Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
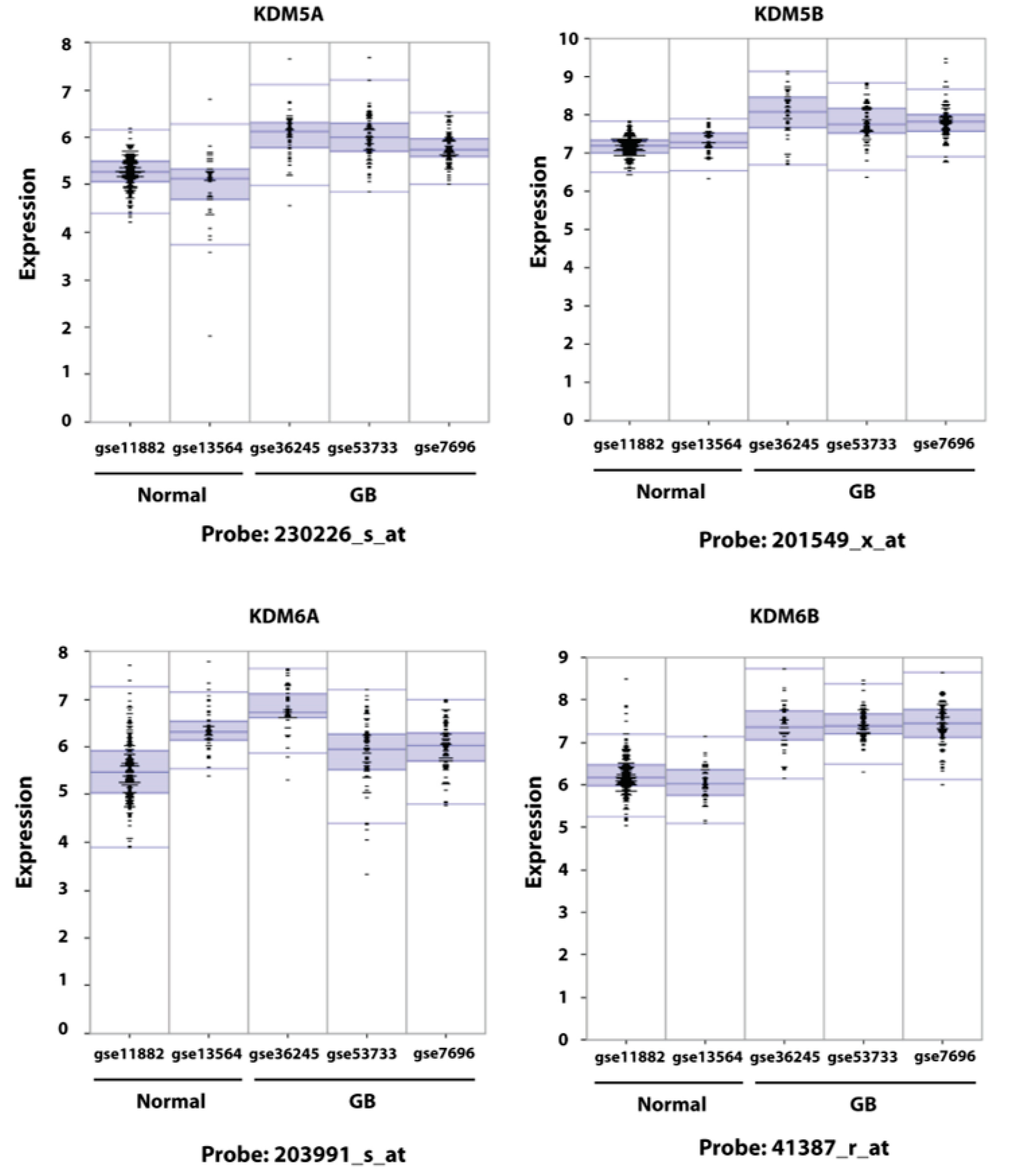
2.1. Expression of KDM6B in GB Cells
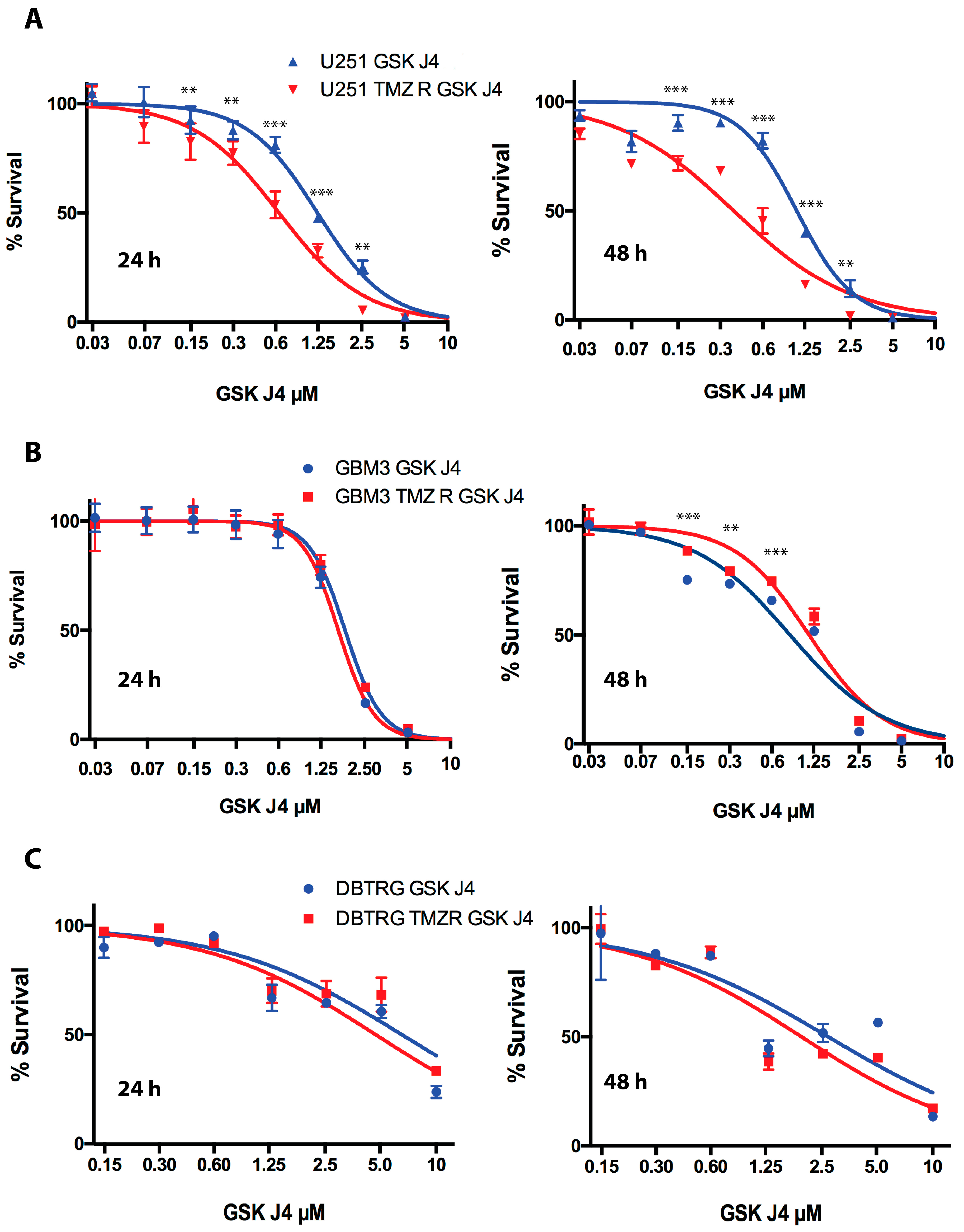
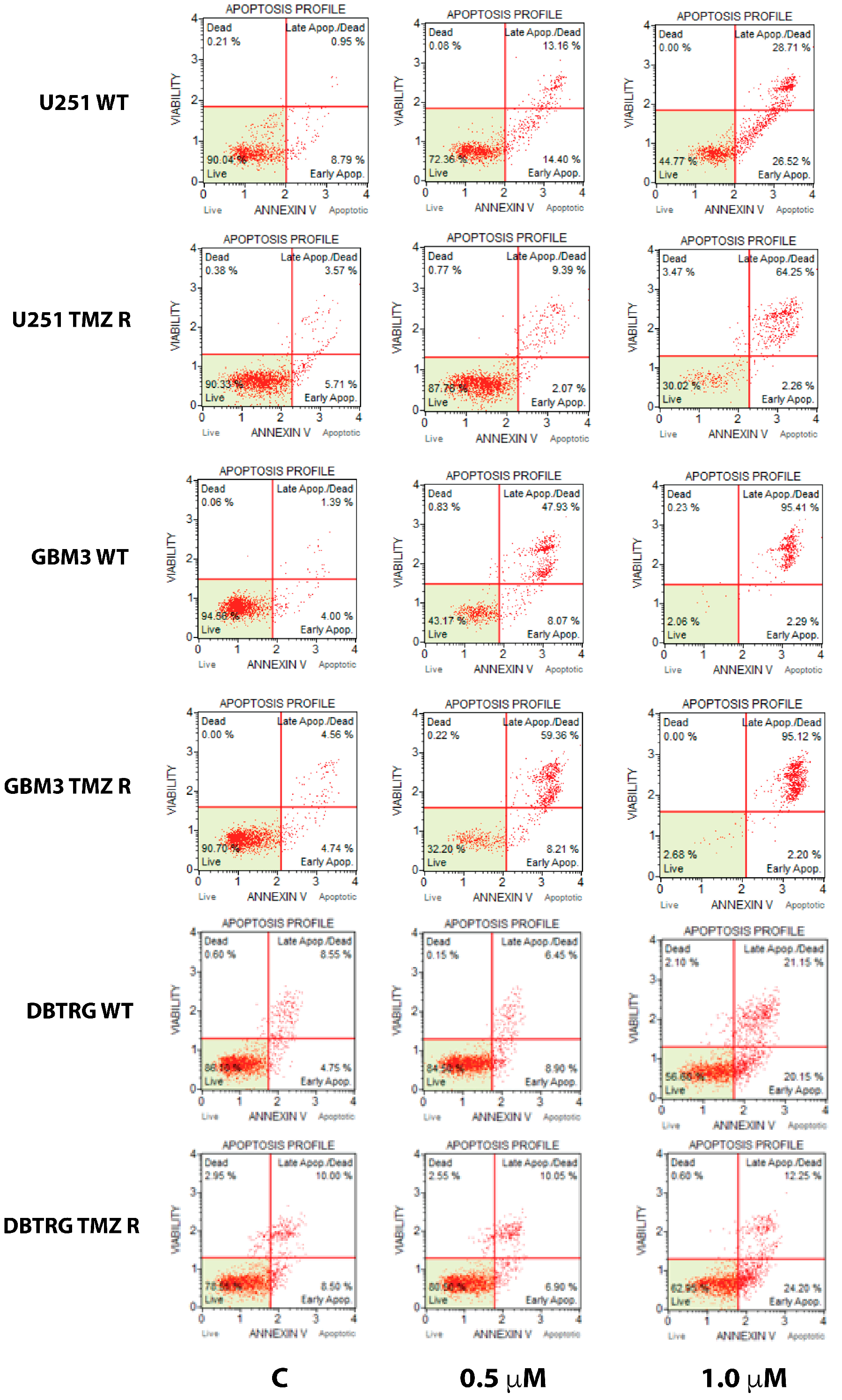
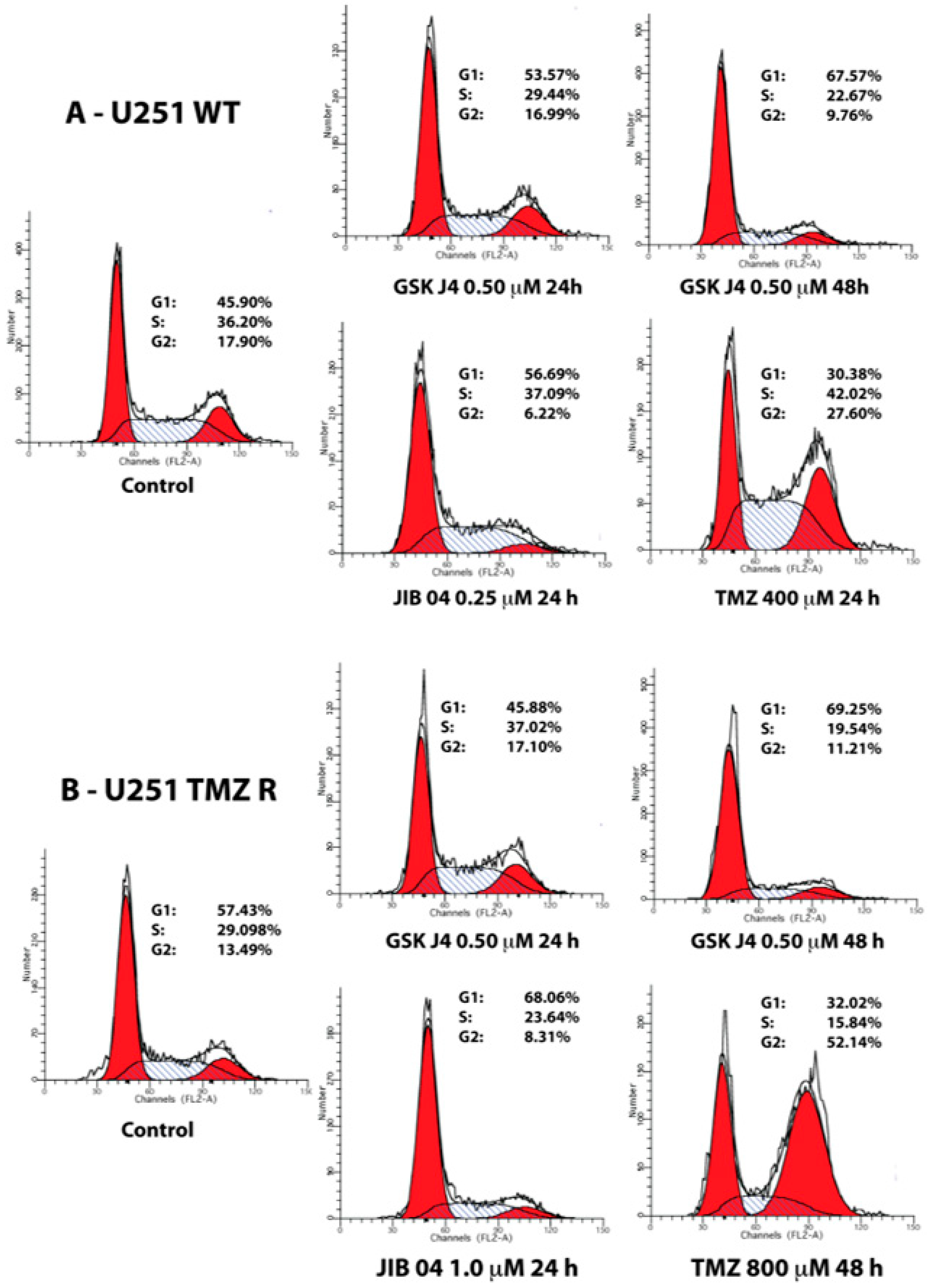
2.2. GSK J4 Inhibits Proliferation, Activates Apoptosis, and Blocks Cell Cycle Progression in Native and TMZ-Resistant Glioblastoma Cells
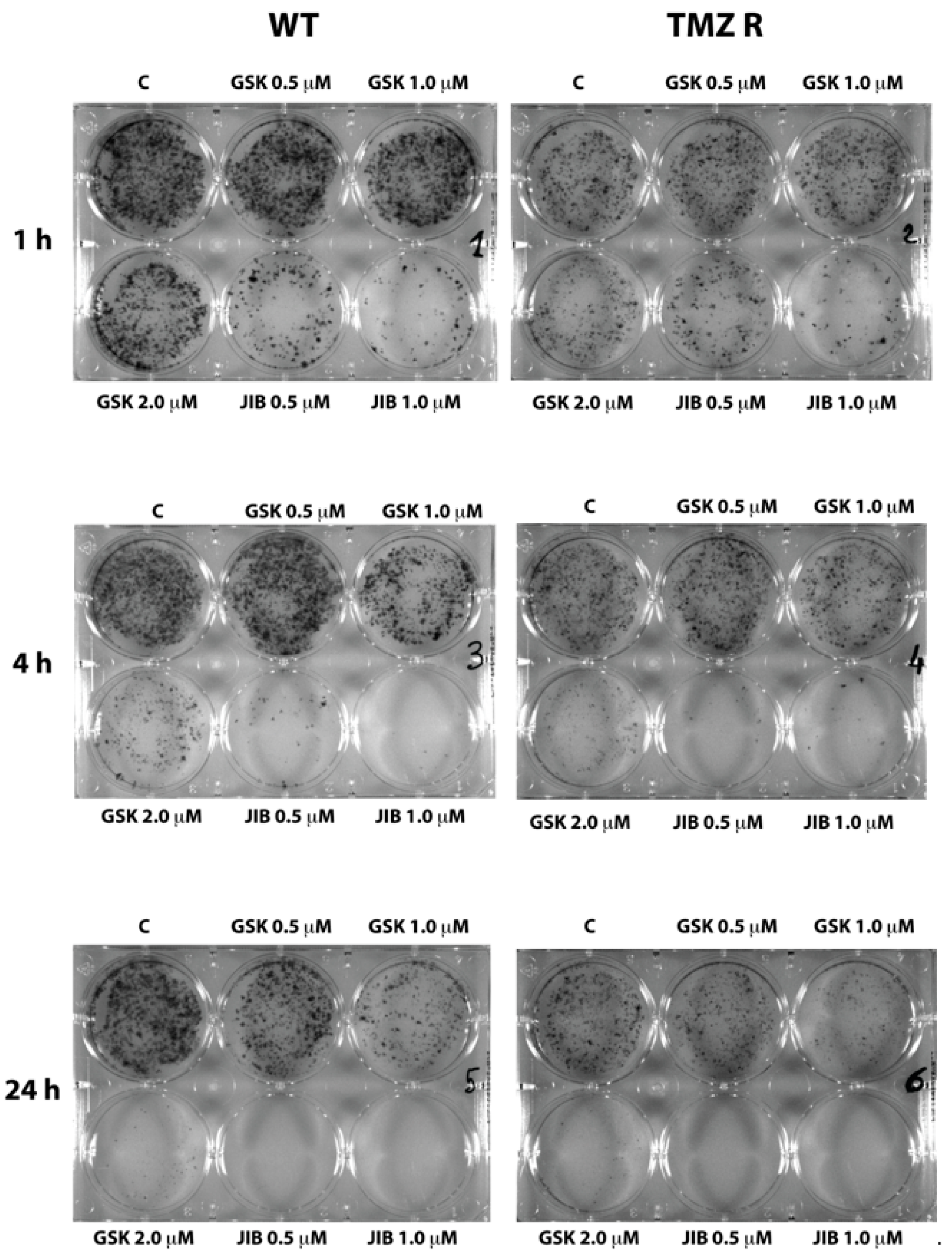
2.3. GSK J4 Reduces Clonogenicity in Native and TMZ-Resistant Glioblastoma Cells
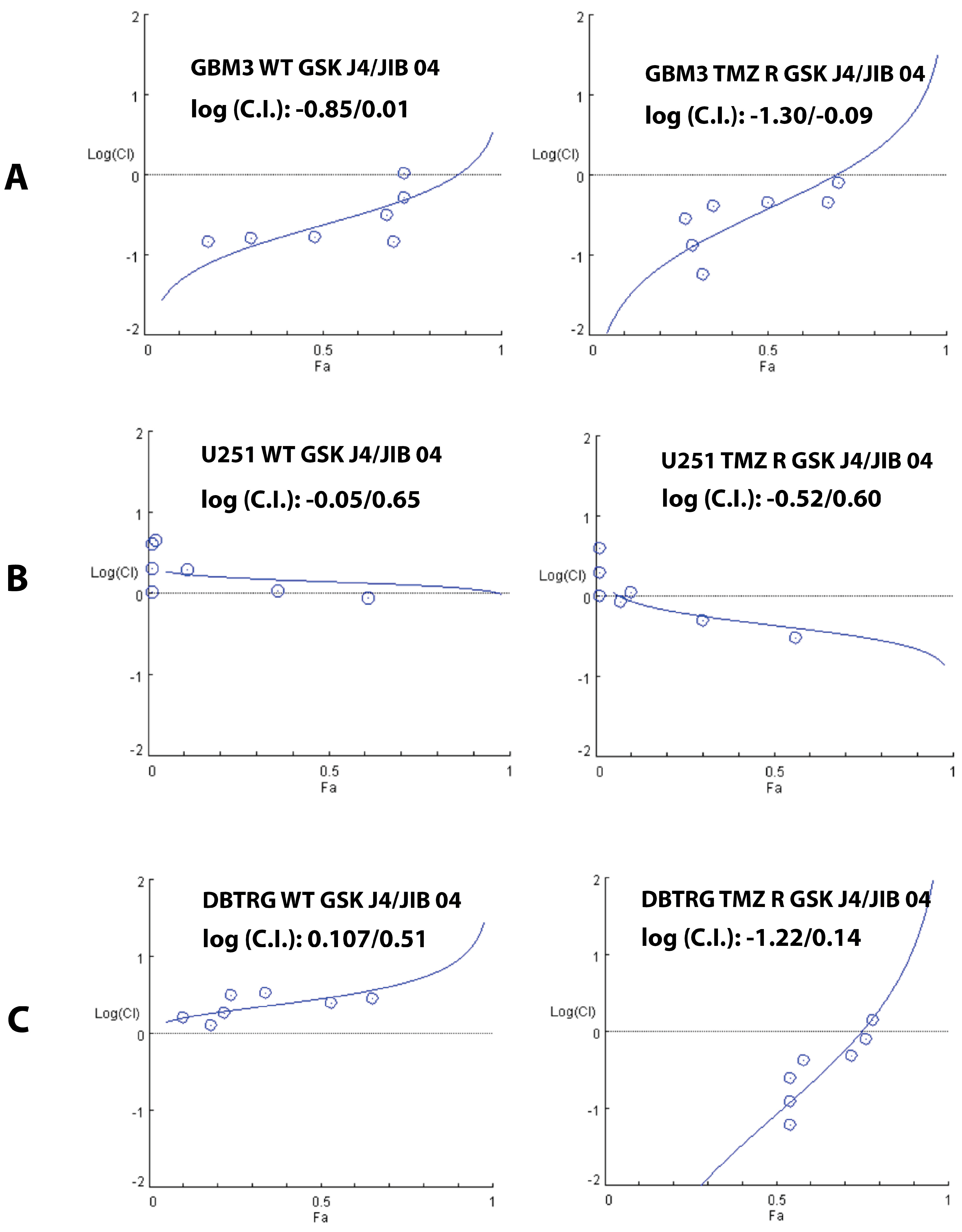
2.4. Cooperation between TMZ, GSK J4, and JIB 04 in Killing Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. In Silico and Statistical Analysis
4.2. Cell Lines, Cell Cultures, and Drugs
4.3. Cellular Analyses
4.4. Cell Cycle Analysis
4.5. Quantitative Real-Time Reverse Transcription-PCR
4.6. Drug Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Cloos, P.A.C.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liu, Y.; Lau, J.L.; Min, J. Epigenetic targets and drug discovery Part 2: Histone demethylation and DNA methylation. Pharmacol. Ther. 2015, 151, 121–140. [Google Scholar] [CrossRef]
- Sadri-Vakili, G.; Cha, J.-H.J. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Wen, K.; Miliç, J.; El-Khodor, B.; Dhana, K.; Nano, J.; Pulido, T.; Kraja, B.; Zaciragic, A.; Bramer, W.M.; Troup, J.; et al. The Role of DNA Methylation and Histone Modifications in Neurodegenerative Diseases: A Systematic Review. PLoS ONE 2016, 11, e0167201. [Google Scholar] [CrossRef]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef]
- Araki, Y.; Mimura, T. The Histone Modification Code in the Pathogenesis of Autoimmune Diseases. Mediat. Inflamm. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.P.; Cao, J.; Zou, M.R.; Sayegh, J.; Yan, Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers 2011, 3, 1383–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotili, D.; Mai, A. Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, J.-H.; Berns, A. Drugging Drug Resistance. Cell 2010, 141, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Chen, X.; Zhang, Q.; Qin, J.; Li, H.; Liu, C.; Calhoun-Davis, T.; Coletta, L.D.; Klostergaard, J.; Fokt, I.; et al. Drug-Tolerant Cancer Cells Show Reduced Tumor-Initiating Capacity: Depletion of CD44+ Cells and Evidence for Epigenetic Mechanisms. PLoS ONE 2011, 6, e24397. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.; Simjee, S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients With High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.P.; Greenslade, M.; Powell, B.; Spiteri, I.; Sottoriva, A.; Kurian, K.M. Current Challenges in Glioblastoma: Intratumour Heterogeneity, Residual Disease, and Models to Predict Disease Recurrence. Front. Oncol. 2015, 5, 251. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ding, K.; Wang, J.; Li, X.; Zhao, P. Chemoresistance caused by the microenvironment of glioblastoma and the corresponding solutions. Biomed. Pharmacother. 2019, 109, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Pollard, S.M.; Beck, S. The good, the bad and the ugly: Epigenetic mechanisms in glioblastoma. Mol. Asp. Med. 2013, 34, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Gusyatiner, O.; Hegi, M.E. Glioma epigenetics: From subclassification to novel treatment options. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of Histone Deacetylation Potentiates the Evolution of Acquired Temozolomide Resistance Linked to MGMT Upregulation in Glioblastoma Xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [Green Version]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Pre-clinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; Carman, C.; Serafim, R.B.; Myronakis, M.; Valente, V.; Price, B.D. Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget 2017, 8, 24518. [Google Scholar] [CrossRef]
- Oberstadt, M.C.; Bien-Möller, S.; Weitmann, K.; Herzog, S.; Hentschel, K.; Rimmbach, C.; Vogelgesang, S.; Balz, E.; Fink, M.; Michael, H.; et al. Epigenetic modulation of the drug resistance genes MGMT, ABCB1 and ABCG2 in glioblastoma multiforme. BMC Cancer 2013, 13, 617. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareddy, G.R.; Nair, B.C.; Krishnan, S.K.; Gonugunta, V.K.; Zhang, Q.; Miyata, N.; Suzuki, T.; Brenner, A.; Brann, D.W.; Vadlamudi, R.K. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013, 4, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.-W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro-Oncology 2011, 13, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banelli, B.; Carra, E.; Barbieri, F.; Würth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D.; et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [Google Scholar] [CrossRef] [Green Version]
- Banelli, B.; Daga, A.; Forlani, A.; Allemanni, G.; Marubbi, D.; Pistillo, M.P.; Profumo, A.; Romani, M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget 2017, 8, 34896–34910. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef]
- Leadem, B.R.; Kagiampakis, I.; Wilson, C.; Cheung, T.K.; Arnott, D.; Trojer, P.; Classon, M.; Easwaran, H.; Baylin, S.B. A KDM5 Inhibitor Increases Global H3K4 Trimethylation Occupancy and Enhances the Biological Efficacy of 5-Aza-2′-Deoxycytidine. Cancer Res. 2018, 78, 1127–1139. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Harmeyer, K.M.; Facompre, N.D.; Herlyn, M.; Basu, D. JARID1 Histone Demethylases: Emerging Targets in Cancer. Trends Cancer 2017, 3, 713–725. [Google Scholar] [CrossRef]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.; Baker, S.J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat. Rev. Cancer 2014, 14, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol. 2006, 7, 241–248. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Taylor, M.D. An epigenetic therapy for diffuse intrinsic pontine gliomas. Nat. Med. 2014, 20, 1378–1379. [Google Scholar] [CrossRef] [PubMed]
- Sui, A.; Xu, Y.; Li, Y.; Hu, Q.; Wang, Z.; Zhang, H.; Yang, J.; Guo, X.; Zhao, W. The pharmacological role of histone demethylase JMJD3 inhibitor GSK-J4 on glioma cells. Oncotarget 2017, 8, 68591–68598. [Google Scholar] [CrossRef] [Green Version]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.-F.; de Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.M.; et al. Stem Cell–Related “Self-Renewal” Signature and High Epidermal Growth Factor Receptor Expression Associated With Resistance to Concomitant Chemoradiotherapy in Glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H.; Gietzelt, J.; Hentschel, B.; Westphal, M.; Simon, M.; et al. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling: Molecular profiles of glioblastoma long-term survivors. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, M.; Sheng, M.; Zhang, P.; Chen, Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.-C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2018, 144, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R.; Sehgal, L.; Havranek, O.; Köhrer, S.; Khashab, T.; Jain, N.; Burger, J.A.; Neelapu, S.S.; Davis, R.E.; Samaniego, F. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 2017, 102, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Pistillo, M.P.; Carosio, R.; Morabito, A.; Banelli, B. Immune Checkpoints and Innovative Therapies in Glioblastoma. Front. Oncol. 2018, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, R.M.R.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 Silencing in Glioblastoma Tumor-Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Griffero, F.; Daga, A.; Marubbi, D.; Capra, M.C.; Melotti, A.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Porcile, C.; Barbieri, F.; et al. Different Response of Human Glioma Tumor-initiating Cells to Epidermal Growth Factor Receptor Kinase Inhibitors. J. Biol. Chem. 2009, 284, 7138–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, S.; Chen, X.; Wang, C.-Y. Histone Demethylase KDM6B Promotes Epithelial-Mesenchymal Transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romani, M.; Daga, A.; Forlani, A.; Pistillo, M.P.; Banelli, B. Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells. Cancers 2019, 11, 878. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060878
Romani M, Daga A, Forlani A, Pistillo MP, Banelli B. Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells. Cancers. 2019; 11(6):878. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060878
Chicago/Turabian StyleRomani, Massimo, Antonio Daga, Alessandra Forlani, Maria Pia Pistillo, and Barbara Banelli. 2019. "Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells" Cancers 11, no. 6: 878. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060878