
YeastNet: Deep-Learning-Enabled Accurate Segmentation of Budding Yeast Cells in Bright-Field Microscopy

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Augmentation
2.3. Non-Trained Method
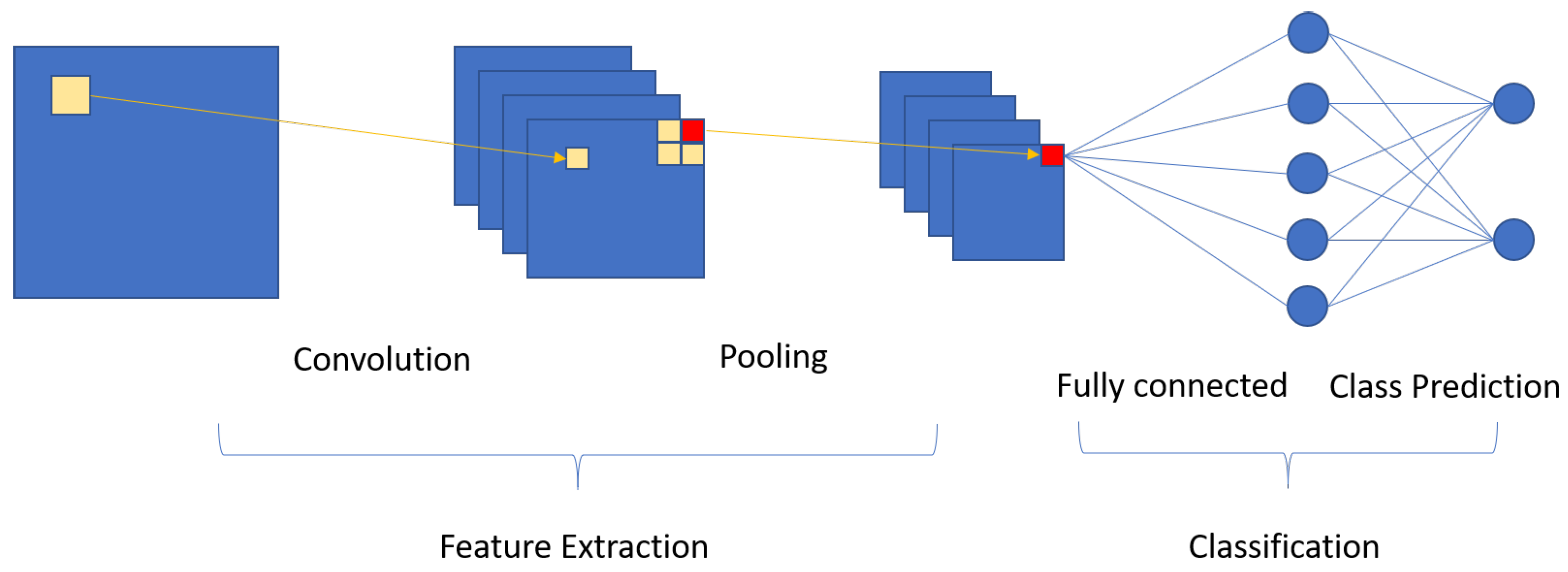
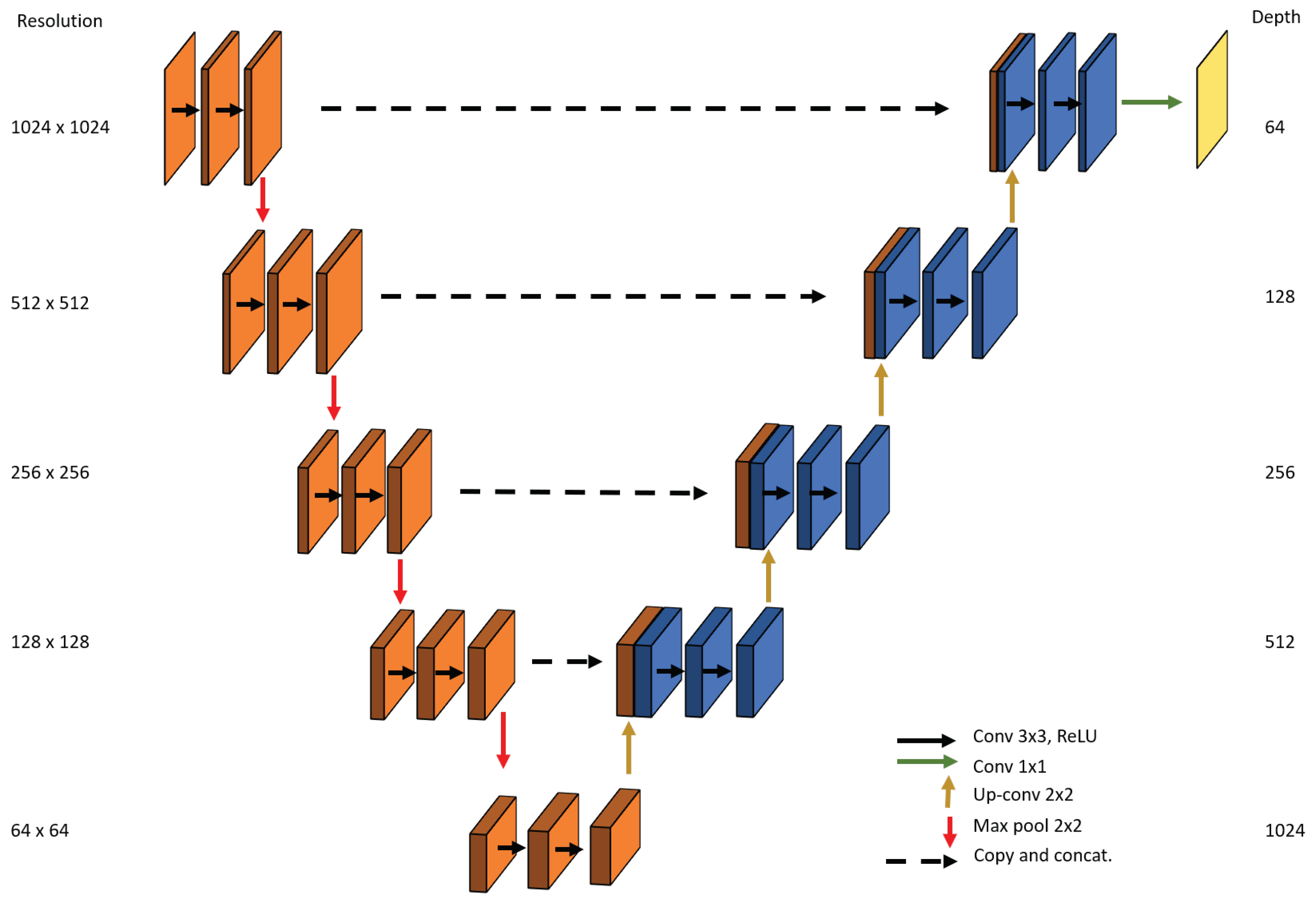
2.4. Proposed Segmentation Model
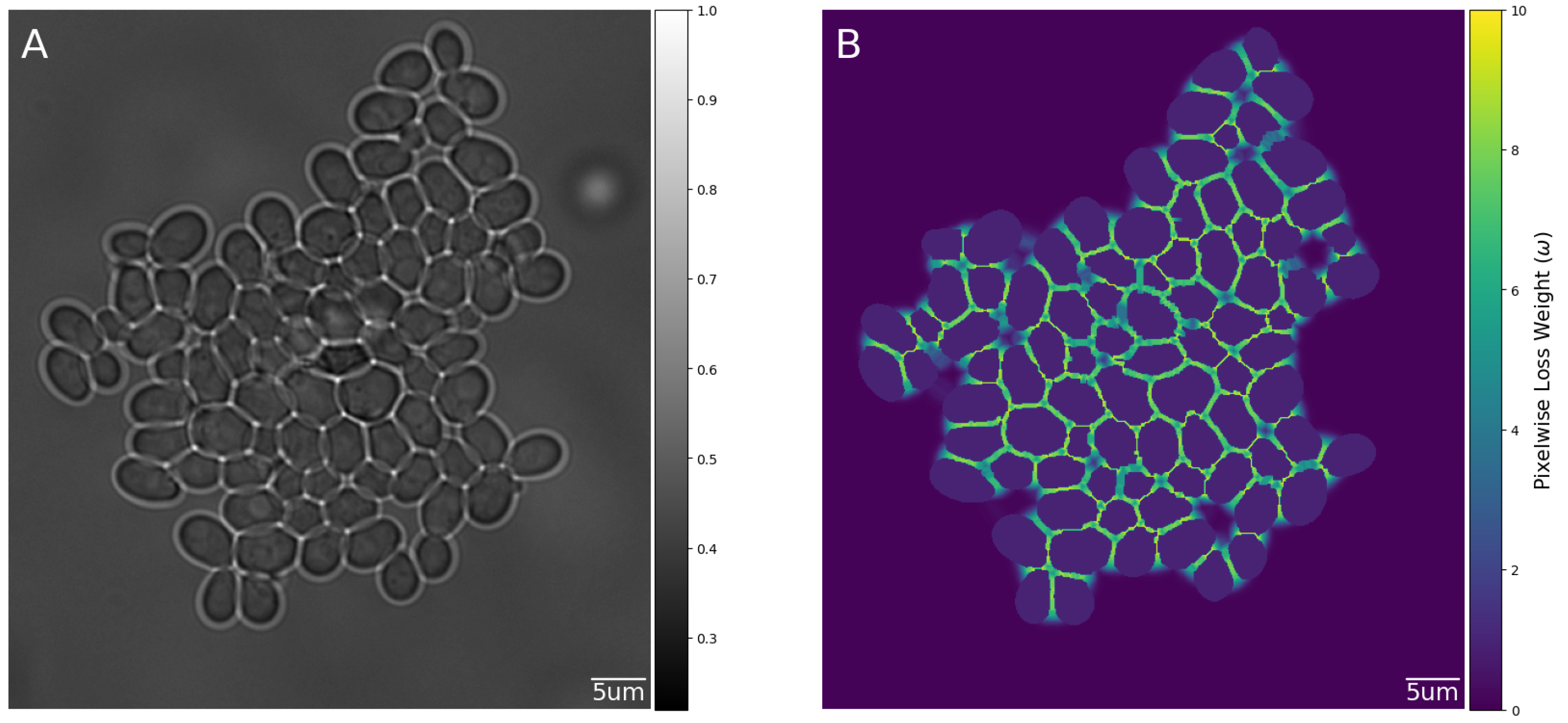
2.5. Weighted Loss Function
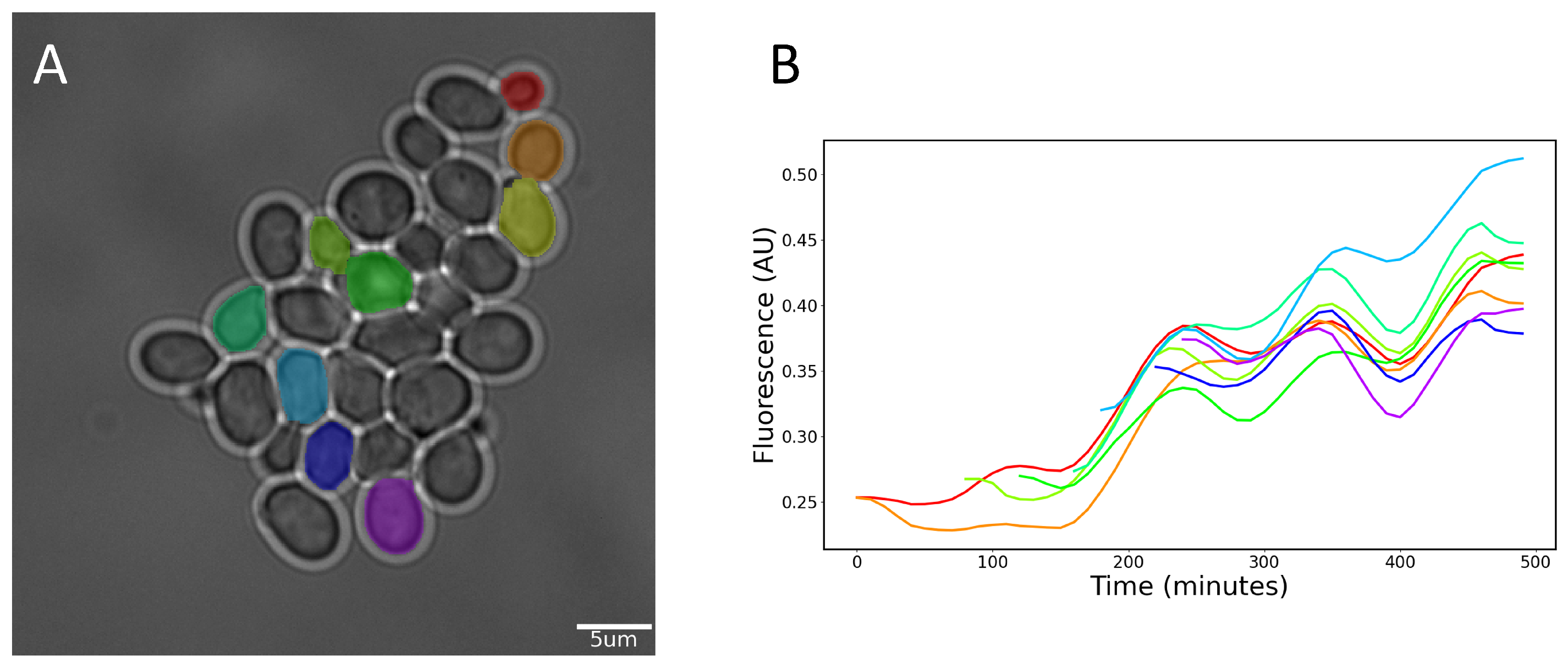
2.6. Cell Tracking
2.7. Training Details
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IoU | Intersection over Union |
| CNN | Convolutional Neural Net |
References
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [Green Version]
- Bintu, L.; Yong, J.; Antebi, Y.E.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.B.; Sternberg, C.; Poulsen, L.K.; Bjørn, S.P.; Givskov, M.; Molin, S. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 1998, 64, 2240–2246. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905. [Google Scholar] [CrossRef]
- Gordon, A.; Colman-Lerner, A.; Chin, T.E.; Benjamin, K.R.; Richard, C.Y.; Brent, R. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat. Methods 2007, 4, 175. [Google Scholar] [CrossRef] [PubMed]
- Prewitt, J.M.; Mendelsohn, M.L. The analysis of cell images. Ann. N. Y. Acad. Sci. 1966, 128, 1035–1053. [Google Scholar] [CrossRef]
- Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Jianzhuang, L.; Wenqing, L.; Yupeng, T. Automatic thresholding of gray-level pictures using two-dimension Otsu method. In Proceedings of the 1991 International Conference on Circuits and Systems, Shenzhen, China, 16–17 June 1991; pp. 325–327. [Google Scholar]
- Bradley, D.; Roth, G. Adaptive thresholding using the integral image. J. Graph. Tools 2007, 12, 13–21. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Tang, C.K.; Shum, H.Y. Lazy snapping. ACM Trans. Graph. (ToG) 2004, 23, 303–308. [Google Scholar] [CrossRef]
- Rother, C.; Kolmogorov, V.; Blake, A. “GrabCut” interactive foreground extraction using iterated graph cuts. ACM Trans. Graph. (TOG) 2004, 23, 309–314. [Google Scholar] [CrossRef]
- Protiere, A.; Sapiro, G. Interactive image segmentation via adaptive weighted distances. IEEE Trans. Image Process. 2007, 16, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, M.; Witkin, A.; Terzopoulos, D. Snakes: Active contour models. Int. J. Comput. Vis. 1988, 1, 321–331. [Google Scholar] [CrossRef]
- Caselles, V.; Kimmel, R.; Sapiro, G. Geodesic active contours. Int. J. Comput. Vis. 1997, 22, 61–79. [Google Scholar] [CrossRef]
- Chan, T.F.; Vese, L.A. Active contours without edges. IEEE Trans. Image Process. 2001, 10, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Beucher, S. Use of watersheds in contour detection. In Proceedings of the International Workshop on Image Processing, Astrophysics, Trieste, 4–8 June 1979. [Google Scholar]
- Meyer, F. Topographic distance and watershed lines. Signal Process. 1994, 38, 113–125. [Google Scholar] [CrossRef]
- Doncic, A.; Eser, U.; Atay, O.; Skotheim, J.M. An algorithm to automate yeast segmentation and tracking. PLoS ONE 2013, 8, e57970. [Google Scholar] [CrossRef]
- Wood, N.E.; Doncic, A. A fully-automated, robust, and versatile algorithm for long-term budding yeast segmentation and tracking. PLoS ONE 2019, 14, e0206395. [Google Scholar] [CrossRef] [Green Version]
- Bredies, K.; Wolinski, H. An active-contour based algorithm for the automated segmentation of dense yeast populations on transmission microscopy images. Comput. Vis. Sci. 2011, 14, 341–352. [Google Scholar] [CrossRef]
- Versari, C.; Stoma, S.; Batmanov, K.; Llamosi, A.; Mroz, F.; Kaczmarek, A.; Deyell, M.; Lhoussaine, C.; Hersen, P.; Batt, G. Long-term tracking of budding yeast cells in brightfield microscopy: CellStar and the Evaluation Platform. J. R. Soc. Interface 2017, 14, 20160705. [Google Scholar] [CrossRef]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, CA, USA, 3–8 December 2012; pp. 1097–1105. [Google Scholar]
- Long, J.; Shelhamer, E.; Darrell, T. Fully convolutional networks for semantic segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, IL, USA, 8–10 June 2015; pp. 3431–3440. [Google Scholar]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional networks for biomedical image segmentation. In Proceedings of the International Conference on Medical Image Computing and Computer Assisted Intervention, Munich, Germany, 5–9 October 2015; pp. 234–241. [Google Scholar]
- Van Valen, D.; Kudo, T.; Lane, K.M.; Macklin, D.N.; Quach, N.T.; DeFelice, M.M.; Maayan, I.; Tanouchi, Y.; Ashley, E.A.; Covert, M.W. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput. Biol. 2016, 12, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Aydin, A.S.; Dubey, A.; Dovrat, D.; Aharoni, A.; Shilkrot, R. CNN based yeast cell segmentation in multi-modal fluorescent microscopy data. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Honolulu, HI, USA, 21–26 July 2017; pp. 753–759. [Google Scholar]
- Badrinarayanan, V.; Kendall, A.; Cipolla, R. Segnet: A deep convolutional encoder-decoder architecture for image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2017, 39, 2481–2495. [Google Scholar] [CrossRef]
- Lu, A.X.; Zarin, T.; Hsu, I.S.; Moses, A.M. YeastSpotter: Accurate and parameter-free web segmentation for microscopy images of yeast cells. Bioinformatics 2019, 35, 4525–4527. [Google Scholar] [CrossRef] [Green Version]
- Ljosa, V.; Caie, P.D.; Ter Horst, R.; Sokolnicki, K.L.; Jenkins, E.L.; Daya, S.; Roberts, M.E.; Jones, T.R.; Singh, S.; Genovesio, A.; et al. Comparison of methods for image-based profiling of cellular morphological responses to small-molecule treatment. J. Biomol. Screen. 2013, 18, 1321–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugagne, J.B.; Lin, H.; Dunlop, M.J. DeLTA: Automated cell segmentation, tracking, and lineage reconstruction using deep learning. PLoS Comput. Biol. 2020, 16, e1007673. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, X.; Xu, M.; Li, Q. RBC semantic segmentation for sickle cell disease based on deformable U-Net. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Granada, Spain, 16–20 September 2018; pp. 695–702. [Google Scholar]
- Dai, J.; Qi, H.; Xiong, Y.; Li, Y.; Zhang, G.; Hu, H.; Wei, Y. Deformable convolutional networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 764–773. [Google Scholar]
- Dietler, N.; Minder, M.; Gligorovski, V.; Economou, A.M.; Joly, D.A.H.L.; Sadeghi, A.; Chan, C.H.M.; Koziński, M.; Weigert, M.; Bitbol, A.F.; et al. A convolutional neural network segments yeast microscopy images with high accuracy. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prangemeier, T.; Wildner, C.; Françani, A.O.; Reich, C.; Koeppl, H. Multiclass yeast segmentation in microstructured environments with deep learning. In Proceedings of the 2020 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Via del Mar, Chile, 27–29 October 2020; pp. 1–8. [Google Scholar]
- Kong, Y.; Li, H.; Ren, Y.; Genchev, G.Z.; Wang, X.; Zhao, H.; Xie, Z.; Lu, H. Automated yeast cells segmentation and counting using a parallel U-Net based two-stage framework. OSA Contin. 2020, 3, 982–992. [Google Scholar] [CrossRef]
- Haralick, R.; Shapiro, L. Computer and Robot Vision; Number v. 1 in Computer and Robot Vision; Addison-Wesley Publishing Company: Reading, MA, USA, 1992. [Google Scholar]
- Uhlendorf, J.; Miermont, A.; Delaveau, T.; Charvin, G.; Fages, F.; Bottani, S.; Batt, G.; Hersen, P. Long-term model predictive control of gene expression at the population and single-cell levels. Proc. Natl. Acad. Sci. USA 2012, 109, 14271–14276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced methods of microscope control using μManager software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676. [Google Scholar] [CrossRef] [Green Version]
- Ricicova, M.; Hamidi, M.; Quiring, A.; Niemistö, A.; Emberly, E.; Hansen, C.L. Dissecting genealogy and cell cycle as sources of cell-to-cell variability in MAPK signaling using high-throughput lineage tracking. Proc. Natl. Acad. Sci. USA 2013, 110, 11403–11408. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H. The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 1955, 2, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Munkres, J. Algorithms for the assignment and transportation problems. J. Soc. Ind. Appl. Math. 1957, 5, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Kachouie, N.; Fieguth, P. Extended-Hungarian-JPDA: Exact Single-Frame Stem Cell Tracking. IEEE Trans. Biomed. Eng. 2007, 54, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In Proceedings of the IEEE International Conference on Computer Vision, Las Condes, Chile, 11–18 December 2015; pp. 1026–1034. [Google Scholar]
- Kreft, M.; Stenovec, M.; Zorec, R. Focus-drift correction in time-lapse confocal imaging. Ann. N. Y. Acad. Sci. 2005, 1048, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S.; et al. An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale. In Proceedings of the International Conference on Learning Representations, Vienna, Austria, 4 May 2021. [Google Scholar]
- Bertels, J.; Eelbode, T.; Berman, M.; Vandermeulen, D.; Maes, F.; Bisschops, R.; Blaschko, M.B. Optimizing the dice score and jaccard index for medical image segmentation: Theory and practice. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Shenzen, China, 13–17 October 2019; pp. 92–100. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Our Dataset | YIT Dataset 1 | YIT Dataset 3 |
|---|---|---|---|
| Non-trainable | 0.562 (0.252) | 0.585 (0.069) | 0.553 (0.060) |
| CellStar | 0.680 (0.184) | 0.701 (0.070) | 0.751 (0.041) |
| YeastSpotter | 0.561 (0.109) | 0.609 (0.072) | 0.677 (0.043) |
| YeastNet | 0.873 (0.020) | 0.681 (0.029) | 0.732 (0.012) |
| YeastNet2 | 0.883 (0.014) | 0.820 (0.020) | 0.855 (0.012) |
| Cell IoU | Tracking Accuracy | |||
|---|---|---|---|---|
| CellStar | YeastNet3 | CellStar | YeastNet3 | |
| Focus 1 | 0.469 (0.144) | 0.888 (0.023) | 0.606 | 0.939 |
| Focus 2 | 0.771 (0.035) | 0.903 (0.029) | 0.858 | 0.922 |
| Focus 3 | 0.806 (0.100) | 0.917 (0.029) | 0.891 | 0.937 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, D.; Li, Y.; Xi, P.; Phenix, H.; Cuperlovic-Culf, M.; Kærn, M. YeastNet: Deep-Learning-Enabled Accurate Segmentation of Budding Yeast Cells in Bright-Field Microscopy. Appl. Sci. 2021, 11, 2692. https://0-doi-org.brum.beds.ac.uk/10.3390/app11062692
Salem D, Li Y, Xi P, Phenix H, Cuperlovic-Culf M, Kærn M. YeastNet: Deep-Learning-Enabled Accurate Segmentation of Budding Yeast Cells in Bright-Field Microscopy. Applied Sciences. 2021; 11(6):2692. https://0-doi-org.brum.beds.ac.uk/10.3390/app11062692
Chicago/Turabian StyleSalem, Danny, Yifeng Li, Pengcheng Xi, Hilary Phenix, Miroslava Cuperlovic-Culf, and Mads Kærn. 2021. "YeastNet: Deep-Learning-Enabled Accurate Segmentation of Budding Yeast Cells in Bright-Field Microscopy" Applied Sciences 11, no. 6: 2692. https://0-doi-org.brum.beds.ac.uk/10.3390/app11062692