established pipelines and criteria (Meyers et al., 2008; Xia et al., 2012, 2015). The same small RNA sequencing data sets were later used to identify small RNAs during the woodland strawberry genome reannotations (v2.0.a2 and v4.0.a2) (Axtell, 2013; Li et al., 2018; Li Y. et al., 2019). In addition to small RNAs, lncRNAs, a class of ncRNAs with length >200 bp, are shown to possess epigenetic regulatory roles in key cellular processes. RNA-Seq data from woodland strawberry flower and fruit tissues were used to identify lncRNAs, leading to 5,884 lncRNAs (Kang and Liu, 2015). In 2017, in updating woodland strawberry genome annotation v2.0.a2, an updated prediction of 4,042 lncRNA was included (Li et al., 2018).
Computational Databases for Rosaceae Species
Computational databases are becoming indispensable tools for research. Below, we discuss databases, highlighting those that are of particular importance to Rosaceae research. Although Table 1 and Supplementary Table 1 provide species-specific websites that accompany the genome-sequencing papers, Table 2 lists highly useful databases with various analysis tools and information.
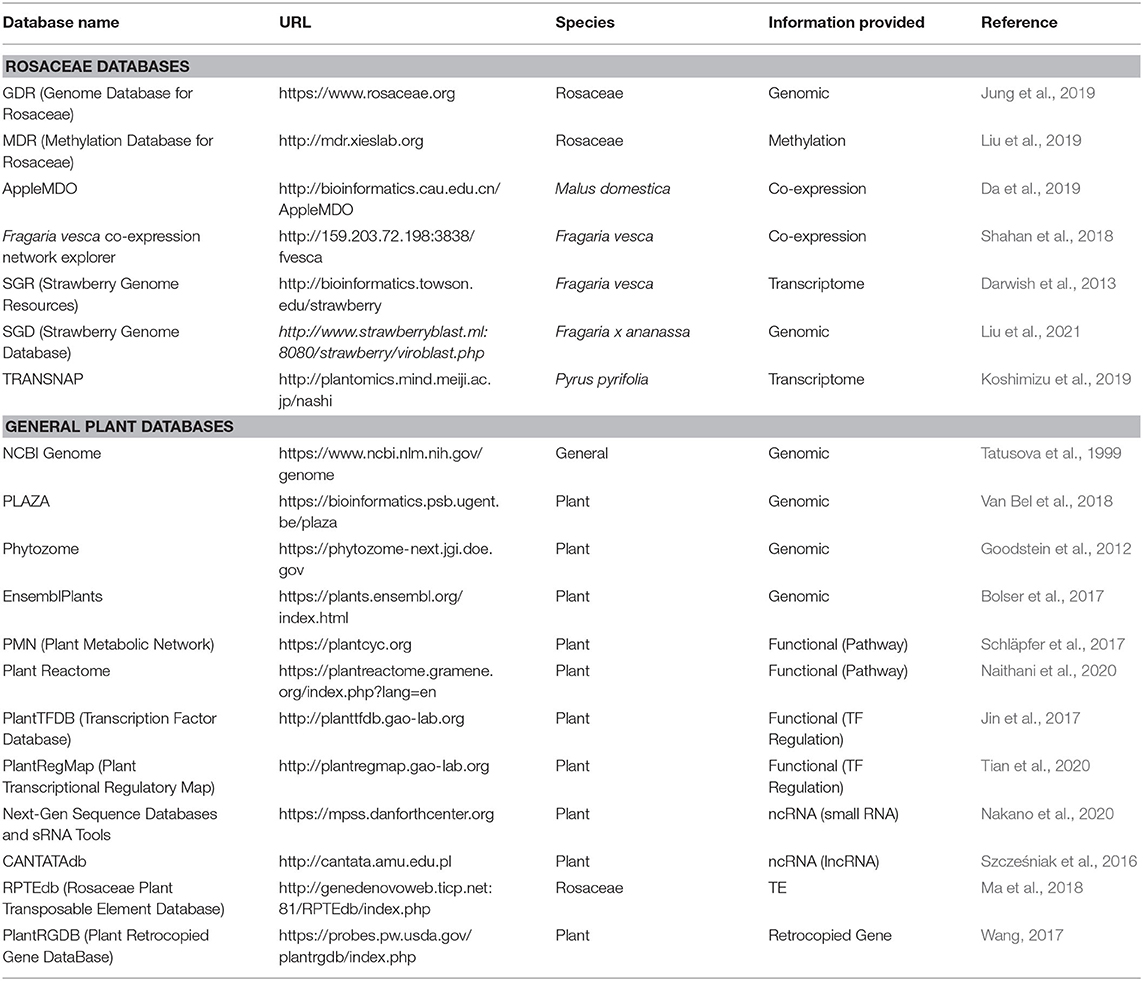
Table 2. List of websites/databases useful for Rosaceae research.
Rosaceae Genome Databases
Genome Database for Rosaceae (GDR) (www.rosaceae.org) (Jung et al., 2019) is, by far, the best resource hub for Rosaceae research. It hosts the most comprehensive and up-to-date collection of genome assembly and annotation versions for widely studied genera, Fragaria, Malus, Prunus, Potentilla, Pyrus, Rosa, and Rubus. For instance, GDR hosts Fragaria vesca genome assemblies of v1.0, v1.1 (an improved pseudochromosome assembly of v1.0), v2.0.a1, and v4.0.a1. Moreover, it incorporates corresponding updated annotations v1.1.a2, v2.0.a2, and v4.0.a2. In addition, GDR serves as the database of record for Rosaceae gene names; standardized gene-naming guideline should be followed to ensure uniformity and clarity (Jung et al., 2015). Besides the genes and genomes, GDR provides genetic maps, markers, germplasm, and trait information as well as an impressive set of tools. For example, the search tools of GDR enable users to search for specific gene sequence, maps, and markers; its MegaSearch tool allows downloading different data types in bulk. With the GDRCyc tool, users can search, visualize, and overlay pathway data. With the Synteny Viewer tool, one can select specific Rosaceae species for comparison, visualize syntenic blocks, and obtain information on syntenic genes.
The NCBI Genome (https://www.ncbi.nlm.nih.gov/genome) (Tatusova et al., 1999) on the other hand collects genomes from a broader range of Rosaceae species (Supplementary Table 1), including lesser-known species, such as Drummond's mountain avens (Dryas drummondii), wood avens (Geum urbanum), and bitterbrush (Purshia tridentate) (Griesmann et al., 2018; Jordan et al., 2018).
Rosaceae Species-Specific Databases
Many genome sequencing or annotation papers of Rosaceae species are accompanied by species-specific websites that provide tools, including BLAST searches for genes of interest. The URLs for these websites are included in Table 1 (or Supplementary Table 1 with a complete list). For instance, the genomes of Yoshino cherry (Cerasus x yedoensis) and sweet cherry (Prunus avium) are both deposited in DBcherry (http://cherry.kazusa.or.jp/) (Shirasawa et al., 2017, 2019). The built-in BLAST enables users to search their sequences of interest against the cherry genomes, and JBrowse is embedded in the database for visualizing the genomic regions. The genomes of garden strawberry (Fragaria x ananassa) and multiflora rose (Rosa multiflora) are available in Strawberry GARDEN (http://strawberry-garden.kazusa.or.jp/) and Rosa multiflora DB (http://rosa.kazusa.or.jp/), respectively (Hirakawa et al., 2014; Nakamura et al., 2018). These two websites as well as the database for cherry are all supported by the Kazusa DNA Research Institute.
Several Rosaceae species have developed species-specific databases with multiple analysis tools and resources, which are summarized in Table 2 and described below. Strawberry Genomic Resources (SGR, http://bioinformatics.towson.edu/strawberry/default.aspx) is a website that integrates different types of woodland strawberry (Fragaria vesca) genomic data (Darwish et al., 2013). It allows users to access the transcriptome analysis of the woodland strawberry early fruit development (Kang et al., 2013). Users can acquire differentially expressed genes between distinct tissues and stages by searching the database and use the eFP browser to visualize RNA-Seq data across tissues and stages for genes of interests (Hawkins et al., 2017). An updated F. vesca eFP browser is hosted at the ePlant (http://bar.utoronto.ca/). In addition, a recent annotation update of the Fragaria x ananassa cv. “Camarosa” genome (v1.0.a2) is accompanied with a separate website, “Strawberry Genome Database” (Table 2), that allows users to search for garden strawberry genes (Liu et al., 2021).
A reference transcriptome of Chinese pear (Pyrus pyrifolia) was constructed by utilizing PacBio, 454, and Sanger sequencing, and it is stored in the database TRANSNAP (http://plantomics.mind.meiji.ac.jp/nashi/) (Koshimizu et al., 2019). The database also includes gene functional annotation performed by BLASTP (Altschul et al., 1990), KAAS (Moriya et al., 2007), and InterProScan (Jones et al., 2014). Users can examine gene-expression patterns generated from GEO (https://www.ncbi.nlm.nih.gov/geo/) microarray data.
The Fragaria vesca gene co-expression network explorer (http://159.203.72.198:3838/fvesca/) was developed to host the non-consensus and consensus co-expression networks generated using RNA-Seq data from flower and fruit tissues of the woodland strawberry (Shahan et al., 2018). Users are able to search for genes of interest and the transcriptional co-expression clusters to which they belong, obtain network statistics, visualize cluster eigengene expression, examine enriched GO terms in the cluster of interest, and download the cluster graphml structure.
AppleMDO (http://bioinformatics.cau.edu.cn/AppleMDO/) is a multidimensional omics database for apple co-expression networks and chromatin states (Da et al., 2019). The global co-expression network was constructed using transcriptomes from a variety of tissues, stages, and stress treatments. The tissue-preferential network was built based on RNA-Seq data sets of different tissues without stress treatments. A combination of ChIP-seq, DNase-seq, and BS-seq data sets were utilized by ChromHMM (Ernst and Kellis, 2012) to predict the chromatin states. Furthermore, AppleMDO offers tools to perform GO analysis and motif scan.
Methylation Database for Rosaceae (http://mdr.xieslab.org/) is a database presenting methylation analyses of Rosaceae species, including woodland strawberry and Chinese rose (Rosa chinensis) (Liu et al., 2019). Using PacBio sequencing data that is publicly available (Edgar et al., 2018; Raymond et al., 2018), DNA N6-methyladenine and N4-methylcytosine modifications were identified for woodland strawberry and Chinese rose with the PacBio SMRT analysis software.
The Rosaceae Plant TE Database (RPTEdb, http://genedenovoweb.ticp.net:81/RPTEdb/index.php) provides information on TEs in five Rosaceae species: woodland strawberry, apple, Japanese apricot (Prunus mume), Chinese white pear, and peach (Ma et al., 2018). The TEs were detected in three ways, de novo identification performed by PILER (Edgar and Myers, 2005) and RepeatModeler (http://www.repeatmasker.org/), signature-based identification achieved by LTR_STRUC (McCarthy and McDonald, 2003) and LTR_FINDER (Xu and Wang, 2007), and similarity-based identification conducted by RepeatMasker (http://www.repeatmasker.org/) using the Repbase database (Jurka et al., 2005; Bao et al., 2015). Users can search and download TEs in each TE family or superfamily and view TE trees constructed using a superfamily of TEs from five Rosaceae species.
Useful Plant Databases for Comparative Genomics, Metabolic Networks, and Others
Although the summary above focuses on Rosaceae databases, many plant databases are also highly useful for Rosaceae research. Table 2 lists some of the most useful ones, such as Plant Transcription Factor Database (http://planttfdb.gao-lab.org/), Plant Transcriptional Regulatory Map (http://plantregmap.gao-lab.org/), CANTATAdb (http://cantata.amu.edu.pl/) for plant lncRNAs, and Plant Retrocopied Gene DataBase (http://probes.pw.usda.gov/plantrgdb) for plant retrocopied genes (Wang, 2017). Below, we highlight four such databases.
PLAZA (https://bioinformatics.psb.ugent.be/plaza/) (Van Bel et al., 2018) and Phytozome (https://phytozome-next.jgi.doe.gov/) (Goodstein et al., 2012) are databases for plant genome comparisons. Currently, Dicots PLAZA 4.5 has integrated genomic resources from 55 species, including four Rosaceae species, apple (Malus domestica), Chinese white pear (Pyrus bretschneideri), peach (Prunus persica), and woodland strawberry (Fragaria vesca). Phytozome v13 has gathered 224 annotated genomes, including three Rosaceae species, woodland strawberry, apple, and peach. The genome assemblies at PLAZA and Phytozome are not always up to date. For instance, older versions of woodland strawberry genome v1.1 and v2.0.a2 are, respectively, hosted at PLAZA and Phytozome at the moment.
Plant Metabolic Network (PMN, https://plantcyc.org/) (Schläpfer et al., 2017) and Plant Reactome (https://plantreactome.gramene.org/index.php?lang=en) (Naithani et al., 2020) are both databases for plant pathways. Plant Metabolic Network is focused on metabolic pathways and hosts the database PlantCyc that contains shared pathways among more than 350 plant species. Additionally, a single-species database was also constructed in PMN, which allows users to access pathways and enzymes for individual species. PpersicaCyc, SweetcherryCyc, MdomesticaCyc, EuropeanpearCyc, Fvesca_VescaCyc, RmultifloraCyc, and RchinensisCyc are developed for Rosaceae family members. Besides the metabolic pathways, Plant Reactome hosts different types of pathways, including gene regulatory pathways, hormone signaling pathways, and others. Users can view and interact with the pathways in the browser and identify chemical compounds and proteins involved in the processes. The database encompasses multiple Rosaceae species, such as peach, woodland strawberry, and apple. Furthermore, the database enables researchers to perform pathway enrichment analysis and species comparison between pathways of rice and those of selected species.
Discussion
As a result of revolutionary improvements in DNA sequencing and analysis software, Rosaceae genome research has seen a dramatic jump in the number of sequenced genomes, transcriptomes, databases, and publications. These genomic data and databases will greatly facilitate the understanding of physiology, growth and development, stress responses, adaptation, and domestication of Rosaceae species, laying the foundation for trait improvement through breeding and genome engineering. This view is also shared by a prior review on the genomes of several commercially important Rosaceae plants (Soundararajan et al., 2019). However, there is still ample room for improvement to fully reap the benefit of the genome sequencing revolution. These include increasing the quality and accuracy of Rosaceae genome assemblies and annotations, in particular, for polyploid and hybrid cultivars; identification and incorporation of AS variants and ncRNA into genome annotations; expansion of pan-genome analyses; and establishing robust molecular markers. Development of user-friendly databases that integrate, organize, and coordinate different data types and species is also essential to increase the accessibility and impact of the ever-increasing genomic data sets. The genomic revolution will likely propel significant research progress and further increase the agronomic value of Rosaceae fruits, seeds, and ornamentals.
Author Contributions
ML and ZL conceived and drafted the manuscript. ML and YX collected information and data. SM provided advice and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research in our labs has been supported by a grant from the National Science Foundation (IOS 1444987) to ZL and SM. ML was supported in part by NSF award DGE-1632976.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.644881/full#supplementary-material
Supplementary Table 1. A comprehensive list of Rosaceae genomes and relevant information.
References
Alioto, T., Alexiou, K. G., Bardil, A., Barteri, F., Castanera, R., Cruz, F., et al. (2020). Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 101, 455–472. doi: 10.1111/tpj.14538
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Axtell, M. J. (2013). ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19, 740–751. doi: 10.1261/rna.035279.112
Baek, S., Choi, K., Kim, G.-B., Yu, H.-J., Cho, A., Jang, H., et al. (2018). Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 19:127. doi: 10.1186/s13059-018-1497-y
Bai, Y., Dougherty, L., and Xu, K. (2014). Towards an improved apple reference transcriptome using RNA-seq. Mol. Genet. Genomics 289, 427–438. doi: 10.1007/s00438-014-0819-3
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. doi: 10.1186/s13100-015-0041-9
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Bolser, D. M., Staines, D. M., Perry, E., and Kersey, P. J. (2017). “Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomic data,” in Plant Genomics Databases: Methods and Protocols, ed A. D. J. van Dijk (New York, NY: Humana Press), 1–31.
Campoy, J. A., Sun, H., Goel, M., Jiao, W.-B. A., Folz-Donahue, K., Kukat, C., et al. (2020). Chromosome-level and haplotype-resolved genome assembly enabled by high-throughput single-cell sequencing of gamete genomes. bioRxiv 2020.04.24.060046. doi: 10.1101/2020.04.24.060046
Cao, K., Peng, Z., Zhao, X., Li, Y., Liu, K., Arus, P., et al. (2020). Pan-genome analyses of peach and its wild relatives provide insights into the genetics of disease resistance and species adaptation. bioRxiv 2020.07.13.200204. doi: 10.1101/2020.07.13.200204
Chagné, D., Crowhurst, R. N., Pindo, M., Thrimawithana, A., Deng, C., Ireland, H., et al. (2014). The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLOS ONE 9:e92644. doi: 10.1371/journal.pone.0092644
Chan, P. P., and Lowe, T. M. (2019). “tRNAscan-SE: searching for tRNA genes in genomic sequences,” in Gene Prediction: Methods and Protocols Methods in Molecular Biology, ed M. Kollmar (New York, NY: Springer), 1–14.
Chen, X., Li, S., Zhang, D., Han, M., Jin, X., Zhao, C., et al. (2019). Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3 (Bethesda). 9, 2051–2060. doi: 10.1534/g3.119.400245
Da, L., Liu, Y., Yang, J., Tian, T., She, J., Ma, X., et al. (2019). AppleMDO: a multi-dimensional omics database for apple co-expression networks and chromatin States. Front. Plant Sci. 10:1333. doi: 10.3389/fpls.2019.01333
Daccord, N., Celton, J.-M., Linsmith, G., Becker, C., Choisne, N., Schijlen, E., et al. (2017). High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106. doi: 10.1038/ng.3886
Darwish, O., Shahan, R., Liu, Z., Slovin, J. P., and Alkharouf, N. W. (2015). Re-annotation of the woodland strawberry (Fragaria vesca) genome. BMC Genomics 16:29. doi: 10.1186/s12864-015-1221-1
Darwish, O., Slovin, J. P., Kang, C., Hollender, C. A., Geretz, A., Houston, S., et al. (2013). SGR: an online genomic resource for the woodland strawberry. BMC Plant Biol. 13:223. doi: 10.1186/1471-2229-13-223
Dirlewanger, E., Graziano, E., Joobeur, T., Garriga-Calderé, F., Cosson, P., Howad, W., et al. (2004). Comparative mapping and marker-assisted selection in Rosaceae fruit crops. Proc. Natl. Acad. Sci. U.S.A.101, 9891–9896. doi: 10.1073/pnas.0307937101
Dong, X., Wang, Z., Tian, L., Zhang, Y., Qi, D., Huo, H., et al. (2020). De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 18, 581–595. doi: 10.1111/pbi.13226
Edgar, R. C., and Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158. doi: 10.1093/bioinformatics/bti1003
Edger, P. P., McKain, M. R., Yocca, A. E., Knapp, S. J., Qiao, Q., and Zhang, T. (2020). Reply to: revisiting the origin of octoploid strawberry. Nat. Genet. 52, 5–7. doi: 10.1038/s41588-019-0544-2
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547. doi: 10.1038/s41588-019-0356-4
Edger, P. P., VanBuren, R., Colle, M., Poorten, T. J., Wai, C. M., Niederhuth, C. E., et al. (2018). Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. GigaScience 7:gix124. doi: 10.1093/gigascience/gix124
Ernst, J., and Kellis, M. (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. doi: 10.1038/nmeth.1906
Feng, C., Wang, J., Harris, A. J., Folta, K. M., Zhao, M., and Kang, M. (2021). Tracing the diploid ancestry of the cultivated octoploid strawberry. Mol. Biol. Evol. 38, 478–485. doi: 10.1093/molbev/msaa238
Formey, D., Sallet, E., Lelandais-Brière, C., Ben, C., Bustos-Sanmamed, P., Niebel, A., et al. (2014). The small RNA diversity from Medicago truncatularoots under biotic interactions evidences the environmental plasticity of the miRNAome. Genome Biol. 15:457. doi: 10.1186/s13059-014-0457-4
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Griesmann, M., Chang, Y., Liu, X., Song, Y., Haberer, G., Crook, M. B., et al. (2018). Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361:eaat1743. doi: 10.1126/science.aat1743
Hawkins, C., Caruana, J., Li, J., Zawora, C., Darwish, O., Wu, J., et al. (2017). An eFP browser for visualizing strawberry fruit and flower transcriptomes. Hortic. Res. 4:17029. doi: 10.1038/hortres.2017.29
Hibrand Saint-Oyant, L., Ruttink, T., Hamama, L., Kirov, I., Lakhwani, D., Zhou, N. N., et al. (2018). A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 4, 473–484. doi: 10.1038/s41477-018-0166-1
Hirakawa, H., Shirasawa, K., Kosugi, S., Tashiro, K., Nakayama, S., Yamada, M., et al. (2014). Dissection of the octoploid strawberry genome by deep sequencing of the genomes of Fragaria species. DNA Res. 21, 169–181. doi: 10.1093/dnares/dst049
Jiang, F., Zhang, J., Wang, S., Yang, L., Luo, Y., Gao, S., et al. (2019). The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic. Res. 6:128. doi: 10.1038/s41438-019-0215-6
Jiao, W.-B., and Schneeberger, K. (2017). The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 36, 64–70. doi: 10.1016/j.pbi.2017.02.002
Jin, J., Tian, F., Yang, D.-C., Meng, Y.-Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jordan, C. Y., Lohse, K., Turner, F., Thomson, M., Gharbi, K., and Ennos, R. A. (2018). Maintaining their genetic distance: little evidence for introgression between widely hybridizing species of Geum with contrasting mating systems. Mol. Ecol. 27, 1214–1228. doi: 10.1111/mec.14426
Jung, S., Bassett, C., Bielenberg, D. G., Cheng, C. H., Dardick, C., Main, D., et al. (2015). A standard nomenclature for gene designation in the Rosaceae. Tree Genet. Genomes 11:108. doi: 10.1007/s11295-015-0931-5
Jung, S., Lee, T., Cheng, C.-H., Buble, K., Zheng, P., Yu, J., et al. (2019). 15 years of GDR: new data and functionality in the genome database for Rosaceae. Nucleic Acids Res. 47, D1137–D1145. doi: 10.1093/nar/gky1000
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kalvari, I., Argasinska, J., Quinones-Olvera, N., Nawrocki, E. P., Rivas, E., Eddy, S. R., et al. (2018). Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342. doi: 10.1093/nar/gkx1038
Kang, C., Darwish, O., Geretz, A., Shahan, R., Alkharouf, N., and Liu, Z. (2013). Genome-scale transcriptomic insights into early-stage fruit development in woodland strawberry Fragaria vesca. Plant Cell 25, 1960–1978. doi: 10.1105/tpc.113.111732
Kang, C., and Liu, Z. (2015). Global identification and analysis of long non-coding RNAs in diploid strawberry Fragaria vesca during flower and fruit development. BMC Genomics 16:815. doi: 10.1186/s12864-015-2014-2
Korbel, J. O., and Lee, C. (2013). Genome assembly and haplotyping with Hi-C. Nat. Biotechnol. 31, 1099–1101. doi: 10.1038/nbt.2764
Korf, I., Flicek, P., Duan, D., and Brent, M. R. (2001). Integrating genomic homology into gene structure prediction. Bioinformatics 17, S140–S148. doi: 10.1093/bioinformatics/17.suppl_1.S140
Koshimizu, S., Nakamura, Y., Nishitani, C., Kobayashi, M., Ohyanagi, H., Yamamoto, T., et al. (2019). TRANSNAP: a web database providing comprehensive information on Japanese pear transcriptome. Sci. Rep. 9:18922. doi: 10.1038/s41598-019-55287-4
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langdon, Q. K., Peris, D., Kyle, B., and Hittinger, C. T. (2018). sppIDer: a species identification tool to investigate hybrid genomes with high-throughput sequencing. Mol. Biol. Evol. 35, 2835–2849. doi: 10.1093/molbev/msy166
Li, H., Huang, C.-H., and Ma, H. (2019). “Whole-genome duplications in pear and apple,” in The Pear Genome Compendium of Plant Genomes, ed. S. S. Korban (Cham: Springer International Publishing), 279–299. doi: 10.1007/978-3-030-11048-2_15
Li, X., Kui, L., Zhang, J., Xie, Y., Wang, L., Yan, Y., et al. (2016). Improved hybrid de novo genome assembly of domesticated apple (Malus x domestica). GigaScience 5:35. doi: 10.1186/s13742-016-0139-0
Li, Y., Pi, M., Gao, Q., Liu, Z., and Kang, C. (2019). Updated annotation of the wild strawberry Fragaria vesca V4 genome. Hortic. Res. 6:61. doi: 10.1038/s41438-019-0174-y
Li, Y., Wei, W., Feng, J., Luo, H., Pi, M., Liu, Z., et al. (2018). Genome re-annotation of the wild strawberry Fragaria vesca using extensive Illumina- and SMRT-based RNA-seq datasets. DNA Res. 25, 61–70. doi: 10.1093/dnares/dsx038
Linsmith, G., Rombauts, S., Montanari, S., Deng, C. H., Celton, J.-M., Guérif, P., et al. (2019). Pseudo-chromosome–length genome assembly of a double haploid “Bartlett” pear (Pyrus communis L.). GigaScience 8: giz138. doi: 10.1093/gigascience/giz138
Liston, A., Wei, N., Tennessen, J. A., Li, J., Dong, M., and Ashman, T.-L. (2020). Revisiting the origin of octoploid strawberry. Nat. Genet. 52, 2–4. doi: 10.1038/s41588-019-0543-3
Liu, C., Feng, C., Peng, W., Hao, J., Wang, J., Pan, J., et al. (2020). Chromosome-level draft genome of a diploid plum (Prunus salicina). GigaScience 9:giaa130. doi: 10.1093/gigascience/giaa130
Liu, D., Mewalal, R., Hu, R., Tuskan, G. A., and Yang, X. (2017). New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 4:17031. doi: 10.1038/hortres.2017.31
Liu, T., Li, M., Liu, Z., Ai, X., and Li, Y. (2021). Reannotation of the cultivated strawberry genome and the establishment of the Strawberry Genome Database Horticult. Res. 8:41. doi: 10.1038/s41438-021-00476-4
Liu, Z., Ma, H., Jung, S., Main, D., and Guo, L. (2020). Developmental mechanisms of fleshy fruit diversity in Rosaceae. Annu. Rev. Plant Biol. 71, 547–573. doi: 10.1146/annurev-arplant-111119-021700
Liu, Z.-Y., Xing, J.-F., Chen, W., Luan, M.-W., Xie, R., Huang, J., et al. (2019). MDR: an integrative DNA N6-methyladenine and N4-methylcytosine modification database for Rosaceae. Hortic. Res. 6:78. doi: 10.1038/s41438-019-0160-4
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F., et al. (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol. 6:26. doi: 10.1186/1748-7188-6-26
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 14, 265–279. doi: 10.1016/j.gpb.2016.05.004
Ma, K., Zhang, Q., Cheng, T., and Wang, J. (2018). Identification of transposons near predicted lncRNA and mRNA pools of Prunus mume using an integrative transposable element database constructed from Rosaceae plant genomes. Mol. Genet. Genomics 293, 1301–1316. doi: 10.1007/s00438-018-1449-y
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
McCarthy, E. M., and McDonald, J. F. (2003). LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19, 362–367. doi: 10.1093/bioinformatics/btf878
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant microRNAs. Plant Cell 20, 3186–3190. doi: 10.1105/tpc.108.064311
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Naithani, S., Gupta, P., Preece, J., D'Eustachio, P., Elser, J. L., Garg, P., et al. (2020). Plant Reactome: a knowledgebase and resource for comparative pathway analysis. Nucleic Acids Res. 48, D1093–D1103. doi: 10.1093/nar/gkz996
Nakamura, N., Hirakawa, H., Sato, S., Otagaki, S., Matsumoto, S., Tabata, S., et al. (2018). Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 25, 113–121. doi: 10.1093/dnares/dsx042
Nakano, M., McCormick, K., Demirci, C., Demirci, F., Gurazada, S. G. R., Ramachandruni, D., et al. (2020). Next-generation sequence databases: RNA and genomic informatics resources for plants. Plant Physiol. 182, 136–146. doi: 10.1104/pp.19.00957
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ou, C., Wang, F., Wang, J., Li, S., Zhang, Y., Fang, M., et al. (2019). A de novo genome assembly of the dwarfing pear rootstock Zhongai 1. Sci. Data 6:281. doi: 10.1038/s41597-019-0291-3
Pinosio, S., Marroni, F., Zuccolo, A., Vitulo, N., Mariette, S., Sonnante, G., et al. (2020). A draft genome of sweet cherry (Prunus avium L.) reveals genome-wide and local effects of domestication. Plant J. 103, 1420–1432. doi: 10.1111/tpj.14809
Raymond, O., Gouzy, J., Just, J., Badouin, H., Verdenaud, M., Lemainque, A., et al. (2018). The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777. doi: 10.1038/s41588-018-0110-3
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genom. Proteom. Bioinform. 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Sánchez-Pérez, R., Pavan, S., Mazzeo, R., Moldovan, C., Cigliano, R. A., Cueto, J. D., et al. (2019). Mutation of a bHLH transcription factor allowed almond domestication. Science 364, 1095–1098. doi: 10.1126/science.aav8197
Sayers, E. W., Cavanaugh, M., Clark, K., Ostell, J., Pruitt, K. D., and Karsch-Mizrachi, I. (2020). GenBank. Nucleic Acids Res. 48, D84–D86. doi: 10.1093/nar/gkaa1023
Schläpfer, P., Zhang, P., Wang, C., Kim, T., Banf, M., Chae, L., et al. (2017). Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 173, 2041–2059. doi: 10.1104/pp.16.01942
Shahan, R., Zawora, C., Wight, H., Sittmann, J., Wang, W., Mount, S. M., et al. (2018). Consensus coexpression network analysis identifies key regulators of flower and fruit development in wild strawberry. Plant Physiol. 178, 202–216. doi: 10.1104/pp.18.00086
Shirasawa, K., Esumi, T., Hirakawa, H., Tanaka, H., Itai, A., Ghelfi, A., et al. (2019). Phased genome sequence of an interspecific hybrid flowering cherry, ‘Somei-Yoshino’ (Cerasus × yedoensis). DNA Res. 26, 379–389. doi: 10.1093/dnares/dsz016
Shirasawa, K., Isuzugawa, K., Ikenaga, M., Saito, Y., Yamamoto, T., Hirakawa, H., et al. (2017). The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 24, 499–508. doi: 10.1093/dnares/dsx020
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. doi: 10.1038/ng.740
Solovyev, V., Kosarev, P., Seledsov, I., and Vorobyev, D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7:S10. doi: 10.1186/gb-2006-7-s1-s10
Soundararajan, P., Won, S. Y., and Kim, J. S. (2019). Insight on Rosaceae Family with genome sequencing and functional genomics perspective. BioMed. Res. Int. 2019:e7519687. doi: 10.1155/2019/7519687
Szcześniak, M. W., Rosikiewicz, W., and Makałowska, I. (2016). CANTATAdb: a collection of plant long non-coding RNAs. Plant Cell Physiol. 57:e8. doi: 10.1093/pcp/pcv201
Tang, H., Lyons, E., and Town, C. D. (2015). Optical mapping in plant comparative genomics. GigaScience 4:3. doi: 10.1186/s13742-015-0044-y
Tatusova, T. A., Karsch-Mizrachi, I., and Ostell, J. A. (1999). Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics 15, 536–543. doi: 10.1093/bioinformatics/15.7.536
Tennessen, J. A., Govindarajulu, R., Ashman, T.-L., and Liston, A. (2014). Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 6, 3295–3313. doi: 10.1093/gbe/evu261
Tian, F., Yang, D.-C., Meng, Y.-Q., Jin, J., and Gao, G. (2020). PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 48, D1104–D1113. doi: 10.1093/nar/gkz1020
Van Bel, M., Diels, T., Vancaester, E., Kreft, L., Botzki, A., Van de Peer, Y., et al. (2018). PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 46, D1190–D1196. doi: 10.1093/nar/gkx1002
VanBuren, R., Wai, C. M., Colle, M., Wang, J., Sullivan, S., Bushakra, J. M., et al. (2018). A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. GigaScience 7:giy094. doi: 10.1093/gigascience/giy094
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839. doi: 10.1038/ng.654
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. doi: 10.1038/ng.2586
Verde, I., Jenkins, J., Dondini, L., Micali, S., Pagliarani, G., Vendramin, E., et al. (2017). The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genomics 18:225. doi: 10.1186/s12864-017-3606-9
Wang, J., Liu, W., Zhu, D., Hong, P., Zhang, S., Xiao, S., et al. (2020a). Chromosome-scale genome assembly of sweet cherry (Prunus avium L.) cv. Tieton obtained using long-read and Hi-C sequencing. Hortic. Res. 7:122. doi: 10.1038/s41438-020-00343-8
Wang, J., Liu, W., Zhu, D., Zhou, X., Hong, P., Zhao, H., et al. (2020b). A de novo assembly of the sweet cherry (Prunus avium cv. Tieton) genome using linked-read sequencing technology. PeerJ 8, e9114. doi: 10.7717/peerj.9114
Wang, Y. (2017). PlantRGDB: a database of plant retrocopied genes. Plant Cell Physiol. 58:e2. doi: 10.1093/pcp/pcw210
Wight, H., Zhou, J., Li, M., Hannenhalli, S., Mount, S. M., and Liu, Z. (2019). Draft genome assembly and annotation of red raspberry Rubus idaeus. bioRxiv 546135. doi: 10.1101/546135
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408. doi: 10.1101/gr.144311.112
Xia, R., Ye, S., Liu, Z., Meyers, B. C., and Liu, Z. (2015). Novel and recently evolved microRNA clusters regulate expansive F-BOX gene networks through phased small interfering RNAs in wild diploid strawberry. Plant Physiol. 169, 594–610. doi: 10.1104/pp.15.00253
Xia, R., Zhu, H., An, Y., Beers, E. P., and Liu, Z. (2012). Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 13:R47. doi: 10.1186/gb-2012-13-6-r47
Xiang, Y., Huang, C.-H., Hu, Y., Wen, J., Li, S., Yi, T., et al. (2017). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 262–281. doi: 10.1093/molbev/msw242
Xu, Z., and Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Xue, H., Wang, S., Yao, J.-L., Deng, C. H., Wang, L., Su, Y., et al. (2018). Chromosome level high-density integrated genetic maps improve the Pyrus bretschneideri ‘DangshanSuli’ v1.0 genome. BMC Genomics 19:833. doi: 10.1186/s12864-018-5224-6
Yuan, H., Yu, H., Huang, T., Shen, X., Xia, J., Pang, F., et al. (2019). The complexity of the Fragaria x ananassa (octoploid) transcriptome by single-molecule long-read sequencing. Hortic. Res. 6:46. doi: 10.1038/s41438-019-0126-6
Zhang, L., Hu, J., Han, X., Li, J., Gao, Y., Richards, C. M., et al. (2019). A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10:1494. doi: 10.1038/s41467-019-09518-x
Keywords: Rosaceae fruits, Rosaceae ornamentals, genome assembly, genome annotation, databases, domestication, origin of species
Citation: Li M, Xiao Y, Mount S and Liu Z (2021) An Atlas of Genomic Resources for Studying Rosaceae Fruits and Ornamentals. Front. Plant Sci. 12:644881. doi: 10.3389/fpls.2021.644881
Received: 22 December 2020; Accepted: 22 February 2021;
Published: 01 April 2021.
Edited by:
Raju Datla, Global Institute for Food Security (GIFS), CanadaReviewed by:
Aikaterini Symeonidi, Technical University of Munich, GermanyJanet Slovin, USDA/ARS Genetic Improvement of Fruits and Vegetables Laboratory, United States
Copyright © 2021 Li, Xiao, Mount and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongchi Liu, zliu@umd.edu