Multimodal Targeted Deep Sequencing of Circulating Tumor Cells and Matched Cell-Free DNA Provides a More Comprehensive Tool to Identify Therapeutic Targets in Metastatic Breast Cancer Patients

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Establishment of an Isolation Protocol for CTC gDNA
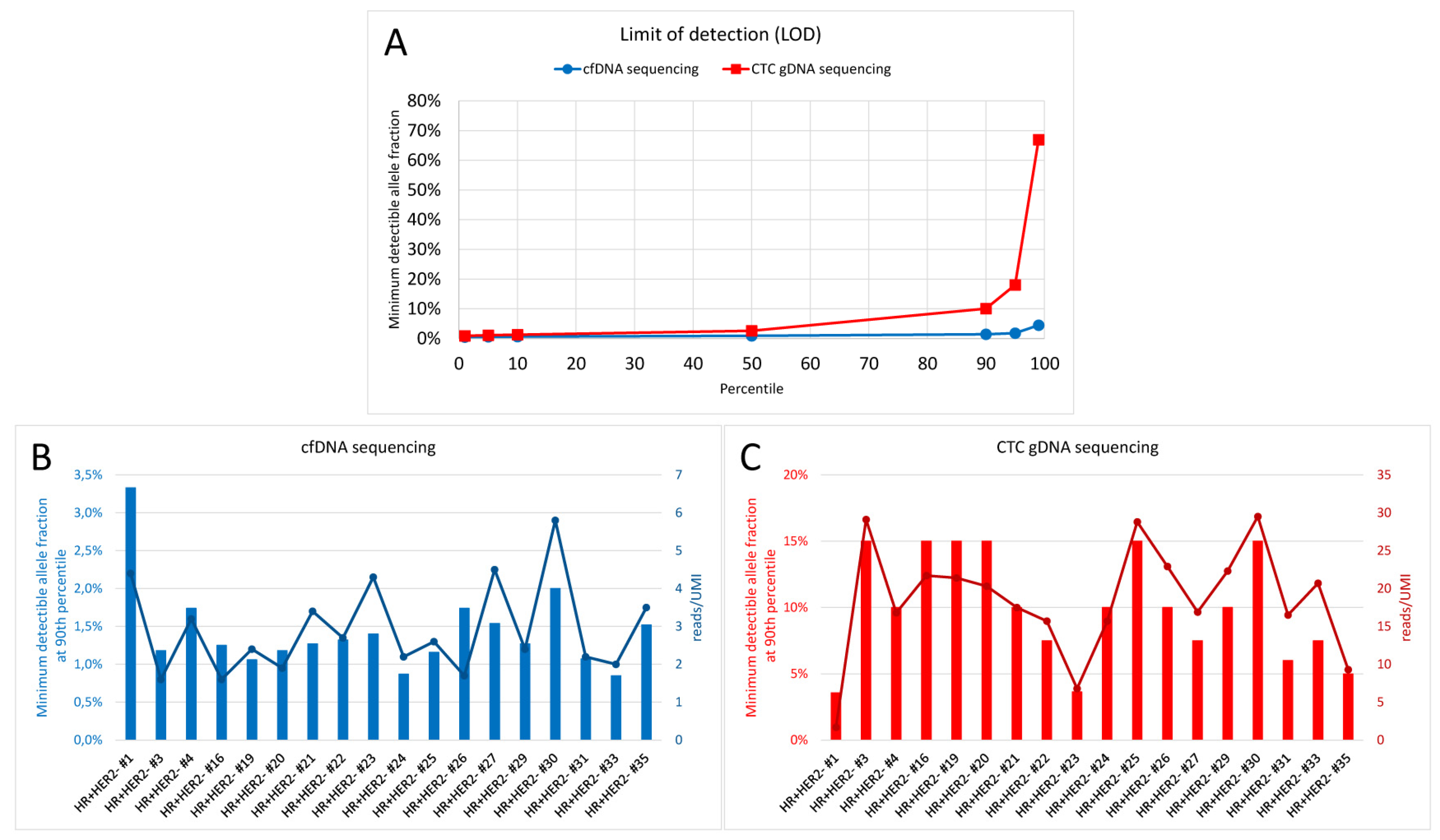
2.2. Sequencing Qualities
2.3. Cohort
2.4. Patient-Specific Variants
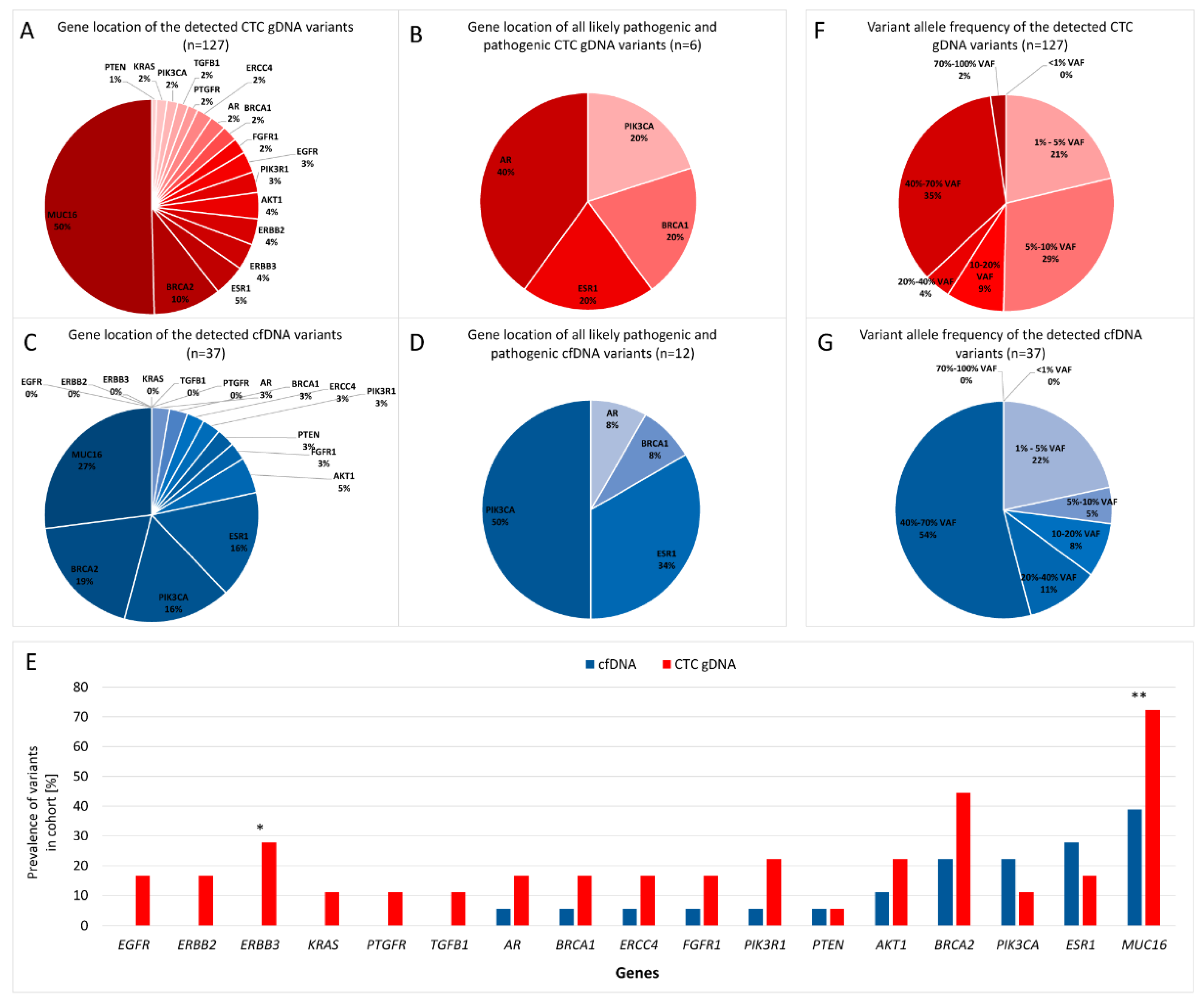
2.5. Localization of the Variants
2.6. Prevalence of Variants
2.7. Variant Allele Frequency of the Variants
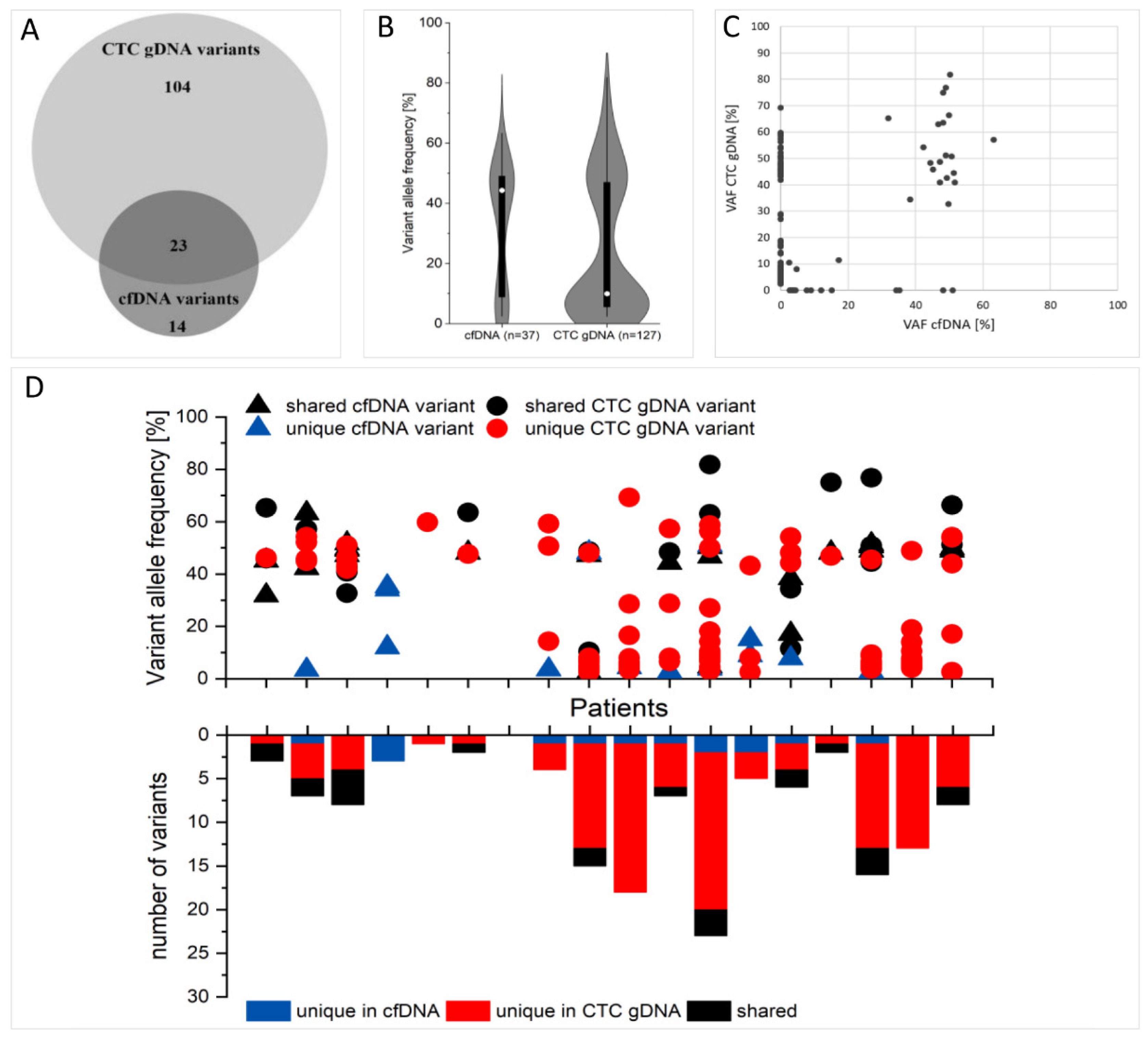
2.8. Concordance of cfDNA and CTC gDNA Variants
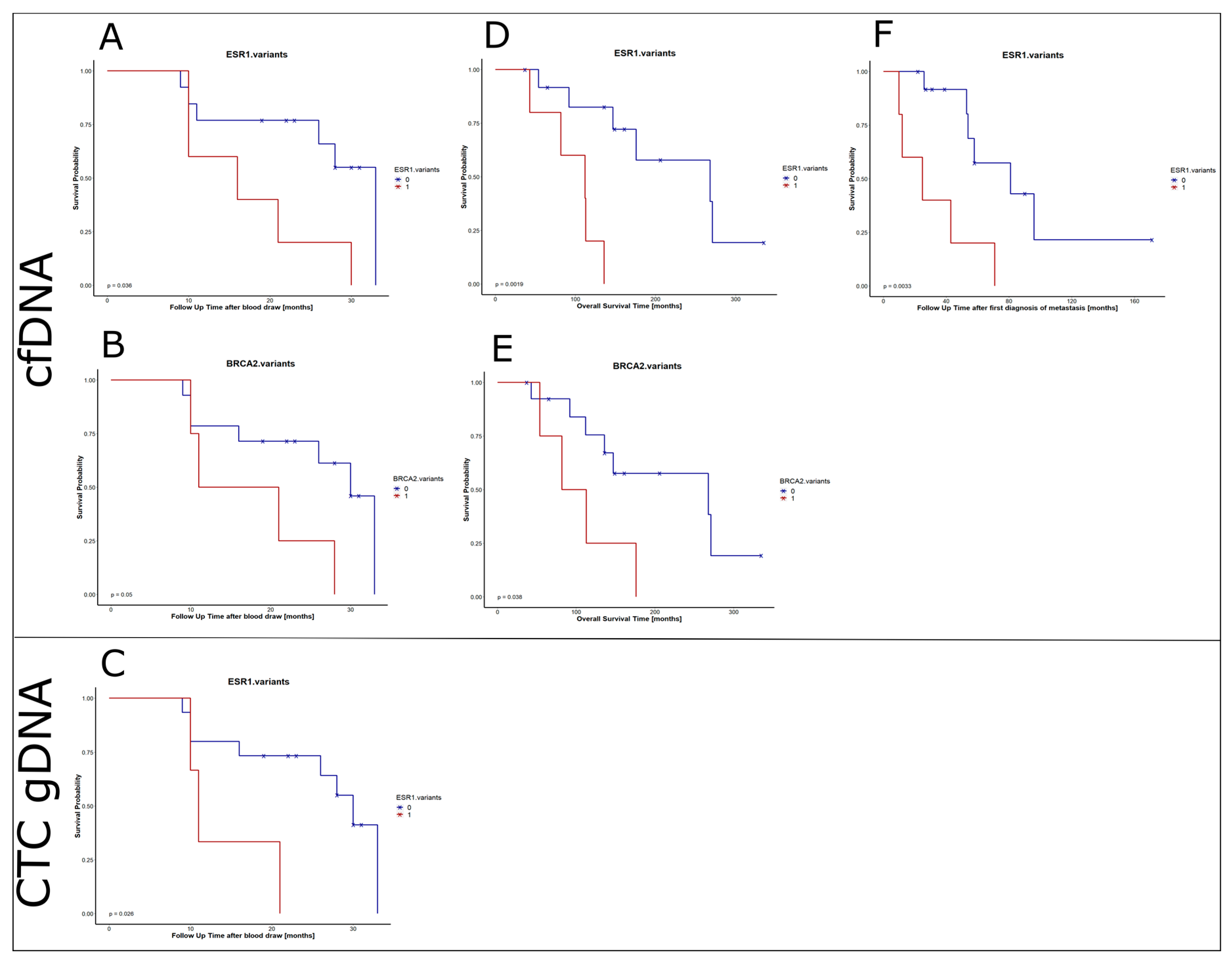
2.9. Prognostic Value
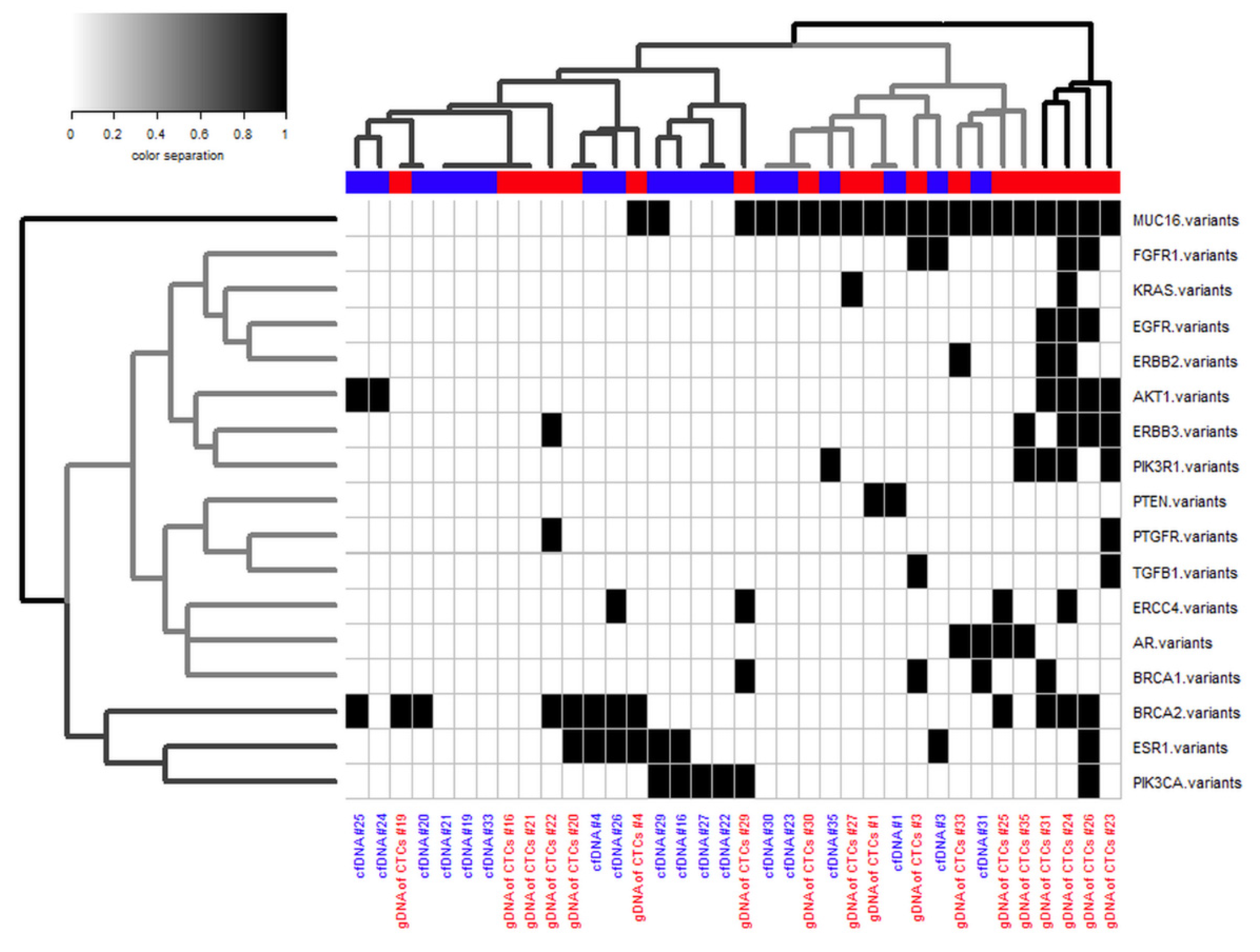
2.10. Cluster Analysis
3. Discussion
3.1. CTC gDNA Isolation and Library Preparation Was Successfully Established
3.2. Sequencing Quality Parameteres Are Essential for Data Evalution
3.3. Characteristics of cfDNA and CTC gDNA Variants in Line with Previous Results
3.4. Variant Analysis in cfDNA and CTCs Is Complementary
4. Materials and Methods
4.1. Patient Population Characteristics and Eligibility Criteria
4.2. Sampling of Blood, Processing of Plasma and Enrichment of Circulating Tumor Cells
4.3. Isolation and Quantification of cfDNA
4.4. Isolation of gDNA from CTCs
4.5. Library Construction
4.6. Sequencing
4.7. Data Analysis/Bioinformatical Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| cfDNA | Cell-free DNA |
| ctDNA | Cell-free tumor DNA |
| CTCs | Circulating tumor cells |
| CTC gDNA | Genomic DNA of CTCs |
| ECOG | Eastern cooperative oncology group |
| ER | Estrogen receptor |
| HR+ | Hormone receptor-positive |
| HER2- | Human epidermal growth factor receptor 2-negative |
| IVA | Ingenuity variant analysis |
| MBC | Metastatic breast cancer |
| NGS | Next-generation sequencing |
| OS | Overall survival |
| UMI | Unique molecular indices |
| PR | Progesterone receptor |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| VAF | Variant allele frequency |
| Vcf | Variant call format |
| WGA | Whole-genome amplification |
List of Genes
| AKT1 | AKT serine/threonine kinase 1 |
| AR | Androgen receptor |
| BRCA1 | BRCA1 DNA repair associated |
| BRCA2 | BRCA2 DNA repair associated |
| EGFR | Epidermal growth factor receptor |
| EpCAM | Epithelial cell adhesion molecule |
| ERCC4 | ERCC excision repair 4, endonuclease noncatalytic subunit |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 coding for the HER2 protein |
| ERBB3 | Erb-b2 receptor tyrosine kinase 3 |
| ESR1 | Estrogen receptor 1 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| KRAS | KRAS proto-oncogene, gtpase |
| MUC16 | Mucin 16, cell-surface associated |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PIK3R1 | Phosphoinositide-3-kinase regulatory subunit 1 |
| PTEN | Phosphatase and tensin homolog |
| PTGFR | Prostaglandin F receptor |
| SOX17 | SRY-box transcription factor 17 |
| TGFB1 | Transforming growth factor beta 1 |
References
- Krawczyk, N.; Fehm, T.; Banys-Paluchowski, M.; Janni, W.; Schramm, A. Liquid Biopsy in Metastasized Breast Cancer as Basis for Treatment Decisions. Oncol. Res. Treat. 2016, 39, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Omoto, Y.; Iwase, H. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget 2017, 8, 52142–52155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lianidou, E.; Pantel, K. Liquid Biopsies. Genes Chromosomes Cancer 2018. [Google Scholar] [CrossRef]
- Appierto, V.; Di Cosimo, S.; Reduzzi, C.; Pala, V.; Cappelletti, V.; Daidone, M.G. How to study and overcome tumor heterogeneity with circulating biomarkers: The breast cancer case. Semin. Cancer Biol. 2017, 44, 106–116. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.-C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Kidess-Sigal, E.; Liu, H.E.; Triboulet, M.M.; Che, J.; Ramani, V.C.; Visser, B.C.; Poultsides, G.A.; Longacre, T.A.; Marziali, A.; Vysotskaia, V.; et al. Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label-free platform: Comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget 2016, 7, 85349–85364. [Google Scholar] [CrossRef] [Green Version]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.-J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.E.; Cristofanilli, M.; Fortina, P. Detection of Activating Estrogen Receptor Gene (ESR1) Mutations in Single Circulating Tumor Cells. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts. Clin. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Keup, C.; Storbeck, M.; Hauch, S.; Hahn, P.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Hoffmann, O.; Kimmig, R.; Kasimir-Bauer, S. Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer. Cancers 2019, 11, 238. [Google Scholar] [CrossRef] [Green Version]
- Heidary, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16, 421. [Google Scholar] [CrossRef] [Green Version]
- Yanagita, M.; Redig, A.J.; Paweletz, C.P.; Dahlberg, S.E.; O’Connell, A.; Feeney, N.; Taibi, M.; Boucher, D.; Oxnard, G.R.; Johnson, B.E.; et al. A Prospective Evaluation of Circulating Tumor Cells and Cell-Free DNA in EGFR-Mutant Non-Small Cell Lung Cancer Patients Treated with Erlotinib on a Phase II Trial. Clin. Cancer Res. 2016, 22, 6010–6020. [Google Scholar] [CrossRef] [Green Version]
- Hodara, E.; Morrison, G.; Cunha, A.; Zainfeld, D.; Xu, T.; Xu, Y.; Dempsey, P.W.; Pagano, P.C.; Bischoff, F.; Khurana, A.; et al. Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Mastoraki, S.; Strati, A.; Tzanikou, E.; Chimonidou, M.; Politaki, E.; Voutsina, A.; Psyrri, A.; Georgoulias, V.; Lianidou, E. ESR1 Methylation: A Liquid Biopsy-Based Epigenetic Assay for the Follow-up of Patients with Metastatic Breast Cancer Receiving Endocrine Treatment. Clin. Cancer Res. 2018, 24, 1500–1510. [Google Scholar] [CrossRef] [Green Version]
- Tzanikou, E.; Markou, A.; Politaki, E.; Koutsopoulos, A.; Psyrri, A.; Mavroudis, D.; Georgoulias, V.; Lianidou, E. PIK3CA hotspot mutations in circulating tumor cells and paired circulating tumor DNA in breast cancer: A direct comparison study. Mol. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.E.; Triboulet, M.; Zia, A.; Vuppalapaty, M.; Kidess-Sigal, E.; Coller, J.; Natu, V.S.; Shokoohi, V.; Che, J.; Renier, C.; et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. NPJ Genom. Med. 2017, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Keup, C.; Benyaa, K.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.-K.; Kimmig, R.; Hahn, P.; Kasimir-Bauer, S. Targeted deep sequencing revealed variants in cell-free DNA of hormone receptor-positive metastatic breast cancer patients. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Beije, N.; Sieuwerts, A.M.; Kraan, J.; Van, N.M.; Onstenk, W.; Vitale, S.R.; van der Vlugt-Daane, M.; Dirix, L.Y.; Brouwer, A.; Hamberg, P.; et al. Estrogen receptor mutations and splice variants determined in liquid biopsies from metastatic breast cancer patients. Mol. Oncol. 2018, 12, 48–57. [Google Scholar] [CrossRef]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Iwase, H. Clinical significance of plasma cell-free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER-positive breast cancer. Mol. Cancer 2018, 17, 67. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.-A.; Iwata, H.; Cortés, J.; de Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2018, e184475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Mu, Z.; Rademaker, A.W.; Austin, L.K.; Strickland, K.S.; Costa, R.L.B.; Nagy, R.J.; Zagonel, V.; Taxter, T.J.; Behdad, A.; et al. Cell-Free DNA and Circulating Tumor Cells: Comprehensive Liquid Biopsy Analysis in Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 560–568. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Fernandez-Garcia, D.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K.; et al. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef]
- Madhavan, D.; Wallwiener, M.; Bents, K.; Zucknick, M.; Nees, J.; Schott, S.; Cuk, K.; Riethdorf, S.; Trumpp, A.; Pantel, K.; et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res. Treat. 2014, 146, 163–174. [Google Scholar] [CrossRef]
- Davis, A.A.; Zhang, Q.; Gerratana, L.; Shah, A.N.; Zhan, Y.; Qiang, W.; Finkelman, B.S.; Flaum, L.; Behdad, A.; Gradishar, W.J.; et al. Association of a novel circulating tumor DNA next-generating sequencing platform with circulating tumor cells (CTCs) and CTC clusters in metastatic breast cancer. Breast Cancer Res. 2019, 21, 137. [Google Scholar] [CrossRef]
- Wang, W.; Liang, M.; Ma, G.; Li, L.; Zhou, W.; Xia, T.; Xie, H.; Wang, S. Plasma cell-free DNA integrity plus circulating tumor cells: A potential biomarker of no distant metastasis breast cancer. Neoplasma 2017, 64, 611–618. [Google Scholar] [CrossRef]
- Chimonidou, M.; Strati, A.; Malamos, N.; Kouneli, S.; Georgoulias, V.; Lianidou, E. Direct comparison study of DNA methylation markers in EpCAM-positive circulating tumour cells, corresponding circulating tumour DNA, and paired primary tumours in breast cancer. Oncotarget 2017, 8, 72054–72068. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, T.K.; Sequist, L.V.; Heymach, J.V.; Riely, G.J.; Jänne, P.A.; Koch, W.H.; Sullivan, J.P.; Fox, D.B.; Maher, R.; Muzikansky, A.; et al. Detection of T790M, the Acquired Resistance EGFR Mutation, by Tumor Biopsy versus Noninvasive Blood-Based Analyses. Clin. Cancer Res. 2016, 22, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, W.M.; Carter, C.; Simmons, J.; Klem, E.; Goodman, N.; Vahidi, B.; Romero, J.; Masterman-Smith, M.; O’Regan, R.; Gogineni, K.; et al. Analysis of tumor template from multiple compartments in a blood sample provides complementary access to peripheral tumor biomarkers. Oncotarget 2016, 7, 26724–26738. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Keup, C.; Mach, P.; Aktas, B.; Tewes, M.; Kolberg, H.-C.; Hauch, S.; Sprenger-Haussels, M.; Kimmig, R.; Kasimir-Bauer, S. RNA Profiles of Circulating Tumor Cells and Extracellular Vesicles for Therapy Stratification of Metastatic Breast Cancer Patients. Clin. Chem. 2018. [Google Scholar] [CrossRef]
- Keup, C.; Hahn, P.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.-K.; Kimmig, R.; Kasimir-Bauer, S.; Benyaa, K. Targeted PCR-Based Deep Sequencing of cfDNA with Unique Molecular Indices by a Customized QIAseq Targeted DNA Panel. Protocols 2018. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keup, C.; Storbeck, M.; Hauch, S.; Hahn, P.; Sprenger-Haussels, M.; Hoffmann, O.; Kimmig, R.; Kasimir-Bauer, S. Multimodal Targeted Deep Sequencing of Circulating Tumor Cells and Matched Cell-Free DNA Provides a More Comprehensive Tool to Identify Therapeutic Targets in Metastatic Breast Cancer Patients. Cancers 2020, 12, 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051084
Keup C, Storbeck M, Hauch S, Hahn P, Sprenger-Haussels M, Hoffmann O, Kimmig R, Kasimir-Bauer S. Multimodal Targeted Deep Sequencing of Circulating Tumor Cells and Matched Cell-Free DNA Provides a More Comprehensive Tool to Identify Therapeutic Targets in Metastatic Breast Cancer Patients. Cancers. 2020; 12(5):1084. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051084
Chicago/Turabian StyleKeup, Corinna, Markus Storbeck, Siegfried Hauch, Peter Hahn, Markus Sprenger-Haussels, Oliver Hoffmann, Rainer Kimmig, and Sabine Kasimir-Bauer. 2020. "Multimodal Targeted Deep Sequencing of Circulating Tumor Cells and Matched Cell-Free DNA Provides a More Comprehensive Tool to Identify Therapeutic Targets in Metastatic Breast Cancer Patients" Cancers 12, no. 5: 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051084