
Soil Bacterial Diversity Is Positively Correlated with Decomposition Rates during Early Phases of Maize Litter Decomposition

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
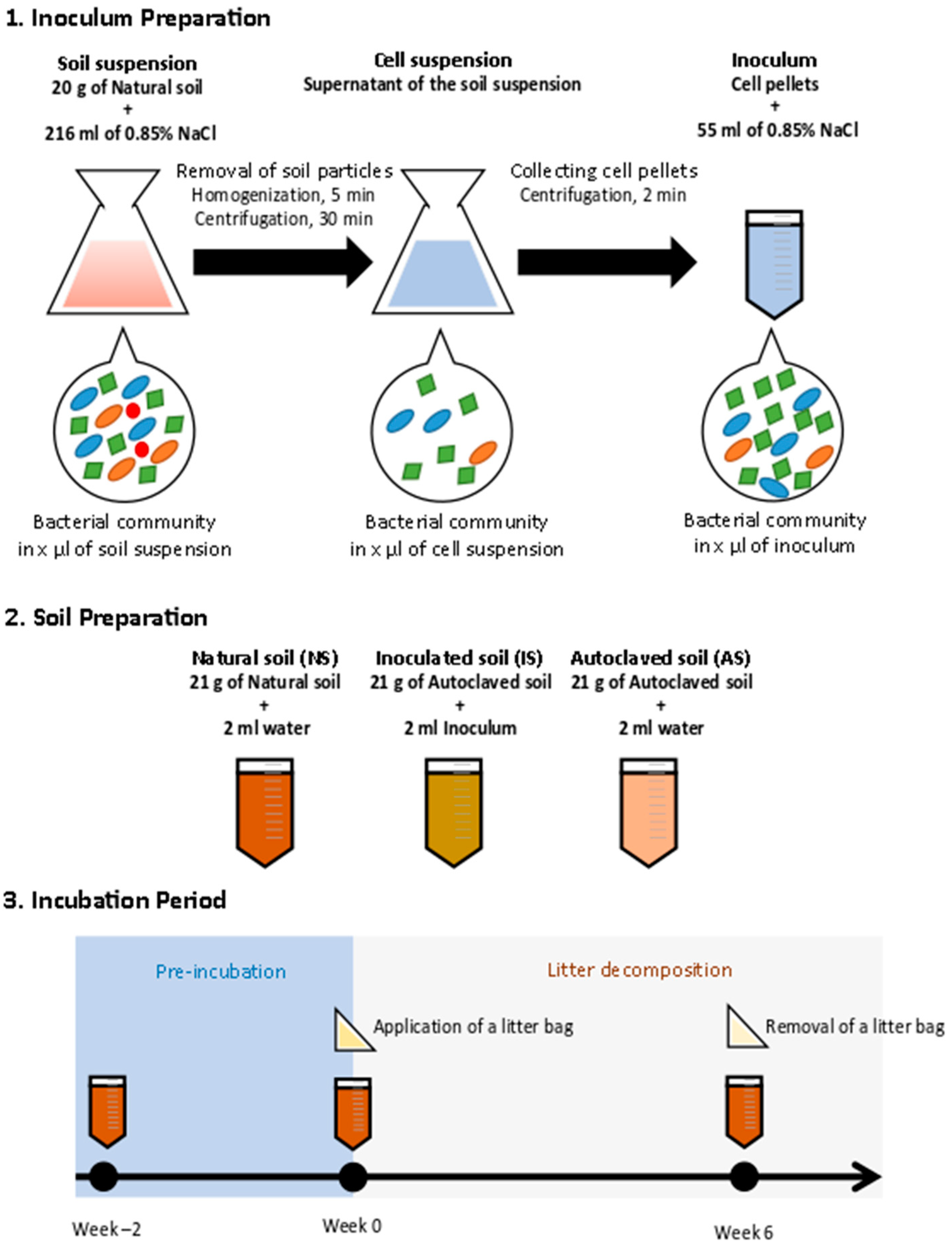
2.1. Sampling
2.2. Incubation Experimental Design
2.3. DNA Extraction and Library Preparation
2.4. Statistics and Bioinformatics
3. Results
3.1. Litter Decomposition
3.2. Dynamics of Bacterial and Fungal Abundance
3.3. Development of Bacterial Communities over Time
3.3.1. Soil Bacteria
3.3.2. Maize Leaf Bacteria
4. Discussion
4.1. Manipulating Soil Bacterial Diversity
4.2. Bacterial Recovery after Disturbance
4.3. Oligotrophic Bacteria and Litter Decomposition
4.4. Litter-Associated Bacteria and Litter Decomposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lugato, E.; Berti, A.; Giardini, L. Soil organic carbon (SOC) dynamics with and without residue incorporation in relation to different nitrogen fertilisation rates. Geoderma 2006, 135, 315–321. [Google Scholar] [CrossRef]
- Finn, D.; Page, K.; Catton, K.; Strounina, E.; Kienzle, M.; Robertson, F.; Armstrong, R.; Dalal, R. Effect of added nitrogen on plant litter decomposition depends on initial soil carbon and nitrogen stoichiometry. Soil Biol. Biochem. 2015, 91, 160–168. [Google Scholar] [CrossRef]
- Novara, A.; Rühl, J.; La Mantia, T.; Gristina, L.; La Bella, S.; Tuttolomondo, T. Litter contribution to soil organic carbon in the processes of agriculture abandon. Solid Earth 2015, 6, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Martens, D.A. Plant residue biochemistry regulates soil carbon cycling and carbon sequestration. Soil Biol. Biochem. 2000, 32, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Grandy, A.S.; Neff, J.C. Molecular C dynamics downstream: The biochemical decomposition sequence and its impact on soil organic matter structure and function. Sci. Total Environ. 2008, 404, 297–307. [Google Scholar] [CrossRef]
- Cornwell, W.K.; Cornelissen, J.H.C.; Amatangelo, K.; Dorrepaal, E.; Eviner, V.T.; Godoy, O.; Hobbie, S.E.; Hoorens, B.; Kurokawa, H.; Pérez-Harguindeguy, N.; et al. Plant species traits are the predominant control on litter decomposition rates within biomes worldwide. Ecol. Lett. 2008, 11, 1065–1071. [Google Scholar] [CrossRef]
- Paterson, E.; Osler, G.; Dawson, L.A.; Gebbing, T.; Sim, A.; Ord, B. Labile and recalcitrant plant fractions are utilised by distinct microbial communities in soil: Independent of the presence of roots and mycorrhizal fungi. Soil Biol. Biochem. 2008, 40, 1103–1113. [Google Scholar] [CrossRef]
- Moore, T.R.; Trofymow, J.A.; Taylor, B.; Prescott, C.; Camiré, C.; Duschene, L.; Fyles, J.; Kozak, L.; Kranabetter, M.; Morrison, I.; et al. Litter decomposition rates in Canadian forests. Glob. Chang. Biol. 1999, 5, 75–82. [Google Scholar] [CrossRef]
- Heim, A.; Frey, B. Early stage litter decomposition rates for Swiss forests. Biogeochemistry 2004, 70, 299–313. [Google Scholar] [CrossRef]
- Esperschütz, J.; Zimmermann, C.; Dümig, A.; Welzl, G.; Buegger, F.; Elmer, M.; Munch, J.C.; Schloter, M. Dynamics of microbial communities during decomposition of litter from pioneering plants in initial soil ecosystems. Biogeosciences 2013, 10, 5115–5124. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Baquerizo, M.; García-Palacios, P.; Milla, R.; Gallardo, A.; Maestre, F.T. Soil characteristics determine soil carbon and nitrogen availability during leaf litter decomposition regardless of litter quality. Soil Biol. Biochem. 2015, 81, 134–142. [Google Scholar] [CrossRef]
- Güsewell, S.; Gessner, M.O. N:P ratios influence litter decomposition and colonization by fungi and bacteria in microcosms. Funct. Ecol. 2009, 23, 211–219. [Google Scholar] [CrossRef]
- Bray, S.R.; Kitajima, K.; Mack, M.C. Temporal dynamics of microbial communities on decomposing leaf litter of 10 plant species in relation to decomposition rate. Soil Biol. Biochem. 2012, 49, 30–37. [Google Scholar] [CrossRef]
- Coûteaux, M.; Bottner, P.; Berg, B. Litter decomposition, climate and liter quality. Trends Ecol. Evol. 1995, 10, 63–66. [Google Scholar] [CrossRef]
- Wickings, K.; Grandy, A.S.; Reed, S.C.; Cleveland, C.C. The origin of litter chemical complexity during decomposition. Ecol. Lett. 2012, 15, 1180–1188. [Google Scholar] [CrossRef]
- Chapman, S.K.; Newman, G.S.; Hart, S.C.; Schweitzer, J.A.; Koch, G.W. Leaf litter mixtures alter microbial community development: Mechanisms for non-additive effects in litter decomposition. PLoS ONE 2013, 8, e62671. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.; Liu, Y.; Freschet, G.T.; Zhang, W.; Yu, X.; Zheng, W.; Guan, X.; Yang, Q.; Chen, L.; Dijkstra, F.A.; et al. Litter carbon and nutrient chemistry control the magnitude of soil priming effect. Funct. Ecol. 2019, 33, 876–888. [Google Scholar] [CrossRef]
- McMahon, S.K.; Williams, M.A.; Bottomley, P.J.; Myrold, D.D. Dynamics of microbial communities during decomposition of carbon-13 labeled ryegrass fractions in soil. Soil Sci. Soc. Am. J. 2005, 69, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Aneja, M.K.; Sharma, S.; Fleischmann, F.; Stich, S.; Heller, W.; Bahnweg, G.; Munch, J.C.; Schloter, M. Microbial colonization of beech and spruce litter—Influence of decomposition site and plant litter species on the diversity of microbial community. Microb. Ecol. 2006, 52, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.; Kermer, R.; Ullmann-Zeunert, L.; Baldwin, I.T.; Seifert, J.; Bastida, F.; Von Bergen, M.; Jehmlich, N. Bacteria dominate the short-term assimilation of plant-derived N in soil. Soil Biol. Biochem. 2016, 96, 30–38. [Google Scholar] [CrossRef]
- Fanin, N.; Moorhead, D.; Bertrand, I. Eco-enzymatic stoichiometry and enzymatic vectors reveal differential C, N, P dynamics in decaying litter along a land-use gradient. Biogeochemistry 2016, 129, 21–36. [Google Scholar] [CrossRef]
- Manzoni, S.; Trofymow, J.A.; Jackson, R.B.; Porporato, A. Stoichiometric controls on carbon, nitrogen, and phosphorus dynamics in decomposing litter. Ecol. Monogr. 2010, 80, 89–106. [Google Scholar] [CrossRef]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Pascault, N.; Ranjard, L.; Kaisermann, A.; Bachar, D.; Christen, R.; Terrat, S.; Mathieu, O.; Lévêque, J.; Mougel, C.; Henault, C.; et al. Stimulation of different functional groups of bacteria by various plant residues as a driver of soil priming effect. Ecosystems 2013, 16, 810–822. [Google Scholar] [CrossRef]
- Herzog, C.; Hartmann, M.; Frey, B.; Stierli, B.; Rumpel, C.; Buchmann, N.; Brunner, I. Microbial succession on decomposing root litter in a drought-prone Scots pine forest. ISME J. 2019, 13, 2346–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purahong, W.; Wubet, T.; Lentendu, G.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Hofrichter, M.; Krüger, D.; Buscot, F. Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 2016, 25, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Z.; Zhao, B.; Zhang, J. Relationship between the chemical structure of straw and composition of main microbial groups during the decomposition of wheat and maize straws as affected by soil texture. Biol. Fertil. Soils 2020, 56, 11–24. [Google Scholar] [CrossRef]
- Pathan, S.I.; Žifčáková, L.; Ceccherini, M.T.; Pantani, O.L.; Větrovský, T.; Baldrian, P. Seasonal variation and distribution of total and active microbial community of β-glucosidase encoding genes in coniferous forest soil. Soil Biol. Biochem. 2017, 105, 71–80. [Google Scholar] [CrossRef]
- Zang, X.; Liu, M.; Wang, H.; Fan, Y.; Zhang, H.; Liu, J.; Xing, E.; Xu, X.; Li, H. The distribution of active β-glucosidase-producing microbial communities in composting. Can. J. Microbiol. 2017, 63, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, K.; Dignac, M.-F.; Rumpel, C.; Bardoux, G.; Sarr, A.; Steffens, M.; Maron, P.-A. Soil microbial diversity affects soil organic matter decomposition in a silty grassland soil. Biogeochemistry 2013, 114, 201–212. [Google Scholar] [CrossRef]
- Griffiths, B.S.; Kuan, H.L.; Ritz, K.; Glover, L.A.; McCaig, A.E.; Fenwick, C. The relationship between microbial community structure and functional stability, tested experimentally in an upland pasture soil. Microb. Ecol. 2004, 47, 104–113. [Google Scholar] [CrossRef]
- Wei, Z.; Yang, T.; Friman, V.-P.; Xu, Y.; Shen, Q.; Jousset, A. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 2015, 6, 8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Elsas, J.D.; Chiurazzi, M.; Mallon, C.A.; Elhottova, D.; Kristufek, V.; Salles, J.F. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. USA 2012, 109, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brankatschk, R.; Töwe, S.; Kleineidam, K.; Schloter, M.; Zeyer, J. Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J. 2011, 5, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Vuko, M.; Cania, B.; Vogel, C.; Kublik, S.; Schloter, M.; Schulz, S. Shifts in reclamation management strategies shape the role of exopolysaccharide and lipopolysaccharide-producing bacteria during soil formation. Microb. Biotechnol. 2020, 13, 584–598. [Google Scholar] [CrossRef]
- Schreiner, K.; Hagn, A.; Kyselková, M.; Moënne-Loccoz, Y.; Welzl, G.; Munch, J.C.; Schloter, M. Comparison of barley succession and take-all disease as environmental factors shaping the rhizobacterial community during take-all decline. Appl. Environ. Microbiol. 2010, 76, 4703–4712. [Google Scholar] [CrossRef] [Green Version]
- Figuerola, E.L.M.; Guerrero, L.D.; Türkowsky, D.; Wall, L.G.; Erijman, L. Crop monoculture rather than agriculture reduces the spatial turnover of soil bacterial communities at a regional scale. Environ. Microbiol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Grayston, S.J.; Wang, S.; Campbell, C.D.; Edwards, A.C. Selective influence of plant species on microbial diversity in the rhizosphere. Soil Biol. Biochem. 1998, 30, 369–378. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, L.; Zhang, Z.; Zhang, J.; Cheng, J.; Wei, G.; Lin, Y. Soil microbial diversity during 30 years of grassland restoration on the Loess Plateau, China: Tight linkages with plant diversity. Land Degrad. Dev. 2019, 30, 1172–1182. [Google Scholar] [CrossRef]
- Dickens, H.E.; Anderson, J.M. Manipulation of soil microbial community structure in bog and forest soils using chloroform fumigation. Soil Biol. Biochem. 1999, 31, 2049–2058. [Google Scholar] [CrossRef]
- Yang, G.; Roy, J.; Veresoglou, S.D.; Rillig, M.C. Soil biodiversity enhances the persistence of legumes under climate change. New Phytol. 2020. [Google Scholar] [CrossRef]
- Maron, P.-A.; Sarr, A.; Kaisermann, A.; Lévêque, J.; Mathieu, O.; Guigue, J.; Karimi, B.; Bernard, L.; Dequiedt, S.; Terrat, S.; et al. High microbial diversity promotes soil ecosystem functioning. Appl. Environ. Microbiol. 2018, 84, e02738-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevors, J.T. Sterilization and inhibition of microbial activity in soil. J. Microbiol. Methods 1996, 26, 53–59. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, X.; Huang, J. Relationship between enantioselective transformation of racemic quizalofop-ethyl and soil bacterial diversity: A destructive approach. Chirality 2019, 31, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, J.M.; Bais, H.P.; Stermitz, F.R.; Thelen, G.C.; Callaway, R.M. Biogeographical variation in community response to root allelochemistry: Novel weapons and exotic invasion. Ecol. Lett. 2004, 7, 285–292. [Google Scholar] [CrossRef]
- Töwe, S.; Wallisch, S.; Bannert, A.; Fischer, D.; Hai, B.; Haesler, F.; Kleineidam, K.; Schloter, M. Improved protocol for the simultaneous extraction and column-based separation of DNA and RNA from different soils. J. Microbiol. Methods 2011, 84, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Bressan, M.; Gattin, I.T.; Desaire, S.; Castel, L.; Gangneux, C.; Laval, K. A rapid flow cytometry method to assess bacterial abundance in agricultural soil. Appl. Soil Ecol. 2015, 88, 60–68. [Google Scholar] [CrossRef]
- Bach, H.-J.; Tomanova, J.; Schloter, M.; Munch, J. Enumeration of total bacteria and bacteria with genes for proteolytic activity in pure cultures and in environmental samples by quantitative PCR mediated amplification. J. Microbiol. Methods 2002, 49, 235–245. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Sninsky, J., White, T., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Schoeler, A.; Jacquiod, S.; Vestergaard, G.; Schulz, S.; Schloter, M. Analysis of soil microbial communities based on amplicon sequencing of marker genes. Biol. Fertil. Soils 2017, 53, 485–489. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 25 January 2021).
- RStudio Team. RStudio: Integrated Development for R; RStudio Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 25 January 2021).
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Lenth, R.V. Least-squares means: The R package lsmeans. J. Stat. Softw. 2016, 69. [Google Scholar] [CrossRef] [Green Version]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Berns, A.E.; Philipp, H.; Narres, H.-D.; Burauel, P.; Vereecken, H.; Tappe, W. Effect of gamma-sterilization and autoclaving on soil organic matter structure as studied by solid state NMR, UV and fluorescence spectroscopy. Eur. J. Soil Sci. 2008, 59, 540–550. [Google Scholar] [CrossRef]
- Simmon, K.E.; Steadman, D.D.; Durkin, S.; Baldwin, A.; Jeffrey, W.H.; Sheridan, P.; Horton, R.; Shields, M.S. Autoclave method for rapid preparation of bacterial PCR-template DNA. J. Microbiol. Methods 2004, 56, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.; Holländer, R.; Mannheim, W. Lipoquinones of some spore-forming rods, lactic-acid bacteria and Actinomycetes. Microbiology 1979, 115, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solyanikova, I.P.; Suzina, N.E.; Egozarjan, N.S.; Polivtseva, V.N.; Mulyukin, A.L.; Egorova, D.O.; El-Registan, G.I.; Golovleva, L.A. Structural and functional rearrangements in the cells of actinobacteria Microbacterium foliorum BN52 during transition from vegetative growth to a dormant state and during germination of dormant forms. Microbiology 2017, 86, 476–486. [Google Scholar] [CrossRef]
- Chase, A.B.; Arevalo, P.; Polz, M.F.; Berlemont, R.; Martiny, J.B.H. Evidence for ecological flexibility in the cosmopolitan genus Curtobacterium. Front. Microbiol. 2016, 7, 1874. [Google Scholar] [CrossRef]
- Yeager, C.M.; Gallegos-Graves, L.V.; Dunbar, J.; Hesse, C.N.; Daligault, H.; Kuske, C.R. Polysaccharide degradation capability of actinomycetales soil isolates from a semiarid grassland of the Colorado Plateau. Appl. Environ. Microbiol. 2017, 83, e03020-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Jia, Y.; Zhang, R.; Lin, Z.; Zhen, Z.; Hu, H.; Yan, Y. Insight into metabolic versatility of an aromatic compounds-degrading Arthrobacter sp. YC-RL1. Front. Microbiol. 2018, 9, 2438. [Google Scholar] [CrossRef] [Green Version]
- Jurburg, S.D.; Nunes, I.; Brejnrod, A.; Jacquiod, S.; Priemé, A.; Sørensen, S.J.; Van Elsas, J.D.; Salles, J.F. Legacy effects on the recovery of soil bacterial communities from extreme temperature perturbation. Front. Microbiol. 2017, 8, 1832. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.C.; Fernandez-Gonzalez, A.; Fernandez-Lopez, M.; Ogaya, R.; Penuelas, J.; Sardans, J.; Lloret, F. Strong functional stability of soil microbial communities under semiarid Mediterranean conditions and subjected to long-term shifts in baseline precipitation. Soil Biol. Biochem. 2014, 69, 223–233. [Google Scholar] [CrossRef]
- Deng, H. A review of diversity-stability relationship of soil microbial community: What do we not know? J. Environ. Sci. 2012, 24, 1027–1035. [Google Scholar] [CrossRef]
- De Vries, M.; Schöler, A.; Ertl, J.; Xu, Z.; Schloter, M. Metagenomic analyses reveal no differences in genes involved in cellulose degradation under different tillage treatments. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [Green Version]
- Velandia, C.A.M.; Izquierdo-García, L.F.; Ongena, M.; Kloepper, J.W.; Cotes, A.M. Soil sterilization, pathogen and antagonist concentration affect biological control of Fusarium wilt of cape gooseberry by Bacillus velezensis Bs006. Plant Soil 2019, 435, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Naether, A.; Foesel, B.U.; Naegele, V.; Wüst, P.K.; Weinert, J.; Bonkowski, M.; Alt, F.; Oelmann, Y.; Polle, A.; Lohaus, G.; et al. Environmental factors affect acidobacterial communities below the subgroup level in grassland and forest soils. Appl. Environ. Microbiol. 2012, 78, 7398–7406. [Google Scholar] [CrossRef] [Green Version]
- Bartram, A.K.; Jiang, X.; Lynch, M.D.J.; Masella, A.P.; Nicol, G.W.; Dushoff, J.; Neufeld, J.D. Exploring links between pH and bacterial community composition in soils from the Craibstone Experimental Farm. FEMS Microbiol. Ecol. 2014, 87, 403–415. [Google Scholar] [CrossRef]
- Yin, C.; Jones, K.L.; Peterson, D.E.; Garrett, K.A.; Hulbert, S.H.; Paulitz, T. Members of soil bacterial communities sensitive to tillage and crop rotation. Soil Biol. Biochem. 2010, 42, 2111–2118. [Google Scholar] [CrossRef]
- Rodrigues, G.R.; Pinto, O.H.B.; Schroeder, L.F.; da Rocha Fernandes, G.; Costa, Y.A.O.; Quirino, F.B.; Kuramae, E.E.; Barreto, C.C. Unraveling the xylanolytic potential of Acidobacteria bacterium AB60 from Cerrado soils. FEMS Microbiol. Lett. 2020, 367, fnaa149. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Zhelezova, A.D.; Chernov, T.I.; Dedysh, S.N. Linking ecology and systematics of acidobacteria: Distinct habitat preferences of the Acidobacteriia and Blastocatellia in tundra soils. PLoS ONE 2020, 15, e0230157. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.J.; Overmann, J. Vicinamibacteraceae fam. nov., the first described family within the subdivision 6 Acidobacteria. Int. J. Syst. Evol. Microbiol. 2018, 68, 2331–2334. [Google Scholar] [CrossRef]
- Vieira, S.; Luckner, M.; Wanner, G.; Overmann, J. Luteitalea pratensis gen. nov., sp. nov. a new member of subdivision 6 Acidobacteria isolated from temperate grassland soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 1408–1414. [Google Scholar] [CrossRef]
- Navarrete, A.A.; Venturini, A.M.; Meyer, K.M.; Klein, A.M.; Tiedje, J.M.; Bohannan, B.J.M.; Nüsslein, K.; Tsai, S.M.; Rodrigues, J.L.M. Differential response of Acidobacteria subgroups to forest-to-pasture conversion and their biogeographic patterns in the Western Brazilian Amazon. Front. Microbiol. 2015, 6, 1443. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Mueth, N.; Hulbert, S.; Schlatter, D.; Paulitz, T.C.; Schroeder, K.; Prescott, A.; Dhingra, A. Bacterial communities on wheat grown under long-term conventional tillage and no-till in the Pacific Northwest of the United States. Phytobiomes J. 2017, 1, 83–90. [Google Scholar] [CrossRef]
- Herber, J.; Klotz, F.; Frommeyer, B.; Weis, S.; Straile, D.; Kolar, A.; Sikorski, J.; Egert, M.; Dannenmann, M.; Pester, M. A single Thaumarchaeon drives nitrification in deep oligotrophic Lake Constance. Environ. Microbiol. 2020, 22, 212–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buresova, A.; Kopecky, J.; Hrdinkova, V.; Kamenik, Z.; Omelka, M.; Sagova-Mareckova, M. Succession of microbial decomposers is determined by litter type, but site conditions drive decomposition rates. Appl. Environ. Microbiol. 2019, 85, e01760-19. [Google Scholar] [CrossRef]
- Koyama, A.; Wallenstein, M.D.; Simpson, R.T.; Moore, J.C. Soil bacterial community composition altered by increased nutrient availability in Arctic tundra soils. Front. Microbiol. 2014, 5, 516. [Google Scholar] [CrossRef] [Green Version]
- Madhaiyan, M.; Saravanan, V.S.; Wirth, J.S.; Alex, T.H.H.; Kim, S.-J.; Weon, H.-Y.; Kwon, S.-W.; Whitman, W.B.; Ji, L. Sphingomonas palmae sp. nov. and Sphingomonas gellani sp. nov., endophytically associated phyllosphere bacteria isolated from economically important crop plants. Antonie Van Leeuwenhoek 2020, 113, 1617–1632. [Google Scholar] [CrossRef]
- Ulrich, A.; Klimke, G.; Wirth, S. Diversity and activity of cellulose-decomposing bacteria, isolated from a sandy and a loamy soil after long-term manure application. Microb. Ecol. 2008, 55, 512–522. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, Q.; Ai, C.; He, P.; Zhou, W. Microbial utilization of rice root residue-derived carbon explored by DNA stable-isotope probing. Eur. J. Soil Sci. 2020, 72, 460–473. [Google Scholar] [CrossRef]
- Montañez, A.; Abreu, C.; Gill, P.R.; Hardarson, G.; Sicardi, M. Biological nitrogen fixation in maize (Zea mays L.) by 15N isotope-dilution and identification of associated culturable diazotrophs. Biol. Fertil. Soils 2009, 45, 253–263. [Google Scholar] [CrossRef]
- Doty, S.L.; Oakley, B.; Xin, G.; Kang, J.W.; Singleton, G.; Khan, Z.; Vajzovic, A.; Staley, J.T. Diazotrophic endophytes of native black cottonwood and willow. Symbiosis 2009, 47, 23–33. [Google Scholar] [CrossRef]
- Islam, R.; Sultana, T.; Cho, J.-C.; Joe, M.M.; Sa, T.M. Diversity of free-living nitrogen-fixing bacteria associated with Korean paddy fields. Ann. Microbiol. 2012, 62, 1643–1650. [Google Scholar] [CrossRef]
- Rao, V.R. Effect of carbon sources on asymbiotic nitrogen fixation in a paddy soil. Soil Biol. Biochem. 1978, 10, 319–321. [Google Scholar] [CrossRef]
- Ragot, S.A.; Kertesz, M.A.; Bünemann, E.K. phoD alkaline phosphatase gene diversity in soil. Appl. Environ. Microbiol. 2015, 81, 7281–7289. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Coronado, M.J.; Hernández, M.; Rodríguez, J.; Arias, M.E. Gas chromatography/mass spectrometry as a suitable alternative technique to evaluate the ability of Streptomyces to degrade lignin from lignocellulosic residues. Rapid Commun. Mass Spectrom. 1998, 12, 1744–1748. [Google Scholar] [CrossRef]
- Wagner, M.R.; Lundberg, D.S.; Del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.G.; Kremling, K.A.; Kovar, L.L.; Buckler, E.S. Quantitative genetics of the maize leaf microbiome. Phytobiomes J. 2018, 2, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.P.; McDonald, I.R.; Wood, A.P. The Family Methylobacteriaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 313–340. [Google Scholar]
- Tamas, I.; Smirnova, A.V.; He, Z.; Dunfield, P.F. The (d)evolution of methanotrophy in the Beijerinckiaceae—A comparative genomics analysis. ISME J. 2014, 8, 369–382. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Li, Z.; Andeer, P.F.; Spaulding, S.; Thomas, B.C.; Singh, A.; Hettich, R.L.; Suttle, K.B.; Probst, A.J.; Tringe, S.G.; et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 2016, 4, e2687. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Week −2 | Week 0 | Week 6 | Tukey’s HSD | ||
|---|---|---|---|---|---|
| Shannon–Weaver diversity index | NS | 6.95 ± 0.18 | 6.47 ± 0.10 | 6.83 ± 0.10 | a |
| IS | 4.92 ± 1.03 | 3.27 ± 0.34 | 4.72 ± 0.30 | b | |
| AS | 5.17 ± 0.77 | 3.33 ± 1.10 | 4.25 ± 0.35 | b | |
| Pielou’s evenness | NS | 0.95 ± 0.00 | 0.94 ± 0.00 | 0.95 ± 0.00 | a |
| IS | 0.74 ± 0.12 | 0.62 ± 0.12 | 0.81 ± 0.04 | b | |
| AS | 0.75 ± 0.09 | 0.60 ± 0.07 | 0.78 ± 0.03 | b | |
| Number of ASVs | NS | 1477 ± 276 | 961 ± 99 | 1361 ± 154 | a |
| IS | 802 ± 283 | 226 ± 129 | 336 ± 41 | b | |
| AS | 997 ± 168 | 346 ± 254 | 228 ± 52 | b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiba, A.; Uchida, Y.; Kublik, S.; Vestergaard, G.; Buegger, F.; Schloter, M.; Schulz, S. Soil Bacterial Diversity Is Positively Correlated with Decomposition Rates during Early Phases of Maize Litter Decomposition. Microorganisms 2021, 9, 357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020357
Chiba A, Uchida Y, Kublik S, Vestergaard G, Buegger F, Schloter M, Schulz S. Soil Bacterial Diversity Is Positively Correlated with Decomposition Rates during Early Phases of Maize Litter Decomposition. Microorganisms. 2021; 9(2):357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020357
Chicago/Turabian StyleChiba, Akane, Yoshitaka Uchida, Susanne Kublik, Gisle Vestergaard, Franz Buegger, Michael Schloter, and Stefanie Schulz. 2021. "Soil Bacterial Diversity Is Positively Correlated with Decomposition Rates during Early Phases of Maize Litter Decomposition" Microorganisms 9, no. 2: 357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020357