 Argel Aguilar-Valles
Argel Aguilar-Valles Brandon Rodrigue
Brandon Rodrigue Edna Matta-Camacho
Edna Matta-Camacho- Department of Neuroscience, Carleton University, Ottawa, ON, Canada
Prenatal infections have been linked to the development of schizophrenia (SCZ) and other neurodevelopmental disorders in the offspring, and work in animal models indicates that this is to occur through the maternal inflammatory response triggered by infection. Several studies in animal models demonstrated that acute inflammatory episodes are sufficient to trigger brain alterations in the adult offspring, especially in the mesolimbic dopamine (DA) system, involved in the pathophysiology of SCZ and other disorders involving psychosis. In the current review, we synthesize the literature on the clinical studies implicating prenatal infectious events in the development of SCZ. Then, we summarize evidence from animal models of maternal immune activation (MIA) and the behavioral and molecular alterations relevant for the function of the DAergic system. Furthermore, we discuss the evidence supporting the involvement of maternal cytokines, such as interleukin 6 (IL-6) and leptin (a hormone with effects on inflammation) in mediating the effects of MIA on the fetal brain, leading to the long-lasting effects on the offspring. In particular, IL-6 has been involved in mediating the effects of MIA animal models in the offspring through actions on the placenta, induction of IL-17a, or triggering the decrease in non-heme iron (hypoferremia). Maternal infection is very likely interacting with additional genetic and environmental risk factors in the development of SCZ; systematically investigating how these interactions produce specific phenotypes is the next step in understanding the etiology of complex psychiatric disorders.
Introduction
We are currently undergoing a SARS-CoV-2 pandemic, which like previous viral outbreaks [e.g., Zika (1)] can leave behind sequelae of health complications, including direct effects in the nervous system (2) and alterations of brain development if infections occur during perinatal stages.
Indeed, maternal infection has been identified as a risk factor for several neurodevelopmental disorders such as cerebral palsy, intellectual disability, autism spectrum disorder (ASD), bipolar disorder (BD), and schizophrenia (SCZ) (3–9). We will focus on reviewing the effects of maternal infection on the dopaminergic neurotransmitter system and the link with psychosis, particularly SCZ.
SCZ is one of the top leading causes of disability worldwide (10) and the seventh most costly medical illness in modern society (11, 12). SCZ is characterized by psychotic symptoms such as delusions and hallucinations (also known as the positive symptom dimension); alterations in drive and volition, including lack of motivation, blunted affect, social withdrawal, and reduction in spontaneous speech (the negative symptom dimension) and alterations in neurocognition, including difficulties in memory, attention, and executive functioning (the cognitive symptom dimension) (13–15).
The positive symptoms of SCZ overlap with different psychiatric disorders. Indeed, psychosis is also frequent during mood episodes in BD, severe depression, substance use disorder and neurodegenerative disorders (16–18). Intriguingly, some SCZ-like psychopathological abnormalities (i.e., paranoid delusional thinking and auditory hallucinations) are expressed in an attenuated form in 5–8% of the otherwise healthy population, especially in individuals with schizotypal or schizoid personality traits (13, 19). This extensive overlapping of symptoms and genetic risk factors with other psychiatric and neurological conditions is suggestive of a common underlying neuropathophysiology for these disorders, which, rather than discrete diagnoses, may represent a continuum that extends to the general population (13, 19, 20).
The Dopamine Theory Of SCZ And Psychosis
The classical dopamine (DA) hypothesis of SCZ (21) states that the hyperactivity of the DA system is responsible for the symptoms of the disorder. More recently, this hypothesis was elaborated to include the proposal that the hyperactivity of the mesolimbic DA system (Figure 1A) contributes to positive symptoms in SCZ. Meanwhile, impaired function of the DA system in the prefrontal cortex (PFC, Figure 1A) contributes to the cognitive symptom dimension (22, 23).
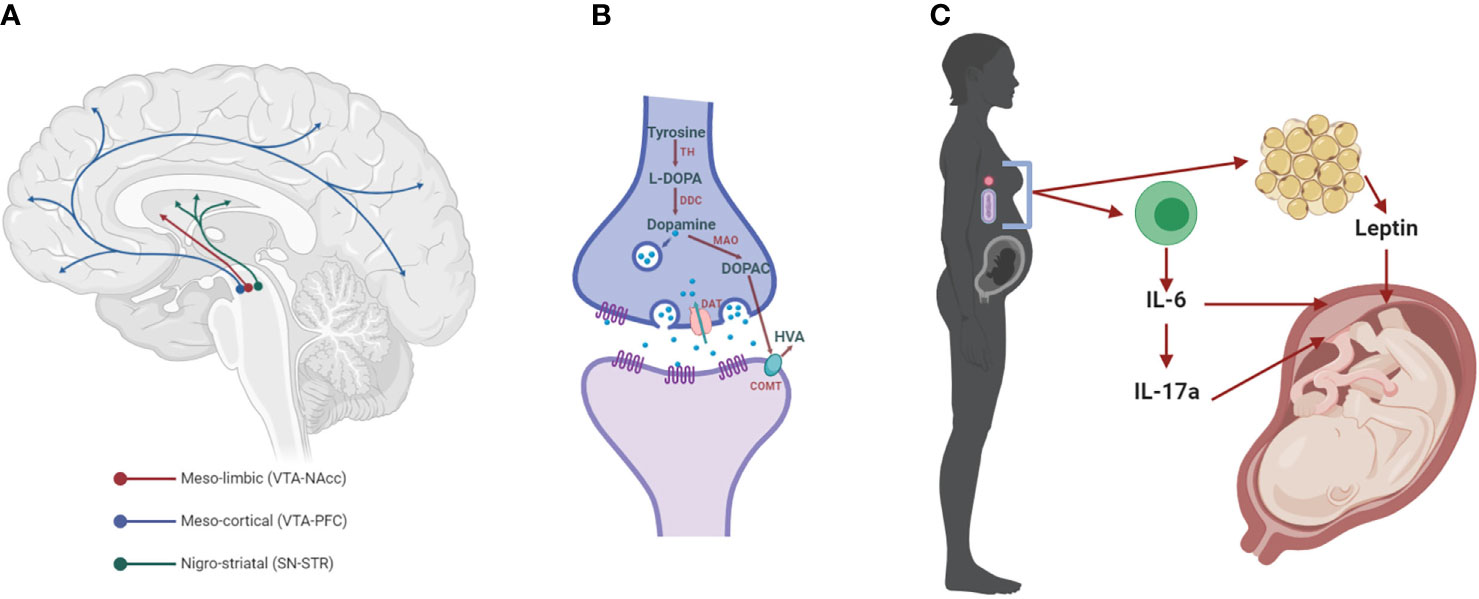
Figure 1 The dopaminergic system and mediators of maternal immune activation. (A) The meso-limbic DA neurons have their cell bodies in the ventral tegmental area (VTA) and terminals innervate the nucleus accumbens (NAcc). Other VTA neurons project to the prefrontal cortex (PFC), constituting the meso-cortical system. The nigro-striatal DA neurons lie in the substantia nigra (SN) and project to the dorsal striatum (STR). (B) Dopaminergic synapse, where dopamine is synthesized by the conversion of tyrosine into L-3,4-dihydroxyphenylalanine (L-DOPA) by the enzyme tyrosine hydroxylase (TH). L-DOPA is then converted to dopamine by the L-DOPA decarboxylase (DDC). Once packaged in synaptic vesicles and released to the extracellular space, dopamine can act on its receptors (DRs) on the post- and pre-synaptic membrane. Dopamine neurotransmission is terminated when the dopamine transporter (DAT) reuptakes the neurotransmitter to the pre-synaptic side, where it can be metabolized into 3,4-dihydroxyphenylacetic acid (DOPAC) by the monoamine oxidase (MAO) and homovanillic acid (HVA) by the catechol-O-methyl transferase (COMT). (C) Maternal immune activation (MIA) with bacteria or viruses leads to the activation of immune cells that release cytokines, including interleukin 6 (IL-6) and, in turn, IL-17a. Both of these cytokines affect brain development in the fetus, increasing the risk for neurodevelopmental disorders, such as SCZ (SCZ). These cytokines can act indirectly on the placenta, or in the case of IL-6 through the induction of hypoferremia, a reduction in circulating non-heme iron. Adipose tissue can also release hormones such as leptin, which affects fetal development.
The DA hypothesis of SCZ derives, in part, from the identification of the mechanisms of action of antipsychotics, many of which act as DA receptor 2 (D2 receptor) blockers (15). Furthermore, pharmacological studies show that a single exposure to amphetamine (AMPH), a stimulant drug that increases extracellular levels of DA in striatal and cortical regions via release and reverse transport (24, 25), evokes or exacerbates positive symptoms in SCZ patients at doses which do not induce psychosis in healthy subjects (26–28). Imaging studies demonstrate that a significant number of non-medicated SCZ patients show marked elevation of AMPH-induced striatal dopamine release in comparison to healthy volunteers (29–31). This response correlates significantly with the emergence or worsening of positive symptoms (31–35).
Understanding the etiology of SCZ is an active area of research. However, evidence accumulated in the last three decades on environmental risk factors that affect early neurodevelopment during pregnancy has led to the proposal of the neurodevelopmental hypothesis for SCZ (36–38). In this sense, accumulating evidence suggests that perinatal insults also contribute to an increase risk of developing BD (9), particularly those cases with psychoses (8).
Neurodevelopmental Etiology Of SCZ And Other Psychiatric Disorders
SCZ has been hypothesized to have a neurodevelopmental origin (22): an outcome of an aberration in developmental processes within the brain, which begins long before the onset of the clinical symptoms (36, 39). There are numerous independent lines of evidence supporting this hypothesis. For example, there is a conspicuous absence of gross physical damage or signs of progressive neurodegeneration in SCZ (22, 39). Besides, children that go on to develop SCZ present behavioral, physical and brain morphological alterations, before the clinical onset of psychosis (15, 36, 39–41).
Finally, individuals who develop SCZ are more likely to have experienced pre- or perinatal adverse events (22, 42), or adolescent disturbances in brain development, compared to control individuals (36, 39, 41, 43). These adverse events include intrauterine growth retardation, pregnancy and birth complications (44), nutritional deficiencies (45, 46) maternal stress (47), and maternal infections (48).
There is also mounting evidence for the role of neurodevelopmental disturbances in the etiology of BD, as thoroughly reviewed by (9). In this regard, there is high co-morbidity between BD and other developmental disorders such as attention-deficit/hyperactivity disorder (ADHD) and ASD (9). Remarkably, there are increased rates of BD due to obstetric complications, cesarean section birth and perinatal infection (8, 9, 49, 50).
Maternal Infection, SCZ, and Other Neurodevelopmental Disorders
Ecological studies, including those based on the subjective report of illness, suggest that SCZ is more prevalent in the offspring of women that were pregnant during periods of influenza epidemics (51, 52), as well as other types of infections, including diphtheria, pneumonia, measles, varicella zoster, mumps and poliovirus (4, 53–56). Similarly, SCZ is more prevalent among individuals born to pregnancies that occur during winter, a season associated with an increased frequency of respiratory infections (36, 51, 57). The main limitation of these studies is that “exposure to infection” was defined solely by the fact that the individual was pregnant during the time of the epidemic (i.e., based on the date of birth of the offspring).
It was later shown that SCZ in the offspring is significantly associated with maternal infections using individual biomarkers of illness in the maternal serum or clinical diagnoses (4). These included respiratory infections (58), influenza (59, 60), rubella (61, 62), Toxoplasma gondii (63, 64), herpes simplex virus-2 (HSV-2) (65, 66), maternal genital or reproductive infections (67), and maternal bacterial infections (68). Some of these studies used a broad definition of psychosis, where both non-affective (e.g., SCZ) and affective (e.g., major depression or BDs with psychotic features) psychiatric disorders were included (62, 66, 69). This suggests that maternal infection may be involved in the development of psychotic features that may not be necessarily restricted to those that characterize SCZ, but several other disorders as well. Indeed, MIA involving influenza has been linked to BD (9) [and Toxoplasma gondii infections to a lesser extent (70)], especially for those patients that also develop psychotic features (8).
What remains unclear from these studies is the critical stage(s) of gestation during which the developing brain may be more vulnerable to this prenatal insult. Indeed, those studies that have tried to dissect a specific trimester of gestation where vulnerability to MIA may be increased, have provided evidence for all three trimesters (58, 59, 68, 69, 71, 72). Overall, effect sizes of prenatal infection across gestation and development of SCZ in the offspring range from 1.5 to 7 for different infections (73), suggesting the existence of additional factors that confer vulnerability or resilience (6).
The wide variety of infections associated with SCZ and BD with psychosis suggests that there may be a common factor underlying increased susceptibility (74). Therefore, it has been hypothesized that maternal immune activation (MIA), and the inflammatory mediators released following all types of infections (4), may be fundamentally involved. Epidemiological studies have provided some evidence supporting this hypothesis. Increased levels of maternal pro-inflammatory cytokines, specifically interleukin (IL)-8 (72), tumor necrosis factor (TNF)α (69, 71, 75, 76), IL-6 (71, 75, 76), C-reactive protein (77) are associated with a higher risk of psychosis or SCZ in the offspring. Several animal models have been developed to investigate the immunological and neurobiological link between MIA and altered behavior in the offspring, with heavy emphasis in behavioral alterations.
Animal Models of Maternal Infection
Initial approaches used prenatal infection with an influenza virus [at gestational day (GD) 9 in mice], followed by the application of a battery of behavioral tests relevant to SCZ in the adult offspring (78). These studies showed that the adult offspring of infected mothers presented, compared to the offspring of control dams, decreased social interaction, reduced exploration in the open field, impaired performance in the novel object test, indicative of impaired working memory as observed in SCZ and diminished PPI of acoustic startle (78). These behavioral alterations are analogous to aspects of SCZ.
Viral Mimetic Poly I:C
Further studies investigated the consequences of MIA using molecular immunogens in rats and mice. The viral mimic polyinosinic:polycytidylic acid (poly I:C) has been used to stimulate the maternal immune system (with one or multiple injections), at several stages of pregnancy in mice or rats, ranging from GD 8.5 until GD 18.5. The effects of these prenatal treatments have been extensively reviewed elsewhere (6, 7, 79–81); thus, we will focus on those consequences more closely relevant for psychosis. Prenatal stimulation with poly I:C induced deficits in an operational measure of sensorimotor gating (82), pre-pulse inhibition of acoustic startle and increased sensitivity to the locomotor activating effects of cocaine, AMPH and methamphetamine, whose locomotor effects depend on the mesolimbic DA system (83–105).
An overall trend regarding these two phenotypes is one where PPI deficits are more consistently observed when MIA occurs at gestational stages earlier than GD 16 in both mice and rats. At the same time, hyper-responsiveness to activators of the mesolimbic dopaminergic system appears when challenging the mothers at any developmental age [reviewed in (79)].
Models of Bacterial Infections
The role of bacterial infection has also been investigated by using the Gram-negative bacterial cell wall component, lipopolysaccharide (LPS). In rats, injections at several stages of gestation, ranging from GD 9 until birth, induced, in the offspring, impairments in PPI, and increases in sensitivity to the locomotor effects of AMPH (106–117). LPS has also been administered either in alternate days (118) or daily throughout pregnancy (119, 120). Similarly to acute LPS administration, these chronic prenatal treatments also induced impairments in PPI (118–120).
Other Models of Inflammation
Turpentine (TURP) is an inflammatory agent whose injection [intramuscular (i.m.)] produces localized necrotic damage (121) and the sequential induction of TNFα and IL-1β at the site of injury, which trigger IL-6 release into the circulation (122, 123). Using TURP at GD 15 or 18 in rats, we found that an earlier challenge with TURP induces greater maternal inflammatory response compared to later in gestation (124). Furthermore, this difference in the inflammatory response during pregnancy correlates with the effect on the offspring, such that treatment at GD 15 induces impairment in PPI and hyper-responsiveness to AMPH, while the same treatment at GD 18 does not affect any of these behaviors (124).
Overall, some of the alterations in behavior induced by either polyI:C or LPS, have been shown to appear in the adult but not in the juvenile offspring (84, 93, 120, 125), as occurs in SCZ patients. Also supporting the validity of the models toward the disorder is the observation that a number of these alterations, including deficits in PPI, were shown to be reversed by either acute or chronic treatment with several antipsychotic drugs in adult or adolescent animals [i.e., haloperidol, chlorpromazine, olanzapine, risperidone or clozapine, which constitute the primary pharmacological treatment for psychotic illness (78, 83–85, 89, 119, 126–132)].
Effects of Maternal Infection on Dopamine Neurotransmission in Mouse Models
Given the central role of DA neurotransmission in SCZ, the findings on the exaggerated locomotor response to AMPH and other drugs that stimulate DAergic neurotransmission following MIA, and the effectiveness of antipsychotic treatments to reverse MIA effects, several studies investigated the effects of prenatal immune activation on this neurotransmitter system. One often used approach is the measurement of tissue DA content and its metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA, Figure 1B).
Prenatal poly I:C treatment at GD 15 induces enhanced release of DA from striatal explants in the adult offspring (83). In addition, poly I:C treatment at GD 9 results in increases DA and DOPAC levels in the PFC and the globus pallidus (GP) and HVA in the nucleus accumbens (NAcc) and GP of adult mice (133). Increases in DA are also found in the NAcc following poly I:C treatment at GD 9 (134). Similarly, several injections of poly I:C (GD 12-17) result in elevated levels of DOPAC and HVA in the adult STR (84).
Prenatal LPS treatment has been shown to have somewhat variable effects on DA. For example, daily administration of LPS throughout the entire pregnancy results in increased DA levels in the NAcc of adult animals (P 120, 170, or 400), but lower DA levels in younger animals (P 39) (119, 120). Interestingly, a single LPS administration at GD 10, results in a decrease of DA in the dSTR (135–140). Decreased DA is also found in the NAcc, PFC, amygdala, hippocampus, and hypothalamus, accompanied by decreased levels of HVA in the NAcc and amygdala (P 120) (140). Similarly, decreased DA levels in the NAcc at P 83 were found when escalating doses of LPS were administered daily from GD 15 until 19 (141).
Using MIA with TURP, we found increases in DA, DOPAC, and HVA in the NAcc, but not in the dorsal STR or the PFC of the male offspring in rats (142, 143).
Prenatal poly I:C treatment (GD 9) results in increased tyrosine hydroxylase (TH) immunoreactivity, the rate-limiting enzyme for the synthesis of DA, in the mesencephalon of embryonic mice at GD 13 and 17 (92), as well as in the NAcc and SN of adult mice (P 120) (93). In the NAcc, TH immunoreactivity was decreased at P 35 (93) but increased at P 70 (90, 93). DAT immunoreactivity is also found to be increased in the fetal mesencephalon (GD 17) (92), but decreased in the dSTR at P 35, as well as in the NAcc at GD 19 and P 35 (93). Immunoreactivity of DA receptors, D1 and D2, is reduced in the adult mice’s PFC (90, 144) and increased in the NAcc (for both D1 and D2) and dSTR (only D1) (93). In contrast, Ozawa et al. reported that DA D2 receptor’s binding is reduced in the STR of adult mice (84). Most of these data are consistent with a scenario of increased synthesis of DA in the adult brain of MIA offspring, particularly in the meso-limbic, but not in the meso-cortical pathway.
The effects of prenatal LPS administration on these markers of DA neurotransmission are rather conflicting. Borrell et al. (118) found increased TH immunoreactivity in the NAcc and bed nucleus of the stria terminalis in adult rats whose mothers were treated with LPS on alternate days during the entire pregnancy. In contrast, Ling et al. reported in several studies that a single dose of LPS at GD 10 leads to a significant decrease in TH immunoreactivity, which was significant in the SN, at several postnatal ages (P 21, 120, 210, 420, 510) as well as in the VTA of post-weanling rats (135–140). As above, these results support the idea that models of bacterial MIA have different outcomes compared to those involving viral mimetics.
Finally, prenatal TURP administration at GD 15 leads to an increase of TH levels in the NAcc, but not in other DA terminal areas such as the dorsal STR or the PFC, nor in the VTA or SN (124, 142, 143).
Overall, poly I:C and TURP induce molecular changes consistent with hyperactivity of mesolimbic DA neurotransmission, which may underlie the hyperactivity in response to AMPH and other behavioral alterations that can be corrected by administration of antipsychotics.
Role of Maternal Cytokines in Inducing MIA Alterations
A more causal role for elevated maternal cytokines in SCZ-related alterations has been established through the administration of exogenous cytokines to pregnant rats or mice. These manipulations have been shown to be sufficient to induce several molecular and behavioral effects in the offspring. For example, prenatal administration of IL-6 in mice (at GD 9, 5 μg, i.p.) results in impairments in PPI and other behaviors in the adult offspring, whereas a similar treatment with IFNγ or TNFα does not affect the offspring (86). Significantly, the effect of an influenza virus and poly I:C treatments on the fetal brain transcriptome overlapped to those of IL-6 administration in utero, supporting the idea that many effects of poly I:C are mediated by this cytokine (145).
Importantly, functional inhibition of poly I:C-induced IL-6 in pregnant mice prevented several of the behavioral effects of prenatal poly I:C in the offspring, including impaired PPI (86). Also, the offspring of IL-6 “knock-out” mothers treated with poly I:C, do not present these alterations (86). Similarly, knock-out of IL-6 receptor in the placental trophoblasts prevented several effects of prenatal poly I:C treatment (146), indicating a crucial role of this organ in mediating the effects of MIA.
We also observed that co-treatment with an anti–IL-6 antibody during gestation and TURP prevented the development of a hyper-active DAergic system (143). This prenatal treatment effectively rescued the exaggerated AMPH-induced hyperlocomotion and behavioral sensitization, elevated DA, and TH in the NAcc in the offspring of TURP-treated mothers (143).
IL-6 can, in turn, act in more than one way to affect neurodevelopment (Figure 1C). One such mechanism is hypoferremia, a reduction in maternal circulating non-heme iron, which characterizes the acute phase response and is triggered by all types of infection (147, 148). Proper iron homeostasis is fundamental for healthy brain development, especially for the DAergic neurons (149). Indeed, we demonstrated that maternal iron supplementation, which counteracts inflammation-induced hypoferremia, prevented the development of exacerbated responses to a single AMPH injection and enhanced behavioral sensitization following repeated exposure to this drug in the offspring (142). Furthermore, maternal iron supplementation during MIA also reversed the increased levels of TH, DA and its metabolites in the NAcc found in the offspring of mothers treated with TURP (142). Notably, iron levels in the placenta were reduced by MIA (but not in the fetal brain), which were rescued by maternal iron supplementation (142), supporting a role for this organ in mediating the effects of MIA in the development of the brain.
Another potential mediator of MIA, downstream of IL-6, is IL-17a (Figure 1C), since blocking the latter cytokine with anti–IL-17a antibodies prevented cortical malformations and the emergence of abnormal behaviors in adult MIA offspring, including impaired social interaction and increases marble-burying behavior (150, 151). Meanwhile, overexpression of the anti-inflammatory cytokine IL-10 in maternal macrophages prevented the MIA-induced deficits in PPI, although in itself, elevated IL-10 also induced other behavioral alterations (91). In addition to IL-6, the hormone leptin has also been implicated on the effects of MIA in the DAergic system.
Leptin
Leptin is the product of ob gene (152), a hormone that regulates food intake and energy expenditure (153–155). Leptin is primarily produced by adipose tissue and secreted into the circulation, where levels correlate positively with body fat mass (156, 157). Leptin has a multitude of physiological roles, including regulation of inflammatory processes (158, 159). For example, leptin treatment induces pro-inflammatory cytokines, including TNFα, IL-1β, IL-6, and IFN-γ (160–162). Inflammatory stimuli (e.g., TNFα, IL-1β, LPS, and TURP) in turn increase leptin synthesis (163–167). During the acute inflammatory response, leptin is involved in the induction of several sickness-type responses, such as anorexia and fever (168–173).
Despite its clear involvement in several aspects of the inflammatory response to infection, the role of leptin in brain development has not yet been extensively studied. We demonstrated that neutralization of leptin during MIA was effective in curtailing several alterations induced by prenatal TURP, including the hype-sensitized locomotor response to AMPH, and increases in DA in the NAcc (Figure 1C) (143). Intriguingly, leptin could affect the development of the dopaminergic system, as constitutive leptin mutant mice have impaired locomotor response to AMPH, and diminished DA release in the NAcc (174). Leptin can also exert impairing effects or the control of cytokines expression in the placenta (175).
Concluding Remarks
MIA alters the development of the dopaminergic system and many other neurotransmitter systems and brain regions (5, 6, 79, 80). Maternal cytokines, particularly IL-6, are central in mediating these effects (5). However, other neuroendocrine factors, such as the adipokine leptin, are potentially involved and deserve further investigation.
Maternal infections and other environmental risk factors for SCZ and neurodevelopmental disorders may independently account for a few clinical cases since exposure to them does not always generate the disorder or are implicated in several psychiatric illnesses (6, 39, 79, 80, 176, 177). In this regard, heterogeneity of response characterizes all known environmental risk factors for psychopathology, including the most overwhelming of traumas (176). Such response heterogeneity is associated with pre-existing genetic (175) or epigenetic (i.e., chromatin modifications) differences (178).
This hypothesis implies that in any given population, individual predisposition is directly responsible for the vulnerability or resilience to the environmental causes of many psychiatric conditions (176), including SCZ (39, 179). Regarding vulnerability, there is a significant interaction between maternal HSV‐2 seropositivity and GRIN2B genetic variation (GRIN2B encodes for a NMDA glutamate receptor) (180). Also, exposure to maternal infection has been reported to increase the risk of SCZ only in cases with a family history of psychiatric disorders (181, 182). Animal models of MIA support this notion, as the effects of poly I:C are enhanced when they occur in mice mutant for genes linked to SCZ and other disorders (183–185). Furthermore, interaction with other environmental risk factors, such as maternal diet, gut microbiota, or experiences of peripubertal trauma, can have a synergistic effect with maternal infection or prevent its detrimental effect (6, 79, 186). Therefore, systematically generating translational models of the interaction between genetic and environmental (or environmental and environmental) risk factors for SCZ and other neurodevelopmental and psychiatric disorders appears to be the next step in understanding the etiology of mental illnesses.
Author Contributions
AA-V, BR, and EM-C wrote and edited the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika Virus and Birth Defects–Reviewing the Evidence for Causality. N Engl J Med (2016) 374(20):1981–7. doi: 10.1056/NEJMsr1604338
2. De Felice FG, Tovar-Moll F, Moll J, Munoz DP, Ferreira ST. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Central Nervous System. Trends Neurosci (2020) 43(6):355–7. doi: 10.1016/j.tins.2020.04.004
3. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron (2009) 64(1):61–78. doi: 10.1016/j.neuron.2009.09.002 S0896-6273(09)00680-1
4. Brown AS, Derkits EJ. Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies. Am J Psychiatry (2010) 167(3):261–80. doi: 10.1176/appi.ajp.2009.09030361
5. Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science (2016) 353(6301):772–7. doi: 10.1126/science.aag3194
6. Meyer U. Neurodevelopmental Resilience and Susceptibility to Maternal Immune Activation. Trends Neurosci (2019) 42(11):793–806. doi: 10.1016/j.tins.2019.08.001
7. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol (2014) 10(11):643–60. doi: 10.1038/nrneurol.2014.187
8. Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M, et al. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry (2014) 171(5):557–63. doi: 10.1176/appi.ajp.2013.13070943
9. Kloiber S, Rosenblat JD, Husain M II, Ortiz A, Berk M, Quevedo J, et al. Neurodevelopmental pathways in bipolar disorder. Neurosci Biobehav Rev (2020) 112:213–26. doi: 10.1016/j.neubiorev.2020.02.005
10. G. B. D. 2017-Disease-Injury-Incidence-Prevalence-Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (Lond Engl) (2018) 392(10159):1789–858. doi: 10.1016/S0140-6736(18)32279-7
11. Freedman R. Schizophrenia. N Engl J Med (2003) 349(18):1738–49. doi: 10.1056/NEJMra035458349/18/1738
12. Chong HY, Teoh SL, Wu DB, Kotirum S, Chiou CF, Chaiyakunapruk N. Global economic burden of schizophrenia: a systematic review. Neuropsychiatr Dis Treat (2016) 12:357–73. doi: 10.2147/NDT.S96649
13. van Os J, Kapur S. Schizophrenia. Lancet (2009) 374(9690):635–45. doi: 10.1016/S0140-6736(09)60995-8
14. Walker E, Kestler L, Bollini A, Hochman KM. Schizophrenia: etiology and course. Annu Rev Psychol (2004) 55:401–30. doi: 10.1146/annurev.psych.55.090902.141950
15. Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron (2006) 52(1):139–53. doi: 10.1016/j.neuron.2006.09.015
16. Charney AW, Mullins N, Park YJ, Xu J. On the diagnostic and neurobiological origins of bipolar disorder. Transl Psychiatry (2020) 10(1):118. doi: 10.1038/s41398-020-0796-8
17. Butala A, Shepard M, Pontone G. Neuropsychiatric aspects of Parkinson disease psychopharmacology: Insights from circuit dynamics. Handb Clin Neurol (2019) 165:83–121. doi: 10.1016/B978-0-444-64012-3.00007-1
18. Deardorff WJ, Grossberg GT. Behavioral and psychological symptoms in Alzheimer’s dementia and vascular dementia. Handb Clin Neurol (2019) 165:5–32. doi: 10.1016/B978-0-444-64012-3.00002-2
19. Taylor JH, Calkins ME, Gur RE. Markers of Psychosis Risk in the General Population. Biol Psychiatry (2020) 88(4):337–48. doi: 10.1016/j.biopsych.2020.02.002
20. Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science (2015) 349(6255):1489–94. doi: 10.1126/science.aaa8954
21. Carlsson A, Lindqvist M. Effect of Chlorpromazine or Haloperidol on Formation of 3methoxytyramine and Normetanephrine in Mouse Brain. Acta Pharmacol Toxicol (Copenh) (1963) 20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x
22. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry (1987) 44(7):660–9. doi: 10.1001/archpsyc.1987.01800190080012
23. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry (1991) 148(11):1474–86. doi: 10.1176/ajp.148.11.1474
24. Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci (1995) 15(5 Pt 2):4102–8. doi: 10.1523/JNEUROSCI.15-05-04102.1995
25. Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci (2008) 28(4):976–89. doi: 10.1523/JNEUROSCI.2796-07.2008
26. Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacol (Berl) (1987) 91(4):415–33. doi: 10.1007/BF00216006
27. Lieberman JA, Sheitman BB, Kinon BJ. Neurochemical sensitization in the pathophysiology of schizophrenia: deficits and dysfunction in neuronal regulation and plasticity. Neuropsychopharmacology (1997) 17(4):205–29. doi: 10.1016/S0893-133X(97)00045-6
28. Yui K, Goto K, Ikemoto S, Ishiguro T, Angrist B, Duncan GE, et al. Neurobiological basis of relapse prediction in stimulant-induced psychosis and schizophrenia: the role of sensitization. Mol Psychiatry (1999) 4(6):512–23. doi: 10.1038/sj.mp.4000575
29. Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U.S.A. (1996) 93(17):9235–40. doi: 10.1073/pnas.93.17.9235
30. Abi-Dargham A, van de Giessen E, Slifstein M, Kegeles LS, Laruelle M. Baseline and amphetamine-stimulated dopamine activity are related in drug-naive schizophrenic subjects. Biol Psychiatry (2009) 65(12):1091–3. doi: 10.1016/j.biopsych.2008.12.007
31. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry (1998) 155(6):761–7. doi: 10.1176/ajp.155.6.761
32. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U.S.A. (2000) 97(14):8104–9. doi: 10.1073/pnas.97.14.8104
33. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry (1999) 46(1):56–72. doi: 10.1016/S0006-3223(99)00067-0
34. Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U.S.A. (1997) 94(6):2569–74. doi: 10.1073/pnas.94.6.2569
35. Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci (2002) 5(3):267–71. doi: 10.1038/nn804
36. Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull (2009) 35(3):528–48. doi: 10.1093/schbul/sbn187
37. Murray RM, Bhavsar V, Tripoli G, Howes O. 30 Years on: How the Neurodevelopmental Hypothesis of Schizophrenia Morphed Into the Developmental Risk Factor Model of Psychosis. Schizophr Bull (2017) 43(6):1190–6. doi: 10.1093/schbul/sbx121
38. Owen MJ, O’Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry (2011) 198(3):173–5. doi: 10.1192/bjp.bp.110.084384
39. Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry (2005) 10(5):434–49. doi: 10.1038/sj.mp.4001642
40. Danielyan A, Nasrallah HA. Neurological disorders in schizophrenia. Psychiatr Clin North Am (2009) 32(4):719–57. doi: 10.1016/j.psc.2009.08.004S0193-953X(09)00075-6
41. Mathalon DH, Rapoport JL, Davis KL, Krystal JH. Neurotoxicity, neuroplasticity, and magnetic resonance imaging morphometry. Arch Gen Psychiatry (2003) 60(8):846–8; author reply 848–9. doi: 10.1001/archpsyc.60.8.846
42. Gilmore JH, van Tol J, Kliewer MA, Silva SG, Cohen SB, Hertzberg BS, et al. Mild ventriculomegaly detected in utero with ultrasound: clinical associations and implications for schizophrenia. Schizophr Res (1998) 33(3):133–40. doi: 10.1016/S0920-9964(98)00073-5
43. Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res (1982) 17(4):319–34. doi: 10.1016/0022-3956(82)90038-3
44. Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol (2011) 93(1):23–58. doi: 10.1016/j.pneurobio.2010.09.003
45. Eyles DW, Trzaskowski M, Vinkhuyzen AAE, Mattheisen M, Meier S, Gooch H, et al. The association between neonatal vitamin D status and risk of schizophrenia. Sci Rep (2018) 8(1):17692. doi: 10.1038/s41598-018-35418-z
46. McGrath JJ, Eyles DW, Pedersen CB, Anderson C, Ko P, Burne TH, et al. Neonatal vitamin D status and risk of schizophrenia: a population-based case-control study. Arch Gen Psychiatry (2010) 67(9):889–94. doi: 10.1001/archgenpsychiatry.2010.110
47. Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, et al. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry (2008) 65(2):146–52. doi: 10.1001/archgenpsychiatry.2007.20
48. Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bull (2011) 37(2):284–90. doi: 10.1093/schbul/sbq146
49. Martelon M, Wilens TE, Anderson JP, Morrison NR, Wozniak J. Are obstetrical, perinatal, and infantile difficulties associated with pediatric bipolar disorder? Bipolar Disord (2012) 14(5):507–14. doi: 10.1111/j.1399-5618.2012.01027.x
50. Chudal R, Sourander A, Polo-Kantola P, Hinkka-Yli-Salomaki S, Lehti V, Sucksdorff D, et al. Perinatal factors and the risk of bipolar disorder in Finland. J Affect Disord (2014) 155:75–80. doi: 10.1016/j.jad.2013.10.026
51. Machon RA, Mednick SA, Schulsinger F. The interaction of seasonality, place of birth, genetic risk and subsequent schizophrenia in a high risk sample. Br J Psychiatry (1983) 143:383–8. doi: 10.1192/bjp.143.4.383
52. Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry (1988) 45(2):189–92. doi: 10.1001/archpsyc.1988.01800260109013
53. Watson CG, Kucala T, Tilleskjor C, Jacobs L. Schizophrenic birth seasonality in relation to the incidence of infectious diseases and temperature extremes. Arch Gen Psychiatry (1984) 41(1):85–90. doi: 10.1001/archpsyc.1984.01790120089011
54. Torrey EF. Stalking the schizovirus. Schizophr Bull (1988) 14(2):223–9. doi: 10.1093/schbul/14.2.223
55. O’Callaghan E, Sham PC, Takei N, Murray G, Glover G, Hare EH, et al. The relationship of schizophrenic births to 16 infectious diseases. Br J Psychiatry (1994) 165(3):353–6. doi: 10.1192/bjp.165.3.353
56. Suvisaari J, Haukka J, Tanskanen A, Hovi T, Lonnqvist J. Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am J Psychiatry (1999) 156(7):1100–2. doi: 10.1176/ajp.156.7.1100
57. Hare EH, Price JS, Slater E. Schizophrenia and season of birth. Br J Psychiatry (1972) 120(554):124–5. doi: 10.1192/bjp.120.554.124-a
58. Brown AS, Schaefer CA, Wyatt RJ, Goetz R, Begg MD, Gorman JM, et al. Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: a prospective birth cohort study. Schizophr Bull (2000) 26(2):287–95. doi: 10.1093/oxfordjournals.schbul.a033453
59. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry (2004) 61(8):774–80. doi: 10.1001/archpsyc.61.8.774
60. Susser ES, Schaefer CA, Brown AS, Begg MD, Wyatt RJ. The design of the prenatal determinants of schizophrenia study. Schizophr Bull (2000) 26(2):257–73. doi: 10.1093/oxfordjournals.schbul.a033451
61. Brown AS, Cohen P, Greenwald S, Susser E. Nonaffective psychosis after prenatal exposure to rubella. Am J Psychiatry (2000) 157(3):438–43. doi: 10.1176/appi.ajp.157.3.438
62. Brown AS, Cohen P, Harkavy-Friedman J, Babulas V, Malaspina D, Gorman JM, et al. A.E. Bennett Research Award. Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol Psychiatry (2001) 49(6):473–86. doi: 10.1016/S0006-3223(01)01068-X
63. Brown AS, Schaefer CA, Quesenberry, Jr. CP, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry (2005) 162(4):767–73. doi: 10.1176/appi.ajp.162.4.767
64. Mortensen PB, Norgaard-Pedersen B, Waltoft BL, Sorensen TL, Hougaard D, Torrey EF, et al. Toxoplasma gondii as a risk factor for early-onset schizophrenia: analysis of filter paper blood samples obtained at birth. Biol Psychiatry (2007) 61(5):688–93. doi: 10.1016/j.biopsych.2006.05.024
65. Buka SL, Cannon TD, Torrey EF, Yolken RH. Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry (2008) 63(8):809–15. doi: 10.1016/j.biopsych.2007.09.022
66. Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Bernstein D, Yolken RH. Maternal infections and subsequent psychosis among offspring. Arch Gen Psychiatry (2001) 58(11):1032–7 doi: 10.1001/archpsyc.58.11.1032
67. Babulas V, Factor-Litvak P, Goetz R, Schaefer CA, Brown AS. Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry (2006) 163(5):927–9. doi: 10.1176/appi.ajp.163.5.927
68. Sorensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull (2009) 35(3):631–7. doi: 10.1093/schbul/sbn121
69. Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun (2001) 15(4):411–20. doi: 10.1006/brbi.2001.0644
70. Bortolato B, Kohler CA, Evangelou E, Leon-Caballero J, Solmi M, Stubbs B, et al. Systematic assessment of environmental risk factors for bipolar disorder: an umbrella review of systematic reviews and meta-analyses. Bipolar Disord (2017) 19(2):84–96. doi: 10.1111/bdi.12490
71. Mac Giollabhui N, Breen EC, Murphy SK, Maxwell SD, Cohn BA, Krigbaum NY, et al. Maternal inflammation during pregnancy and offspring psychiatric symptoms in childhood: Timing and sex matter. J Psychiatr Res (2019) 111:96–103. doi: 10.1016/j.jpsychires.2019.01.009
72. Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, et al. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry (2004) 161(5):889–95. doi: 10.1176/appi.ajp.161.5.889
73. Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol Med (2013) 43(2):239–57. doi: 10.1017/S0033291712000736
74. Gilmore JH, Jarskog LF. Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res (1997) 24(3):365–7. doi: 10.1016/S0920-9964(96)00123-5
75. Allswede DM, Buka SL, Yolken RH, Torrey EF, Cannon TD. Elevated maternal cytokine levels at birth and risk for psychosis in adult offspring. Schizophr Res (2016) 172(1-3):41–5. doi: 10.1016/j.schres.2016.02.022
76. Goldstein JM, Cherkerzian S, Seidman LJ, Donatelli JA, Remington AG, Tsuang MT, et al. Prenatal maternal immune disruption and sex-dependent risk for psychoses. Psychol Med (2014) 44(15):3249–61. doi: 10.1017/S0033291714000683
77. Canetta S, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, Leiviska J, Kellendonk C, et al. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am J Psychiatry (2014) 171(9):960–8. doi: 10.1176/appi.ajp.2014.13121579
78. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci (2003) 23(1):297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003
79. Haddad F, Patel S, Schmid S. Maternal Immune Activation by Poly I:C as a preclinical Model for Neurodevelopmental Disorders: A focus on Autism and Schizophrenia. Neurosci Biobehav Rev (2020) 113:546–67. doi: 10.1016/j.neubiorev.2020.04.012
80. Bergdolt L, Dunaevsky A. Brain changes in a maternal immune activation model of neurodevelopmental brain disorders. Prog Neurobiol (2019) 175:1–19. doi: 10.1016/j.pneurobio.2018.12.002
81. Meyer U. Prenatal Poly(I:C) Exposure and Other Developmental Immune Activation Models in Rodent Systems. Biol Psychiatry (2013) 75(4):307–15. doi: 10.1016/j.biopsych.2013.07.011
82. Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacol (Berl) (2001) 156(2-3):194–215. doi: 10.1007/s002130100799
83. Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology (2003) 28(10):1778–89. doi: 10.1038/sj.npp.1300248
84. Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry (2006) 59(6):546–54. doi: 10.1016/j.biopsych.2005.07.031
85. Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res (2005) 39(3):311–23. doi: 10.1016/j.jpsychires.2004.08.008
86. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci (2007) 27(40):10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
87. Meyer U, Schwendener S, Feldon J, Yee BK. Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Exp Brain Res (2006) 173(2):243–57. doi: 10.1007/s00221-006-0419-5
88. Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci (2006) 26(18):4752–62. doi: 10.1523/JNEUROSCI.0099-06.2006
89. Meyer U, Spoerri E, Yee BK, Schwarz MJ, Feldon J. Evaluating Early Preventive Antipsychotic and Antidepressant Drug Treatment in an Infection-Based Neurodevelopmental Mouse Model of Schizophrenia. Schizophr Bull (2008) 36(3):607–23. doi: 10.1093/schbul/sbn131
90. Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology (2008) 33(2):441–56. doi: 10.1038/sj.npp.1301413
91. Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry (2008) 13(2):208–21. doi: 10.1038/sj.mp.4002042
92. Meyer U, Engler A, Weber L, Schedlowski M, Feldon J. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience (2008) 154(2):701–9. doi: 10.1016/j.neuroscience.2008.04.031
93. Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci (2010) 30(4):1270–87. doi: 10.1523/JNEUROSCI.5408-09.2010
94. Luan W, Hammond LA, Vuillermot S, Meyer U, Eyles DW. Maternal Vitamin D Prevents Abnormal Dopaminergic Development and Function in a Mouse Model of Prenatal Immune Activation. Sci Rep (2018) 8(1):9741. doi: 10.1038/s41598-018-28090-w
95. Borcoi AR, Patti CL, Zanin KA, Hollais AW, Santos-Baldaia R, Ceccon LM, et al. Effects of prenatal immune activation on amphetamine-induced addictive behaviors: Contributions from animal models. Prog Neuropsychopharmacol Biol Psychiatry (2015) 63:63–9. doi: 10.1016/j.pnpbp.2015.05.015
96. Missault S, Van den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, et al. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun (2014) 42:138–46. doi: 10.1016/j.bbi.2014.06.013
97. Willi R, Harmeier A, Giovanoli S, Meyer U. Altered GSK3beta signaling in an infection-based mouse model of developmental neuropsychiatric disease. Neuropharmacology (2013) 73:56–65. doi: 10.1016/j.neuropharm.2013.05.012
98. Piontkewitz Y, Arad M, Weiner I. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull (2011) 37(6):1257–69. doi: 10.1093/schbul/sbq040
99. Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry (2011) 70(9):842–51. doi: 10.1016/j.biopsych.2011.06.007
100. Lins BR, Hurtubise JL, Roebuck AJ, Marks WN, Zabder NK, Scott GA, et al. Prospective Analysis of the Effects of Maternal Immune Activation on Rat Cytokines during Pregnancy and Behavior of the Male Offspring Relevant to Schizophrenia. eNeuro (2018) 5(4):ENEURO.0249-18.2018. doi: 10.1523/ENEURO.0249-18.2018
101. Chou S, Jones S, Li M. Adolescent olanzapine sensitization is correlated with hippocampal stem cell proliferation in a maternal immune activation rat model of schizophrenia. Brain Res (2015) 1618:122–35. doi: 10.1016/j.brainres.2015.05.036
102. Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology (2010) 35(12):2462–78. doi: 10.1038/npp.2010.129
103. da Silveira VT, Medeiros DC, Ropke J, Guidine PA, Rezende GH, Moraes MF, et al. Effects of early or late prenatal immune activation in mice on behavioral and neuroanatomical abnormalities relevant to schizophrenia in the adulthood. Int J Dev Neurosci (2017) 58:1–8. doi: 10.1016/j.ijdevneu.2017.01.009
104. Labouesse MA, Langhans W, Meyer U. Abnormal context-reward associations in an immune-mediated neurodevelopmental mouse model with relevance to schizophrenia. Transl Psychiatry (2015) 5:e637. doi: 10.1038/tp.2015.129
105. Richetto J, Calabrese F, Meyer U, Riva MA. Prenatal versus postnatal maternal factors in the development of infection-induced working memory impairments in mice. Brain Behav Immun (2013) 33:190–200. doi: 10.1016/j.bbi.2013.07.006
106. Fortier ME, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res (2004) 38(3):335–45. doi: 10.1016/j.jpsychires.2003.10.001
107. Fortier ME, Luheshi GN, Boksa P. Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res (2007) 181(2):270–7. doi: 10.1016/j.bbr.2007.04.016
108. Liu X, Lee JG, Yee SK, Bresee CJ, Poland RE, Pechnick RN. Endotoxin exposure in utero increases ethanol consumption in adult male offspring. Neuroreport (2004) 15(1):203–6. doi: 10.1097/00001756-200401190-00039
109. Girard S, Tremblay L, Lepage M, Sebire G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol (2010) 184(7):3997–4005. doi: 10.4049/jimmunol.0903349
110. Girard S, Kadhim H, Beaudet N, Sarret P, Sebire G. Developmental motor deficits induced by combined fetal exposure to lipopolysaccharide and early neonatal hypoxia/ischemia: a novel animal model for cerebral palsy in very premature infants. Neuroscience (2009) 158(2):673–82. doi: 10.1016/j.neuroscience.2008.10.032
111. Lante F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R, et al. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus (2008) 18(6):602–9. doi: 10.1002/hipo.20421
112. Lante F, Meunier J, Guiramand J, Maurice T, Cavalier M, de Jesus Ferreira MC, et al. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radic Biol Med (2007) 42(8):1231–45. doi: 10.1016/j.freeradbiomed.2007.01.027
113. Wang H, Meng XH, Ning H, Zhao XF, Wang Q, Liu P, et al. Age- and gender-dependent impairments of neurobehaviors in mice whose mothers were exposed to lipopolysaccharide during pregnancy. Toxicol Lett (2010) 192(2):245–51. doi: 10.1016/j.toxlet.2009.10.030
114. Golan H, Stilman M, Lev V, Huleihel M. Normal aging of offspring mice of mothers with induced inflammation during pregnancy. Neuroscience (2006) 141(4):1909–18. doi: 10.1016/j.neuroscience.2006.05.045
115. Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology (2005) 48(6):903–17. doi: 10.1016/j.neuropharm.2004.12.023
116. Hava G, Vered L, Yael M, Mordechai H, Mahoud H. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev Psychobiol (2006) 48(2):162–8. doi: 10.1002/dev.20116
117. Basta-Kaim A, Szczesny E, Leskiewicz M, Glombik K, Slusarczyk J, Budziszewska B, et al. Maternal immune activation leads to age-related behavioral and immunological changes in male rat offspring - the effect of antipsychotic drugs. Pharmacol Rep (2012) 64(6):1400–10. doi: 10.1016/s1734-1140(12)70937-4
118. Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology (2002) 26(2):204–15. doi: 10.1016/S0893-133X(01)00360-8
119. Romero E, Ali C, Molina-Holgado E, Castellano B, Guaza C, Borrell J. Neurobehavioral and Immunological Consequences of Prenatal Immune Activation in Rats. Influence of Antipsychotics. Neuropsychopharmacology (2006) 32(8):1791–804. doi: 10.1038/sj.npp.1301292
120. Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry (2008) 15(4):372–83 . doi: 10.1038/mp.2008.44
121. Wusteman M, Wight DG, Elia M. Protein metabolism after injury with turpentine: a rat model for clinical trauma. Am J Physiol (1990) 259(6 Pt 1):E763–9. doi: 10.1152/ajpendo.1990.259.6.E763
122. Turnbull AV, Prehar S, Kennedy AR, Little RA, Hopkins SJ. Interleukin-6 is an afferent signal to the hypothalamo-pituitary-adrenal axis during local inflammation in mice. Endocrinology (2003) 144(5):1894–906. doi: 10.1210/en.2002-220964
123. Aguilar-Valles A, Poole S, Mistry Y, Williams S, Luheshi GN. Attenuated fever in rats during late pregnancy is linked to suppressed interleukin-6 production after localized inflammation with turpentine. J Physiol (2007) 583(Pt 1):391–403. doi: 10.1113/jphysiol.2007.132829
124. Aguilar-Valles A, Luheshi GN. Alterations in cognitive function and behavioral response to amphetamine induced by prenatal inflammation are dependent on the stage of pregnancy. Psychoneuroendocrinology (2011) 36(5):634–48. doi: 10.1016/j.psyneuen.2010.09.006
125. Zuckerman L, Weiner I. Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacol (Berl) (2003) 169(3-4):308–13. doi: 10.1007/s00213-003-1461-7
126. Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci U.S.A. (2000) 97(14):7673–5. doi: 10.1073/pnas.97.14.7673
127. MacDowell KS, Munarriz-Cuezva E, Caso JR, Madrigal JL, Zabala A, Meana JJ, et al. Paliperidone reverts Toll-like receptor 3 signaling pathway activation and cognitive deficits in a maternal immune activation mouse model of schizophrenia. Neuropharmacology (2017) 116:196–207. doi: 10.1016/j.neuropharm.2016.12.025
128. Meyer U, Knuesel I, Nyffeler M, Feldon J. Chronic clozapine treatment improves prenatal infection-induced working memory deficits without influencing adult hippocampal neurogenesis. Psychopharmacol (Berl) (2010) 208(4):531–43. doi: 10.1007/s00213-009-1754-6
129. Meyer U, Spoerri E, Yee BK, Schwarz MJ, Feldon J. Evaluating early preventive antipsychotic and antidepressant drug treatment in an infection-based neurodevelopmental mouse model of schizophrenia. Schizophr Bull (2010) 36(3):607–23. doi: 10.1093/schbul/sbn131
130. Patrich E, Piontkewitz Y, Peretz A, Weiner I, Attali B. Maternal immune activation produces neonatal excitability defects in offspring hippocampal neurons from pregnant rats treated with poly I:C. Sci Rep (2016) 6:19106. doi: 10.1038/srep19106
131. Piontkewitz Y, Bernstein HG, Dobrowolny H, Bogerts B, Weiner I, Keilhoff G. Effects of risperidone treatment in adolescence on hippocampal neurogenesis, parvalbumin expression, and vascularization following prenatal immune activation in rats. Brain Behav Immun (2012) 26(2):353–63. doi: 10.1016/j.bbi.2011.11.004
132. Richtand NM, Ahlbrand R, Horn P, Stanford K, Bronson SL, McNamara RK. Effects of risperidone and paliperidone pre-treatment on locomotor response following prenatal immune activation. J Psychiatr Res (2011) 45(9):1194–201. doi: 10.1016/j.jpsychires.2011.02.007
133. Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, et al. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol (2009) 12(4):513–24. doi: 10.1017/S1461145708009206
134. Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science (2013) 339(6123):1095–9. doi: 10.1126/science.1228261
135. Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, et al. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord (2002) 17(1):116–24. doi: 10.1002/mds.10078
136. Ling ZD, Chang Q, Lipton JW, Tong CW, Landers TM, Carvey PM. Combined toxicity of prenatal bacterial endotoxin exposure and postnatal 6-hydroxydopamine in the adult rat midbrain. Neuroscience (2004) 124(3):619–28. doi: 10.1016/j.neuroscience.2003.12.017
137. Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp Neurol (2004) 190(2):373–83. doi: 10.1016/j.expneurol.2004.08.006
138. Ling Z, Zhu Y, Tong C, Snyder JA, Lipton JW, Carvey PM. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp Neurol (2006) 199(2):499–512. doi: 10.1016/j.expneurol.2006.01.010
139. Ling Z, Zhu Y, Tong CW, Snyder JA, Lipton JW, Carvey PM. Prenatal lipopolysaccharide does not accelerate progressive dopamine neuron loss in the rat as a result of normal aging. Exp Neurol (2009) 216(2):312–20. doi: 10.1016/j.expneurol.2008.12.004
140. Wang S, Yan JY, Lo YK, Carvey PM, Ling Z. Dopaminergic and serotoninergic deficiencies in young adult rats prenatally exposed to the bacterial lipopolysaccharide. Brain Res (2009) 1265:196–204. doi: 10.1016/j.brainres.2009.02.022
141. Bakos J, Duncko R, Makatsori A, Pirnik Z, Kiss A, Jezova D. Prenatal immune challenge affects growth, behavior, and brain dopamine in offspring. Ann N Y Acad Sci (2004) 1018:281–7. doi: 10.1196/annals.1296.033
142. Aguilar-Valles A, Flores C, Luheshi GN. Prenatal inflammation-induced hypoferremia alters dopamine function in the adult offspring in rat: relevance for schizophrenia. PloS One (2010) 5(6):e10967. doi: 10.1371/journal.pone.0010967
143. Aguilar-Valles A, Jung S, Poole S, Flores C, Luheshi GN. Leptin and interleukin-6 alter the function of mesolimbic dopamine neurons in a rodent model of prenatal inflammation. Psychoneuroendocrinology (2012) 37(7):956–69. doi: 10.1016/j.psyneuen.2011.11.003
144. Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun (2008) 22(4):469–86. doi: 10.1016/j.bbi.2007.09.012
145. Garbett KA, Hsiao EY, Kalman S, Patterson PH, Mirnics K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry (2012) 2:e98. doi: 10.1038/tp.2012.24
146. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun (2017) 62:11–23. doi: 10.1016/j.bbi.2016.11.007
147. Grieger TA, Kluger MJ. Fever and survival: the role of serum iron. J Physiol (1978) 279:187–96. doi: 10.1113/jphysiol.1978.sp012339
148. Kluger MJ, Rothenburg BA. Fever and reduced iron: their interaction as a host defense response to bacterial infection. Science (1979) 203(4378):374–6. doi: 10.1126/science.760197
149. Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr (2003) 23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739
150. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (2016) 351(6276):933–9. doi: 10.1126/science.aad0314
151. Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature (2017) 549(7673):528–32. doi: 10.1038/nature23910
152. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi: 10.1038/372425a0
153. Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science (1995) 269(5223):546–9. doi: 10.1126/science.7624778
154. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (1995) 269(5223):543–6. doi: 10.1126/science.7624777
155. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science (1995) 269(5223):540–3. doi: 10.1126/science.7624776
156. Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin Levels Reflect Body Lipid-Content in Mice - Evidence for Diet-Induced Resistance to Leptin Action. Nat Med (1995) 1(12):1311–4. doi: 10.1038/nm1295-1311
157. Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med (1995) 1(11):1155–61. doi: 10.1038/nm1195-1155
158. Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci (2014) 15(4):6184–223. doi: 10.3390/ijms15046184
159. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol (2011) 11(2):85–97. doi: 10.1038/nri2921
160. Zarkesh-Esfahani H, Pockley G, Metcalfe RA, Bidlingmaier M, Wu Z, Ajami A, et al. High-dose leptin activates human leukocytes via receptor expression on monocytes. J Immunol (2001) 167(8):4593–9. doi: 10.4049/jimmunol.167.8.4593
161. Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest (2004) 114(1):57–66. doi: 10.1172/JCI200421134
162. Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, et al. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci U.S.A. (2004) 101(21):8138–43. doi: 10.1073/pnas.0305683101
163. Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ, et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med (1997) 185(1):171–5. doi: 10.1084/jem.185.1.171
164. Grunfeld C, Zhao C, Fuller J, Pollock A, Moser A, Friedman J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. A role for leptin in the anorexia of infection. J Clin Invest (1996) 97(9):2152–7. doi: 10.1172/JCI118653
165. Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C. IL-1 beta mediates leptin induction during inflammation. Am J Physiol (1998) 274(1 Pt 2):R204–8. doi: 10.1152/ajpregu.1998.274.1.R204
166. Gualillo O, Eiras S, Lago F, Dieguez C, Casanueva FF. Elevated serum leptin concentrations induced by experimental acute inflammation. Life Sci (2000) 67(20):2433–41. doi: 10.1016/S0024-3205(00)00827-4
167. Mastronardi CA, Yu WH, Srivastava VK, Dees WL, McCann SM. Lipopolysaccharide-induced leptin release is neurally controlled. Proc Natl Acad Sci U States America (2001) 98(25):14720–5. doi: 10.1073/pnas.251543598
168. Sachot C, Poole S, Luheshi GN. Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. J Physiol (2004) 561(Pt 1):263–72. doi: 10.1113/jphysiol.2004.074351
169. Harden LM, du Plessis I, Poole S, Laburn HP. Interleukin-6 and leptin mediate lipopolysaccharide-induced fever and sickness behavior. Physiol Behav (2006) 89(2):146–55. doi: 10.1016/j.physbeh.2006.05.016
170. Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci U.S.A. (1999) 96(12):7047–52. doi: 10.1073/pnas.96.12.7047
171. Turek VF, Olster DH, Gililland KR, Sheehy M, Ettenberg A, Carlisle HJ. The effects of melanocortin agonists and antagonists on leptin-induced fever in rats. J Thermal Biol (2004) 29(7-8):423–30. doi: 10.1016/j.jtherbio.2004.08.011
172. Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet (1996) 13(1):18–9. doi: 10.1038/ng0596-18
173. Inoue W, Poole S, Bristow AF, Luheshi GN. Leptin induces cyclooxygenase-2 via an interaction with interleukin-1beta in the rat brain. Eur J Neurosci (2006) 24(8):2233–45. doi: 10.1111/j.1460-9568.2006.05105.x
174. Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron (2006) 51(6):811–22. doi: 10.1016/j.neuron.2006.09.006
175. Lappas M, Permezel M, Rice GE. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-kappaB, peroxisomal proliferator-activated receptor-gamma and extracellularly regulated kinase 1/2. Endocrinology (2005) 146(8):3334–42. doi: 10.1210/en.2005-0406
176. Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci (2006) 7(7):583–90. doi: 10.1038/nrn1925
177. DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, et al. The developmental neurobiology of autism spectrum disorder. J Neurosci (2006) 26(26):6897–906. doi: 10.1523/JNEUROSCI.1712-06.2006
178. Richetto J, Meyer U. Epigenetic Modifications in Schizophrenia and Related Disorders: Molecular Scars of Environmental Exposures and Source of Phenotypic Variability. Biol Psychiatry (2020). doi: 10.1016/j.biopsych.2020.03.008
179. Dean B, Boer S, Gibbons A, Money T, Scarr E. Recent advances in postmortem pathology and neurochemistry in schizophrenia. Curr Opin Psychiatry (2009) 22(2):154–60. doi: 10.1097/YCO.0b013e328323d52e
180. Demontis D, Nyegaard M, Buttenschon HN, Hedemand A, Pedersen CB, Grove J, et al. Association of GRIN1 and GRIN2A-D with schizophrenia and genetic interaction with maternal herpes simplex virus-2 infection affecting disease risk. Am J Med Genet B Neuropsychiatr Genet (2011) 156B(8):913–22. doi: 10.1002/ajmg.b.31234
181. Borglum AD, Demontis D, Grove J, Pallesen J, Hollegaard MV, Pedersen CB, et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry (2014) 19(3):325–33. doi: 10.1038/mp.2013.2
182. Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry (2009) 166(9):1025–30. doi: 10.1176/appi.ajp.2009.08010031
183. Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry (2010) 68(12):1172–81. doi: 10.1016/j.biopsych.2010.09.022
184. Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci (2013) 33(18):7654–66. doi: 10.1523/JNEUROSCI.0091-13.2013
185. Hemmerle AM, Ahlbrand R, Bronson SL, Lundgren KH, Richtand NM, Seroogy KB. Modulation of schizophrenia-related genes in the forebrain of adolescent and adult rats exposed to maternal immune activation. Schizophr Res (2015) 168(1-2):411–20. doi: 10.1016/j.schres.2015.07.006
Keywords: maternal infection, schizophrenia, dopamine, animal models, cytokines, IL-6, iron, leptin
Citation: Aguilar-Valles A, Rodrigue B and Matta-Camacho E (2020) Maternal Immune Activation and the Development of Dopaminergic Neurotransmission of the Offspring: Relevance for Schizophrenia and Other Psychoses. Front. Psychiatry 11:852. doi: 10.3389/fpsyt.2020.00852
Received: 06 May 2020; Accepted: 04 August 2020;
Published: 21 August 2020.
Edited by:
Grazia Rutigliano, University of Pisa, ItalyReviewed by:
Urs Meyer, ETH Zürich, SwitzerlandFelice Iasevoli, University of Naples Federico II, Italy
Copyright © 2020 Aguilar-Valles, Rodrigue and Matta-Camacho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Argel Aguilar-Valles, argel.aguilavalles@carleton.ca