Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer


Division of Medical Oncology & Hematology, Bras Family Drug Development Program, Princess Margaret Cancer Centre, University Health Network, Toronto, ON M5G 2M9, Canada
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(6), 1607; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061607
Submission received: 1 June 2020
/
Revised: 12 June 2020
/
Accepted: 15 June 2020
/
Published: 17 June 2020
(This article belongs to the Special Issue Preclinical and Clinical Advances in Ovarian Cancer)
Abstract
:Platinum chemotherapy remains the cornerstone of treatment for epithelial ovarian cancer (OC) and Poly (ADP-ribose) polymerase inhibitors (PARPi) now have an established role as maintenance therapy. The mechanisms of action of these agents is, in many ways, complementary, and crucially reliant on the intracellular DNA Damage Repair (DDR) response. Here, we review mechanisms of primary and acquired resistance to treatment with platinum and PARPi, examining the interplay between both classes of agents. A key resistance mechanism appears to be the restoration of the Homologous Recombination (HR) repair pathway, through BRCA reversion mutations and epigenetic upregulation of BRCA1. Alterations in non-homologous end-joint (NHEJ) repair, replication fork protection, upregulation of cellular drug efflux pumps, reduction in PARP1 activity and alterations to the tumour microenvironment have also been described. These resistance mechanisms reveal molecular vulnerabilities, which may be targeted to re-sensitise OC to platinum or PARPi treatment. Promising therapeutic strategies include ATR inhibition, epigenetic re-sensitisation through DNMT inhibition, cell cycle checkpoint inhibition, combination with anti-angiogenic therapy, BET inhibition and G-quadruplex stabilisation. Translational studies to elucidate mechanisms of treatment resistance should be incorporated into future clinical trials, as understanding these biologic mechanisms is crucial to developing new and effective therapeutic approaches in advanced OC.
1. Introduction
Epithelial Ovarian Cancer (EOC) is the seventh most common cancer in women and the leading cause of gynecologic cancer death worldwide [1]. For the last three decades, platinum-based chemotherapy has been the cornerstone of systemic treatment for EOC [2,3,4]. Standard front-line treatment for advanced EOC consists of cytoreductive surgery, with the goal of no residual disease (R0), and platinum-based chemotherapy [5]. At the time of relapse, if the time interval since the last dose of platinum chemotherapy (Treatment Free Interval-Platinum; TFIp) is more than six months, the disease is considered platinum sensitive, and standard treatment consists of re-challenge with platinum [5]. The majority of high-grade serous ovarian cancers (HGSC) are initially platinum sensitive. However, even with optimal treatment, HGSC will typically follow a frequent relapse-response pattern, before eventually becoming platinum resistant [5]. Maintenance treatment has emerged as an important strategy to prolong the period between treatment responses and disease relapse [6]. The Poly-ADP Ribose Polymerase inhibitors (PARPi) have demonstrated impressive activity in the first [7,8,9] and second-line [10,11] maintenance settings. This benefit does is not limited to patients with germline or somatic BRCA mutation (BRCAm), or patients with other forms of Homologous Recombination Deficiency (HRD) [12]. Unfortunately, as with platinum chemotherapy, many if not most patients will eventually acquire resistance to PARPi treatment. Outcomes in platinum resistant EOC are extremely poor, with median survival of only 12 months [5]. There is consequently an urgent need to elucidate the mechanisms of platinum and PARPi resistance in EOC to improve patient stratification for therapeutic strategies that target molecular vulnerabilities to overcome treatment resistance. In both cases, mechanisms appear to be complex and inter-related. Resistance to platinum chemotherapy is strongly predictive of resistance to PARPi treatment. Cross resistance between two different therapeutic classes points to overlap in biologic mechanisms of susceptibility and resistance [13]. This article will review the literature regarding mechanisms of treatment resistance to platinum and PARPi in EOC and highlight planned strategies and clinical trials that may effectively re-sensitise tumours to these agents.
2. Alterations in DNA Damage Repair Can Drive Treatment Resistance
The DNA Damage Repair (DDR) response is designed to detect DNA damage, and initiate cell repair in order to maintain genomic integrity within the cell [14]. It consists of a complex network of inter-related signalling pathways. The “master sensors” (ATM, ATR, and DNA-PKs) are large serine/threonine kinases, which sense DNA damage and initiate repair signalling cascades by phosphorylating key proteins such as BRCA1, CHK1, CHK2, p53 and RAD17 [15]. The activation of signalling-transduction pathways promotes activation of DNA-damage-dependent cell checkpoints that slow or halt cell cycle progression, allowing more time for DNA repair [14]. To date, six main DDR pathways have been described; Homologous Recombination (HR), Non-Homologous End Joining (NHEJ), Base Excision Repair (BER), Nucleotide Excision Repair (NER), and Fanconi Anaemia (FA) pathway, and Mismatch Repair (MMR) [14].
Alterations in these DDR pathways have dual significance in OC. In oncogenesis, alterations in DDR leading to genomic instability are a hallmark of cancer development [16]. Germline mutations in HR pathway genes (BRCA 1/2) result in a 14–44% cumulative lifetime risk of EOC [17], with the contribution of other genes becoming increasingly apparent [18]. However, these same alterations in DDR pathways present molecular vulnerabilities, which are targetable by anti-cancer therapy (Table 1). Both platinum chemotherapy and PARPi are genotoxic agents, which exploit defects in DDR pathways to affect cancer cell death [19]. Alterations in DDR (including upregulation or downregulation of key effectors) can drive sensitivity or resistance to these agents (Table 2) [20].
2.1. HRD Conveys Sensitivity to Platinum and PARPi
Platinum chemotherapy exerts its cytotoxic effect by binding with DNA to form mono-adducts. These mono-adducts then evolve through second covalent binding to a DNA crosslink (commonly intra-strand, but also inter-strand), resulting in structural change to the DNA double helix. Platinum treatment leads to an accumulation of DNA double-strand breaks (DSBs) [21]. Tumours with HRD demonstrate a higher degree of chromosomal instability and are fatally unable to repair DSBs, enhancing sensitivity to platinum agents [21].
The PARP family of proteins (especially PARP1) are essential for sensing single-strand DNA breaks (SSBs), initiating repair via predominantly the BER pathway [22]. PARPi are able to block the activity of PARP1 for the repair of SSBs, and trap PARP1 on damaged DNA, posing an insurmountable block to the replisome [22]. The accumulation of SSBs leads to the development of fatal DSBs. Cells require a functional HR repair pathway to resolve these replisome blocks and resume cell-cycle progression, and to repair DSBs [23]. Consequently, PARPi exploit the concept of synthetic lethality in HR-deficient tumours, in order to effect cancer cell death [23].
Some form of HRD is likely to be present at baseline in more than 50% of HGSC potentially underlying the initial sensitivity of this type of tumour to DNA damaging drugs, such as platinum chemotherapy [24]. HR functions to repair DSBs or stalled replication forks during the S and G2 phases of the cell cycle [25]. The pathway is initiated by BRCA1, and uses a sister chromatid as a template for DNA repair, via recruitment of the MRN complex, BRCA2 and RAD51 [19]. Deregulation of HR repair pathway results in chromosomal instability—a phenotype referred to as ‘BRCAness’—which increases the sensitivity of the tumour to DNA damaging drugs [23]. HRD may be the most clinically relevant molecular stratification in OC, and it is an important predictive biomarker for response to both platinum chemotherapy and PARPi [26]. However, the definitive classification of HRD has proven to be elusive. A number of HR assays are undergoing validation (i.e., BROCA, Myriad, Foundation Medicine, HRD detect) [27]. These assays are necessarily limited in their capacity to consider clinically relevant changes such as epigenetic alterations, or to characterise for spatial or temporal tumour heterogeneity within an individual patient. The complexity of defining HRD is increased by the fact that a small subset of patients with HRD never demonstrate a response to platinum and PARPi treatment, whilst some patients without HRD respond well to these same treatments. Any discussion of the key role of HRD in platinum and PARPi mechanisms of action must also consider that multiple and overlapping DDR pathways are likely involved in determining the response, and HRD alone is probably insufficient to predict response to treatment.
2.2. Reactivation of HR Is a Mechanism of Acquired Resistance
Restoration of the function HR pathway in HRD tumours is a key mechanism of acquired platinum and PARPi resistance, which has been demonstrated in vitro and in vivo (Table 3). Restoration of HR may be achieved by secondary mutations, which restore the open reading frame of the BRCA gene, functionally restoring protein activity (BRCA reversion) [28,29,30]. BRCA reversion mutations have been described in both progression biopsies and cell-free DNA (cfDNA), in patients with acquired resistance to PARPi [31,32,33,34,35]. Some cfDNA studies have been able to demonstrate a polyclonality of multiple reversion mutations within a single patient, illustrating that treatment exerts profound selective pressure to restore BRCA1/2 protein activity and overcome PARPi sensitivity [31]. Reversion mutations in HR genes have also been described as acquired resistance mechanisms to platinum chemotherapy [33]. Reversion mutations in BRCA and other HR genes have also been observed in PARPi resistant tumours, including RAD51C/RAD51D reversion mutation in patients with acquired resistance to rucaparib [36]. Other mechanisms that restore BRCA function have been reported in platinum and PARPi resistant cancers, including the loss of BRCA1 promoter methylation [28,37]. Additionally, functional restoration of mutant BRCA1 protein may be achieved through interaction with HSP90, which can promote RAD51 loading onto DNA following DNA damage [38]. Interestingly, in the absence of BRCA reversion mutation, an increase in BRCA1 protein expression has been observed in patients with BRCA1 C-terminal domain mutations [39], suggesting alternative mechanisms to BRCA reversion mutation for reactivating HR. The OC genome appears to display a significant degree of adaptability in response to selective pressure of treatment, with reactivation of HR occurring by multiple different mechanisms, to enable cancer cell survival in the presence of Platinum and PARPi treatments.
2.3. Non-Homologous End Joining (NHEJ)
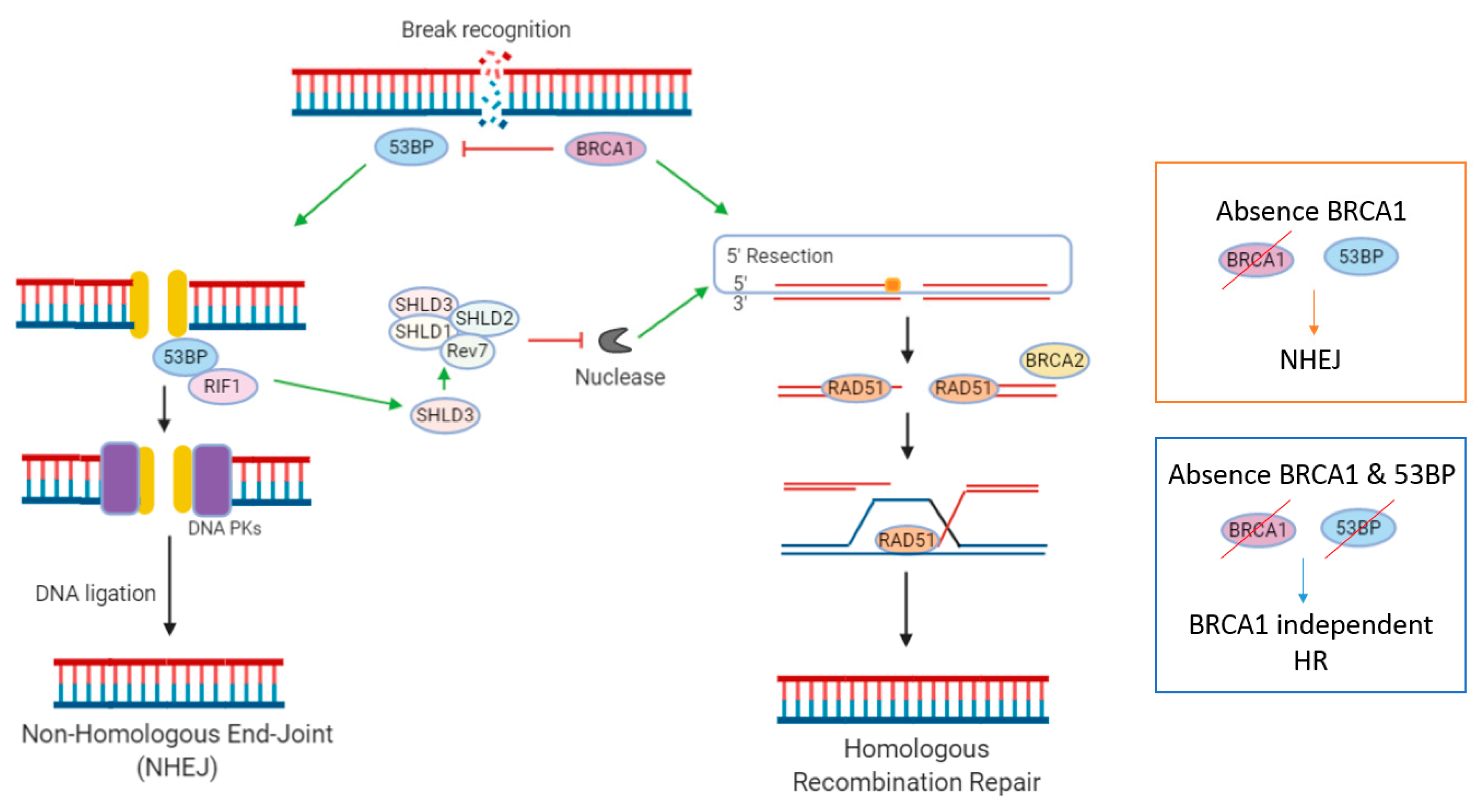
NHEJ is the second important DDR pathway involved in the repair of DSBs. In contrast to HR, which initiates repair during S and G2 phases of the cell cycle, NHEJ occurs throughout interphase [19]. Multiple regulatory mechanisms dictate whether DSBs are repaired via the NHEJ or HR pathway. A key mechanism appears to be the antagonism between HRR-promoting factor BRCA1 and NHEJ-promoting factor 53BP1 [40] (Figure 1). 53BP1 promotes DSB repair by NHEJ in G1 phase cells, by forming 53BP1-RIF1 complexes that protect DSB ends from exonuclease processing [40]. The loss of BRCA1 in HRD tumours can be overcome by concomitant loss of 53BP1, resulting in HR reactivation, rendering cells resistant to the synthetically lethal approach of PARPi (Figure 1) [40]. This re-wiring of DDR pathways via loss of 53BP1 has been described in numerous in vitro studies [41,42,43]. This was first reported by three landmark papers, in which it was observed from mouse models that HRD caused by BRCA1m could be reversed by concomitant loss of 53BP1 (TP53BP1), a protein involved in NHEJ repair [44,45,46]. Interestingly, loss of 53BP1 does not seem to restore HR function in BRCA2m cells, a finding which may be relevant when considering predictive biomarkers and patient stratification.
PARPi-resistance CRISPR/Cas9 screens have identified that the Shieldin (SHLD) complex is actively involved in inhibiting DNA resection [40,42]. Here, SHLD proteins (Rev7, SHLD1, SHLD2, SHLD3) are recruited to DSBs via SHLD3 in a 53BP1 and RIF1-dependent manner. SHLD then blocks nucleases at DSBs, thereby inhibiting 5′ end resection. Resection at 5′ends of DSBs is essential for initiation of repair by the HR pathway. When resection is lost, HR can be re-initiated in a BRCA-independent fashion, via recruitment of PALB2. CRISPR/Cas9 screens have also identified DYNLL1 and HELB as, respectively, 53BP1-dependent and 53BP1-independent, mediators of PARPi resistance [40].
2.4. Nucleotide Excision Repair
The Nucleotide Excision Repair (NER) pathway is a highly conserved mechanism to remove “bulky lesions”, which distort the DNA double helix, including the intra-strand crosslinks formed by platinum adducts [19]. The NER pathway is altered in about 8% of EOC at baseline [47]. The NER pathway defect has been associated with improved Overall Survival (OS) and Progression Free Survival (PFS), suggesting that NER pathway inactivation could confer platinum sensitivity [47]. Interestingly, however, in vitro data demonstrates that NER alteration does not confer sensitivity to PARPi, suggesting that PARP and platinum sensitivity do not always occur in parallel [48]. ERCC1 and XPF are the two components of NER that are potentially most important for removal of platinum-DNA adducts [49]. ERCC1 complexed with XPF is involved in the 5′ cleavage of DNA strands that carry platinum adduct. This complex also plays a role in the HR pathway, repairing intra-strand crosslink. There is conflicting data regarding the validity of ERCC1 as a predictive biomarker of platinum resistance [47,49,50,51,52,53,54,55,56,57]. Polymorphisms in other NER components (i.e., XPA, XPB, XPF, XPD) have shown no correlation with platinum resistance [58].
2.5. Replication Fork Protection
In a normal cell cycle, replication forks may stall in response to replication stress, prolonging S-phase arrest so that DNA repair can occur [59]. In addition to their role in HR, BRCA1 and BRCA2 bind to stalled replication forks, prohibiting the recruitment of MRE11 nuclease, protecting forks from excessive nuclease degradation and stabilising the replication fork [60].
Loss of replication fork protection elicits genomic instability, which initially promotes tumourigenesis. However, replication fork stabilisation by BRCA-independent mechanisms has been described as a mechanism of acquired treatment resistance, promoting cancer cell progression through the cell cycle even in the presence of DNA damage and replication stress. For example, overexpression of RAD51 has been reported in BRCAm cells, restoring RAD51 loading and replication fork protection [59,61,62,63]. SLFN11 is another regulator of S-phase, recruited to stressed replication forks, and able to open chromatin and block replication, prolonging S-phase arrest in the presence of DNA damage. Loss of SLFN11 has been described as a mechanism of acquired platinum resistance [64,65]. Pax2 transactivation domain-interacting protein (PTIP) and Chromodomain Helicase DNA Binding Protein 4 (CHD4) promote recruitment of MRE11 nuclease to replication forks [59]. Lower PTIP expression and CHD4, prohibiting MRE11-dependent onceolytic degradation of nascent DNA, have also been associated with replication fork stabilisation and platinum resistance [59].
The Fanconi Anaemia (FA) repair pathway is important for removing intra-strand DNA crosslinks, formed by platinum adducts [66]. Additionally, this pathway coordinates DNA replication, fine-tuning mitotic checkpoints to ensure error-free chromosomal segregation [66]. The FA pathway is important for replication fork stabilisation, and mutations in FA pathway genes may have similar effects to BRCA1 and BRCA2 mutation, in promoting progression of cancer cells through the cell cycle, even in a setting of DNA damage and replication stress [59].
2.6. Reduced Cellular Availability of Drugs
Reduced cellular availability of chemotherapeutic drugs has been reported as a mechanism of acquired resistance to both platinum chemotherapy and PARPi in ovarian cancer. The copper transporters CTR1, CTR2, ATP7A, and ATP7B regulate intracellular concentration of platinum by mediating its uptake and efflux in cells [67,68,69]. Overexpression of ABCB1, the gene that encodes Multidrug Resistance protein 1 (MDR1), an ATP-binding cassette member involved in the cellular efflux of chemotherapeutic drugs, has also been reported as an acquired resistance mechanism to PARPi [70,71,72]. Upregulation of MDR1 has been described in an engineered PARPi-resistant human ovarian cancer cell line [72], as a result of chromosomal translocations involving the ABCB1 gene [73]. A Whole Genome Sequencing (WGS) study of ovarian cancer cells from matched primary and recurrent ascites, demonstrated upregulation of ABCB1 (through promoter fusion and translocation involving the 5′ region of the gene) in approximately 8% of recurrent HGSOC samples [28]. Most PARPi are MDR1 substrates [74], and this data suggests that prior paclitaxel chemotherapy may precondition tumours to be resistant to PARPi via upregulation of MDR1. The co-administration of MDR1 inhibitors with PARPi is a strategy that has not yet been explored in clinical trials [75], but future research may focus on targeting patients with ABCB1 mutations involved in PARPi resistance. Alteration in intracellular proteins that are able to bind and sequester platinum (including metallothioneins and glutathione [76,77]), and altered expression of pro-survival or anti-survival proteins have also been described as resistance mechanisms which reduce cellular availability of drugs. Proteomic analysis of paired primary and recurrent OC cells from ascites has revealed that RELA and STAT5 proteins cooperate in inducing the anti-apoptotic Bcl-X promoter activity and synergistically enhance Bcl-xL expression in chemo-resistant ovarian cancer cells [78].
3. Immunosuppressive Tumour Microenvironment
These genomic alterations in DNA damage response, must be considered in conjunction with the tumour microenvironment when attempting to elucidate a broad mechanistic understanding of resistance mechanisms in ovarian cancer. Detailed discussions of biomarkers of response to immunotherapy and anti-angiogenic therapy are beyond the scope of this article. However, broadly speaking, immunosuppressive changes within the tumour microenvironment have been associated with resistance to chemotherapy. The increased infiltration of immunosuppressive CD163+ macrophages [79,80], and increased infiltration of regulatory FOXP3+ T cells l [81], has been shown to favour tumour growth, in contrast to the presence of tumour infilitrating lymphocytes (TILs) which is positively correlated with survival [82]. PD1, PD-L1 expression and Tumour Mutational Burden have not demonstrated consistent validity as predictive biomarkers for immune checkpoint inhibition in ovarian cancer. Retinoic acid-inducible gene-I (RIG-I) overexpression is associated with poor-prognosis platinum resistant and refractory cancers [83]. Importantly, RIG-I overexpression was also associated with local immunosuppressive changes such as increased interferon production, and a distinct immune-regulatory signature involving immune checkpoint molecules (PD-L1/PD-1), the RNA editing enzyme ADAR1 and regulatory FOXP3+ T cells [83].
Stromal activation, including extensive stromal desmoplasia, has also been reported to be associated with acquired treatment resistance [28]. It appears to be a key phenotypic characteristic of the chemo-resistant “mesenchymal” molecular subgroup defined in The Cancer Genome Atlas (TCGA) network study [84].
4. Targeting Molecular Vulnerabilities to Overcome Treatment Resistance
4.1. Targeting ATR
Ataxia telangiectasia and Rad3-related (ATR) protein is a key kinase at the heart of the DDR, responsible for sensing replication stress and signalling to S and G2/M checkpoints to initiate repair [15]. ATR inhibitors may be able to reduce the rate of DNA repair in cells, thereby increasing DNA damage and causing cell death [85]. However, single agent ATR inhibition appears to be less effective than synergistic combinations, that increase the effect of synthetic lethality in tumours with DNA repair deficiencies [86]. Combined with PARPi, the simultaneous inhibition of two repair pathways during S-phase, with abrogation of the S/G2 cell cycle checkpoint, leads to accumulation of DSBs in actively replicating cancer cells and cell death in the M-phase [85,86]. The DUETTE study (NCT04239014), will assess the combination of Ceralasertib (AZD6738)—a potent, selective ATR inhibitor—in combination with Olaparib, as second maintenance therapy in patients who have platinum-sensitive OC, who have acquired resistance from prior PARPi treatment. Another ongoing clinical trial is combining the ATR inhibitor M6620 with the PARP inhibitor veliparib to evaluate if this combination can impair DNA repair and induce the ‘BRCAness’ phenotype in solid tumours, including ovarian cancer, which may increase sensitivity to platinum chemotherapy (NCT02723864) [87].
4.2. Epigenetic Resensitisation
It has been observed that the acquisition of a treatment resistant phenotype is associated with the accumulation of epigenetic changes. These include transcriptional silencing of tumour suppressor and DNA repair genes, including BRCA1, TP53, PTEN and MLH1 [88,89]. It is proposed that epigenetic modulators may be able to re-sensitise tumours to platinum-chemotherapy. The DNA methyltransferase (DNMT) inhibitors have not been effective as a single agent treatment in platinum resistant OC. However, in combination they may be able to enhance sensitivity to platinum by altering epigenetic regulation of gene expression. Mixed results have been observed in clinical trials. Decitabine administered one week prior to carboplatin initially demonstrated no activity [90]. It was proposed that this may have been related to the dosing schedule. At low dose, administered continuously in combination with carboplatin, decitabine was shown to reduce DNA methylation of genes in cancer pathways and apoptosis [91,92]. Azacitadine in combination with carboplatin was investigated in a phase Ib/IIa trial, with an objective response rate (ORR) of 13.8% observed in a platinum resistant OC population [93]. In a randomised phase II study [94], assessing Guadecitabine in combination with carboplatin, the ORR and clinical benefit rate was 15% and 45%, respectively. Correlative analyses of this study, including transcriptomic analysis, demonstrated that inhibiting DNA methylation can sensitise OC cells to platinum drugs, in part by altering gene expression patterns related to DNA repair and immune activation, an approach warranting further investigation [95].
The DNA damage initiated by DNMT inhibitors is repaired by the BER pathway, of which PARP1 plays a central role. Additionally, it has been proposed that DNMT treatment induces a “BRCAness” phenotype [96,97]. In light of this, there may be a mechanistic rationale to investigate the combination of DNMT inhibitors with PARPi. This therapeutic approach is yet to be explored in clinical trials.
Histone Deacetylase (HDAC) inhibitors are another class of epigenetic modulator that have not shown significant anti-tumour activity as single agents in platinum resistant OC. A phase II study [98], assessing the combination of Belinostat with carboplatin (n = 29), was stopped early due to lack of activity. Another phase II study assessing Belinostat in combination with carboplatin (n = 35) and paclitaxel [99] demonstrated an ORR 43%. The discrepancy between anti-tumour activity observed between these studies may be related to heterogeneity in study populations, small sample size and potentially the independent cytotoxic effect of paclitaxel.
4.3. Cell Cycle Checkpoint Inhibitors
The cell cycle checkpoint regulators, CHK1/2, halt cell division to allow DNA damage to be repaired prior to DNA replication [100,101]. Cell cycle checkpoint inhibition may be able to prevent progression of cancer cells through cell-cycle, halting replication and tumour growth.
Prexasertib is a CHK1/2 inhibitor, which demonstrated an ORR of 29% in non-gBRCAm carriers in a phase II study in recurrent (predominantly platinum-resistant) HGSOC cohort (NCT02203513) [102]. The results from the germline BRCAm cohort of this study is awaited.
CHK1/2 inhibitors are also being tested in combination with platinum chemotherapy (NCT02797977) and PARP inhibitors (NCT03057145), to assess possible synergy, and if combination treatment can amplify the effects of DNA damage and increase CHK1/2 inhibition-related apoptosis.
WEE-1 inhibitors act on WEE-1 kinase, a G2 cell-cycle checkpoint regulator, in order to abrogate G2 cell cycle arrest, and enhance cancer cell apoptosis in the setting of DNA damage [103]. A phase II proof-of-concept trial [104] assessed the combination of WEE-1 inhibitor, (AZD1775) and carboplatin in TP53 mutated platinum resistant OC, demonstrating an ORR of 43%. Correlative transcriptomic analyses revealed that BRCA1, MYC and CCNE1 alterations were present in one patient with an exceptional prolonged response, highlighting the influence that DDR alterations may have on response to treatment.
A recent randomised phase II study assessed the combination of AZD1775 and gemcitabine, versus gemcitabine monotherapy, in platinum resistant HGSOC [105]. Impressively, a survival benefit was observed, with median Overall Survival 11.5 months with combination treatment, compared to 7.2 months in the gemcitabine arm (Hazard Ratio 0.56, 95% Confidence Interval 0.34–0.92; p = 0.022). A partial response was observed in 13 (21%) of the patients in this study. Given the survival benefit observed, this approach warrants further investigation in a phase III trial.
4.4. BET Inhibitors
Bromodomains are small protein domains that recognise and bind to acetylated histone tails, modify chromatin structure, and lead to upregulation of target genes to drive oncogenesis [106]. BET inhibitors reversibly bind to bromodomains of BET proteins, blocking interaction with acetylated histones and transcription factors. Inhibition of BET interferes with BRCA1 and RAD51 expression [107]. BET inhibition has been demonstrated to induce HRD in ovarian cancer cell lines, by decreasing transcription of BRCA1 and RAD51, depleting the DNA double stand break resection protein CtIP (C-terminal binding protein (CtBP) interacting protein) and downregulating the G2-M cell-cycle checkpoint regulator WEE1 and the DNA-damage response factor TOPBP1 [107,108,109,110]. BET and PARP inhibition has demonstrated a synergistic effect on reducing xenograft tumour growth in HR-proficient ovarian cancer mouse models [107,108]. These effects have been shown to be independent of BRCA1/2, TP53, RAS, and BRAF mutation status. Combining PARP and BET inhibitors may thus help overcome not only primary resistance but also the development of secondary resistance.
4.5. Anti-Angiogenic Therapies
Preclinical studies have demonstrated that anti-angiogenic therapy can induce a hypoxic tumour microenvironment, which is associated with downregulation of HR genes [111]. This is the mechanistic rationale for investigating the combination of anti-angiogenic therapy with PARPi, to enhance their synthetically lethal effects.
A single arm phase II study [112] assessing the combination of olaparib and cediranib demonstrated an ORR of 20% in a platinum resistant OC cohort. The observed benefit appeared to be higher in patients with germline BRCAm. The Evolve study [113] was a proof of concept clinical-translational phase II trial of cediranib-olaparib in ovarian cancer, including patients who had acquired resistance to prior PARPi therapy, enrolled into platinum sensitive (n = 10), platinum resistant (n = 10) and exploratory (n = 10) cohorts. Two partial responses were observed amongst a cohort of 10 patients with platinum-resistant OC. The 16-week PFS was 54.5% (31.8–93.6) in PS, 50% (26.9–92.9) in PR and 36% (15.6–82.8) in PE, respectively. OS at one year was 81.8% (61.9–100) in PS, 64.8% (39.3–100) in PR and 39.1% (14.7–100) in PE. Correlative analyses identified mechanisms of PARPi resistance in ~77% of evaluable patients with matched pre-post PARP inhibitor progression biopsies, such as reversion mutations in BRCA1/2 and other HR genes, MDR1 upregulation, CCNE amplification and RIG-I like receptor downregulation [113]. The anti-tumour activity of this combination will be assessed further in the OVC2 trial (NCT02502266), a randomised phase III study evaluating Olaparib+Cediranib vs. chemotherapy in Platinum Resistant OC. Importantly, this study demonstrates the feasibility of integrating translational objectives into clinical trial protocol design. This is critical, as further translational studies to exploring mechanisms of treatment resistance will be crucial to developing more effective therapeutic strategies.
4.6. G-Quadruplex Stabilisation
G-quadruplex (G4) structures can potentially form at over 700,000 sequences in the human genome, including telomeres, rDNA, the immunoglobulin heavy chain switch regions, minisatellite and microsatellite repeats [114,115,116]. G4 structures increase the tendency for DNA damage to occur, by impeding DNA polymerase and DNA damage repair processes. Chromosome breaks induced by G4 structures will activate diverse repair pathways, such as HR, TLS, NHEJ pathways and pol θ-mediated alternative end-joining pathways [117,118,119,120]. CX-5461, is a novel, synthetically derived small molecule, which selectively kills HR deficient cancer cells, through stabilising G4 structures and inducing replication-dependent DNA damage. Phase 1 studies of CX-5461 in solid tumours have been completed [121], with further investigation planned in an ovarian cancer cohort with acquired resistance to treatment from prior PARPi and/or platinum exposure.
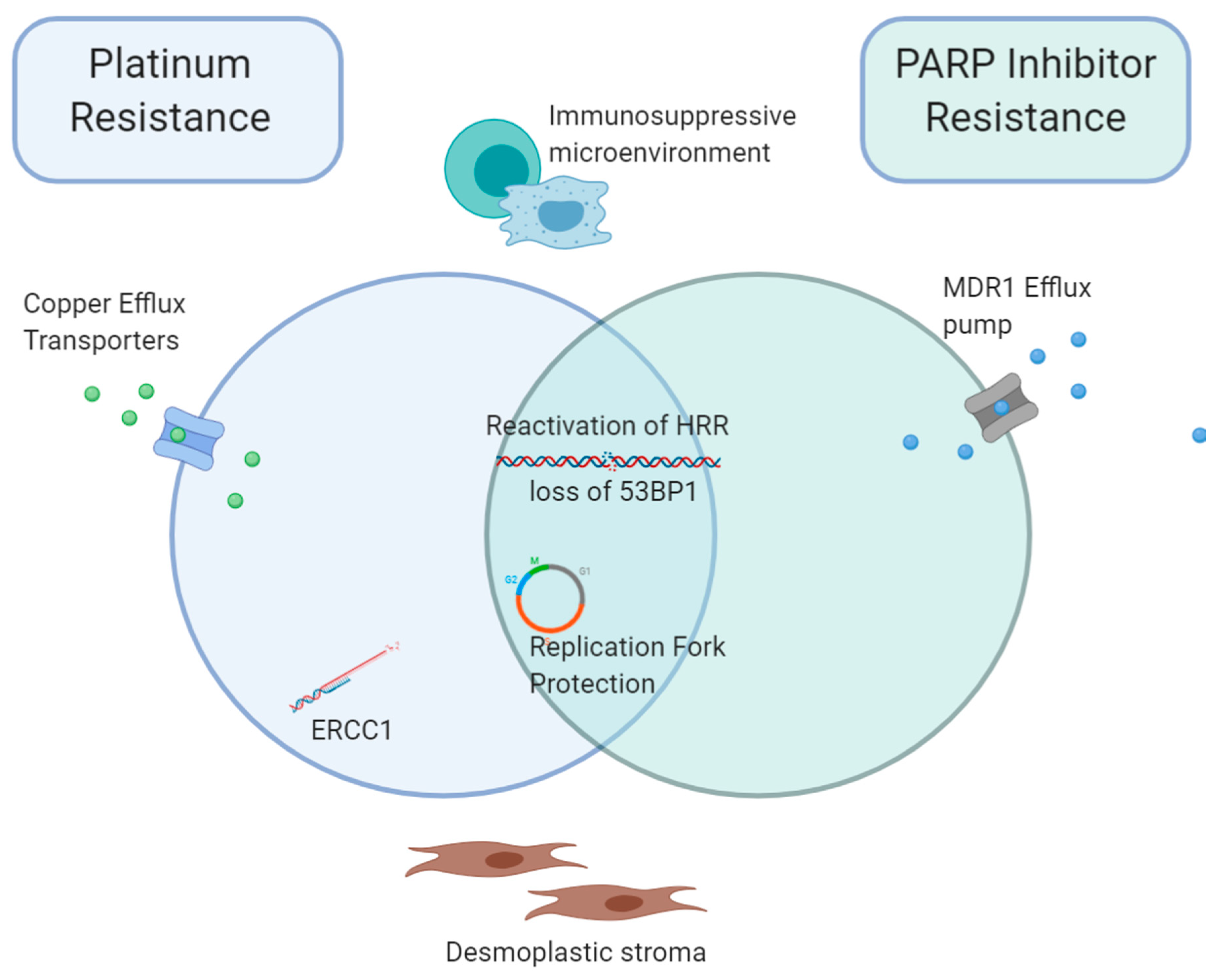
5. Overlap between Acquired Platinum and PARPi Resistance Mechanisms
Clearly, there is significant overlap between mechanisms of resistance to platinum chemotherapy, and PARPi, with DDR alterations playing a key role. However, it must be noted that patients who progress on PARPi maintenance often retain sensitivity to platinum chemotherapy. Acquired resistance to these classes of agents clearly does not develop entirely in parallel (Figure 2). It is not yet clear whether patients who progress on PARPi, then respond to platinum chemotherapy, may retain some sensitivity to PARPi and benefit from second maintenance therapy with PARPi. This question is currently being explored in the OReO study (NCT03106987; ENGOT-ov38/OReO), a Phase IIIb study of olaparib maintenance retreatment in patients with epithelial ovarian cancer previously treated with a PARPi and responding to repeat platinum chemotherapy. It will also be addressed in the olaparib monotherapy arm of the DUETTE study (NCT04239014). With the success of PARPi as first and second-line maintenance treatment, a new patient population with platinum sensitive relapsed OC is emerging, for whom no standard of care exists. Hence, the results of these studies are eagerly awaited, for their capacity to define a new standard of care in this patient population of unmet needs.
6. Biomarkers of Resistance to PARPi and Platinum
Predicting platinum or PARPi resistance in individual patients remains challenging, as resistance mechanisms are multifactorial and evolve over time. As discussed earlier, whilst current HRD assays have their limitations, HRD is a powerful biomarker for predicting the initial response to both platinum chemotherapy and PARPi [26]. At time of relapse, the Therapy Free Interval (TFI), defined as the time between last treatment and documented relapse, remains the most widely used clinical predictor of likely response [122]. Recently, however, the biological relevance of using a six-month TFI to define platinum resistance has been challenged, and much effort has been put into defining and validating molecular biomarkers of treatment resistance [123]. The detection of somatic mutations linked to acquired resistance mechanisms (such as BRCA reversion mutation) in ctDNA is a promising non-invasive biomarker, which warrants investigation in future prospective studies.
7. Conclusions
Platinum chemotherapy remains the cornerstone of systemic therapy for OC, with PARPi playing a key role as maintenance treatment. Resistance to these treatments has important prognostic implications. Whilst research has improved our knowledge thus far, our global mechanistic understanding is still limited. In particular, the complex interactions, compensatory changes, and relative contributions between DDR pathways and the tumour microenvironment remain to be fully described. The incorporation of translational objectives into clinical trial protocol design will enable further elucidation of these important mechanistic questions, in order to improve patient stratification, and develop therapeutic approaches that effectively target molecular vulnerabilities in treatment-resistant OC.
Funding
This research received no external funding.
Conflicts of Interest
A.M.O. is on the steering committee of G.S.K., A.Z., Clovis, Tesaro and Merck (uncompensated), and is P.I. on clinical trials for A.Z., G.S.K., and Clovis. M.M, A.M. and K.K. have no COI to disclose.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, G.; Lawton, F.; Redman, C.; Kelly, K. Response of patients in phase II studies of chemotherapy in ovarian cancer: Implications for patient treatment and the design of phase II trials. Br. J. Cancer 1989, 59, 650–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, M.E.; Fryatt, I.; Wiltshaw, E.; Dawson, T. Treatment of relapsed carcinoma of the ovary with cisplatin or carboplatin following initial treatment with these compounds. Gynecol. Oncol. 1990, 36, 207–211. [Google Scholar] [CrossRef]
- Markman, M.; Rothman, R.; Hakes, T.; Reichman, B.; Hoskins, W.; Rubin, S.; Jones, W.; Almadrones, L.; Lewis, J.L., Jr. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J. Clin. Oncol. 1991, 9, 389–393. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Madariaga, A.; Rustin, G.J.S.; Buckanovich, R.J.; Trent, J.C.; Oza, A.M. Wanna Get Away? Maintenance Treatments and Chemotherapy Holidays in Gynecologic Cancers. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, e152–e166. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Eng. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Tomao, F.; Bardhi, E.; Di Pinto, A.; Sassu, C.M.; Biagioli, E.; Petrella, M.C.; Palaia, I.; Muzii, L.; Colombo, N.; Panici, P.B. Parp inhibitors as maintenance treatment in platinum sensitive recurrent ovarian cancer: An updated meta-analysis of randomized clinical trials according to BRCA mutational status. Cancer Treat. Rev. 2019, 80, 101909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- McCuaig, J.M.; Stockley, T.L.; Shaw, P.; Fung-Kee-Fung, M.; Altman, A.D.; Bentley, J.; Bernardini, M.Q.; Cormier, B.; Hirte, H.; Kieser, K.; et al. Evolution of genetic assessment for BRCA-associated gynaecologic malignancies: A Canadian multisociety roadmap. J. Med. Genet. 2018, 55, 571–577. [Google Scholar] [CrossRef]
- Gee, M.E.; Faraahi, Z.; McCormick, A.; Edmondson, R.J. DNA damage repair in ovarian cancer: Unlocking the heterogeneity. J. Ovarian Res. 2018, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Kubelac, P.; Genestie, C.; Auguste, A.; Mesnage, S.; Le Formal, A.; Pautier, P.; Gouy, S.; Morice, P.; Bentivegna, E.; Maulard, A.; et al. Changes in DNA Damage Response Markers with Treatment in Advanced Ovarian Cancer. Cancers 2020, 12, 707. [Google Scholar] [CrossRef] [Green Version]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Shar-yin, N.H.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Madariaga, A.; Lheureux, S.; Oza, A.M. Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations. Cancers 2019, 11, 416. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Hay, T.; Matthews, J.R.; Pietzka, L.; Lau, A.; Cranston, A.; Nygren, A.O.; Douglas-Jones, A.; Smith, G.C.; Martin, N.M.; O’Connor, M.; et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009, 69, 3850–3855. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, P.; Gay, L.M.; Lele, S.; Elvin, J.A. BRCA1 reversion mutation acquired after treatment identified by liquid biopsy. Gynecol. Oncol. Rep. 2017, 21, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Mendez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; Garcia-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer. Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M. Reversion Mutations with Clinical Use of PARP Inhibitors: Many Genes, Many Versions. Cancer Discov. 2017, 7, 937–939. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 Recovery as a Resistance Mechanism After Exceptional Response to Poly (ADP-ribose) Polymerase Inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Bowden, N.A. Nucleotide excision repair: Why is it not used to predict response to platinum-based chemotherapy? Cancer Lett. 2014, 346, 163–171. [Google Scholar] [CrossRef]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.; Alblihy, A.; Moseley, P.; Chan, S.Y.T.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, A.; Zhao, Y.; Xiang, J.; Yu, D.; Liang, Z.; Xu, C.; Zhang, Q.; Li, J.; Duan, P. The association of polymorphisms in nucleotide excision repair genes with ovarian cancer susceptibility. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcy, K.M.; Tian, C.; Reed, E. A Gynecologic Oncology Group study of platinum-DNA adducts and excision repair cross-complementation group 1 expression in optimal, stage III epithelial ovarian cancer treated with platinum-taxane chemotherapy. Cancer Res. 2007, 67, 4474–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmegna, B.; Uboldi, S.; Frapolli, R.; Licandro, S.A.; Panini, N.; Galmarini, C.M.; Badri, N.; Spanswick, V.J.; Bingham, J.P.; Kiakos, K.; et al. Increased sensitivity to platinum drugs of cancer cells with acquired resistance to trabectedin. Br. J. Cancer 2015, 113, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hu, P.; Cao, Y.; Wang, G.Y.; Wang, N.; Zhou, R.M. Predicting the outcome of platinum-based chemotherapies in epithelial ovarian cancer using the 8092C/A polymorphism of ERCC1: A meta-analysis. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2014, 19, 128–134. [Google Scholar]
- Rubatt, J.M.; Darcy, K.M.; Tian, C.; Muggia, F.; Dhir, R.; Armstrong, D.K.; Bookman, M.A.; Niedernhofer, L.J.; Deloia, J.; Birrer, M.; et al. Pre-treatment tumor expression of ERCC1 in women with advanced stage epithelial ovarian cancer is not predictive of clinical outcomes: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Krivak, T.C.; Darcy, K.M.; Tian, C.; Armstrong, D.; Baysal, B.E.; Gallion, H.; Ambrosone, C.B.; DeLoia, J.A. Relationship between ERCC1 polymorphisms, disease progression, and survival in the Gynecologic Oncology Group Phase III Trial of intraperitoneal versus intravenous cisplatin and paclitaxel for stage III epithelial ovarian cancer. J. Clin. Oncol. 2008, 26, 3598–3606. [Google Scholar] [CrossRef] [Green Version]
- Steffensen, K.D.; Smoter, M.; Waldstrom, M.; Grala, B.; Bodnar, L.; Stec, R.; Szczylik, C.; Jakobsen, A. Resistance to first line platinum paclitaxel chemotherapy in serous epithelial ovarian cancer: The prediction value of ERCC1 and Tau expression. Int. J. Oncol. 2014, 44, 1736–1744. [Google Scholar] [CrossRef]
- Mouw, K.W.; D’Andrea, A.D.; Konstantinopoulos, P.A. Nucleotide excision repair (NER) alterations as evolving biomarkers and therapeutic targets in epithelial cancers. Oncoscience 2015, 2, 942–943. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Schlacher, K. PARPi focus the spotlight on replication fork protection in cancer. Nat. Cell Biol. 2017, 19, 1309–1310. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Li, T.; Peng, J.; Zeng, F.; Zhang, K.; Liu, J.; Li, X.; Ouyang, Q.; Wang, G.; Wang, L.; Liu, Z.; et al. Association between polymorphisms in CTR1, CTR2, ATP7A, and ATP7B and platinum resistance in epithelial ovarian cancer. Int. J. Clin. Pharmacol. Ther. 2017, 55, 774–780. [Google Scholar] [CrossRef]
- Li, Y.Q.; Yin, J.Y.; Liu, Z.Q.; Li, X.P. Copper efflux transporters ATP7A and ATP7B: Novel biomarkers for platinum drug resistance and targets for therapy. IUBMB Life 2018, 70, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Briz, O.; Perez-Silva, L.; Al-Abdulla, R.; Abete, L.; Reviejo, M.; Romero, M.R.; Marin, J.J.G. What “The Cancer Genome Atlas”database tells us about the role of ATP-binding cassette (ABC) proteins in chemoresistance to anticancer drugs. Expert Opin. Drug Metab. Toxicol. 2019, 15, 577–593. [Google Scholar] [CrossRef]
- Tian, C.; Ambrosone, C.B.; Darcy, K.M.; Krivak, T.C.; Armstrong, D.K.; Bookman, M.A.; Davis, W.; Zhao, H.; Moysich, K.; Gallion, H.; et al. Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 124, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.; Paulo, A. Small molecule inhibitors of multidrug resistance gene (MDR1) expression: Preclinical evaluation and mechanisms of action. Curr. Cancer Drug Targets 2013, 13, 814–828. [Google Scholar] [CrossRef]
- Kleih, M.; Bopple, K.; Dong, M.; Gaissler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, B.; Ramadoss, S.; Chaudhuri, G.; Janzen, C.; Sen, S.; Mascharak, P.K. Carbon monoxide sensitizes cisplatin-resistant ovarian cancer cell lines toward cisplatin via attenuation of levels of glutathione and nuclear metallothionein. J. Inorg. Biochem. 2019, 191, 29–39. [Google Scholar] [CrossRef]
- Jinawath, N.; Vasoontara, C.; Jinawath, A.; Fang, X.; Zhao, K.; Yap, K.-L.; Guo, T.; Lee, C.S.; Wang, W.; Balgley, B.M. Oncoproteomic analysis reveals co-upregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Ojalvo, L.S.; Thompson, E.D.; Wang, T.L.; Meeker, A.K.; Shih, I.M.; Fader, A.N.; Cimino-Mathews, A.; Emens, L.A. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum. Pathol. 2018, 74, 135–147. [Google Scholar] [CrossRef]
- Martin de la Fuente, L.; Westbom-Fremer, S.; Arildsen, N.S.; Hartman, L.; Malander, S.; Kannisto, P.; Masback, A.; Hedenfalk, I. PD-1/PD-L1 expression and tumor-infiltrating lymphocytes are prognostically favorable in advanced high-grade serous ovarian carcinoma. Virchows Arch. Int. J. Pathol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Fiegl, H.; Zeimet, A.G.; Wieser, V.; Marth, C.; Sprung, S.; Sopper, S.; Hartmann, G.; Reimer, D.; Boesch, M. High RIG-I expression in ovarian cancer associates with an immune-escape signature and poor clinical outcome. Int. J. Cancer 2020, 146, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- O’Sullivan Coyne, G.H.; Do, K.T.; Kummar, S.; Takebe, N.; Quinn, M.F.; Piha-Paul, S.A.; Bruns, A.; Juwara, L.; Sharon, E.; Piekarz, R. Phase I trial of the triplet M6620 (formerly VX970)+ veliparib+ cisplatin in patients with advanced solid tumors. Am. Soc. Clin. Oncol. 2018, 36 (Suppl. 15), 2549. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. JNCI J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Li, M.; Balch, C.; Montgomery, J.S.; Jeong, M.; Chung, J.H.; Yan, P.; Huang, T.H.; Kim, S.; Nephew, K.P. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genom. 2009, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Glasspool, R.; Brown, R.; Gore, M.; Rustin, G.; McNeish, I.; Wilson, R.; Pledge, S.; Paul, J.; Mackean, M.; Hall, G. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer 2014, 110, 1923–1929. [Google Scholar] [CrossRef]
- Matei, D.; Shen, C.; Fang, F.; Schilder, J.; Li, M.; Arnold, A.; Zeng, Y.; Pilrose, J.; Kulesavage, C.; Balch, C. A phase II study of decitabine and carboplatin in recurrent platinum (Pt)-resistant ovarian cancer (OC). J. Clin. Oncol. 2011, 29 (Suppl. 15), 5011. [Google Scholar] [CrossRef]
- Fang, F.; Zuo, Q.; Pilrose, J.; Wang, Y.; Shen, C.; Li, M.; Wulfridge, P.; Matei, D.; Nephew, K.P. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget 2014, 5, 3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Hu, W.; Iyer, R.; Kavanagh, J.J.; Coleman, R.L.; Levenback, C.F.; Sood, A.K.; Wolf, J.K.; Gershenson, D.M.; Markman, M. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2011, 117, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Matulonis, U.A.; Alvarez Secord, A.; Nemunaitis, J.; Roman, L.D.; Blagden, S.P.; Banerjee, S.; McGuire, W.P.; Ghamande, S.; Birrer, M.J.; et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin. Cancer Res. 2020, 26, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and epigenomic signatures in ovarian cancer associated with resensitization to platinum drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Kogan, A.A.; Mclaughlin, L.J.; Topper, M.; Muvarak, N.; Stojanovic, L.; Creed, T.M.; Bentzen, S.; Civin, C.I.; Baer, M.R.; Kingsbury, T.J. DNA Demethylating Agents Generate a Brcaness Effect in Multiple Sporadic Tumor Types: Prediction for Sensitivity to PARP Inhibitors in AML. Blood 2017, 130 (Suppl. 1), 3347. [Google Scholar]
- Wiegmans, A.P.; Yap, P.-Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in expression of key DNA damage repair genes after epigenetic-induced BRCAness dictate synthetic lethality with PARP1 inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; DiSilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S.; Damstrup, L.; Finkler, N.J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Lichenstein, H.S.; Knoblach, P.; Penson, R.T. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 979–986. [Google Scholar] [CrossRef]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Cabanero, M.; Cristea, M.C.; Mantia-Smaldone, G.; Olawaiye, A.; Ellard, S.; Weberpals, J.I.; Wahner Hendrickson, A.E.; Fleming, G.F.; Welch, S.; et al. A randomized double-blind placebo-controlled phase II trial comparing gemcitabine monotherapy to gemcitabine in combination with adavosertib in women with recurrent, platinum resistant epithelial ovarian cancer: A trial of the Princess Margaret, California, Chicago and Mayo Phase II Consortia. J. Clin. Oncol. 2019, 37 (Suppl. 15), 5518. [Google Scholar]
- Dawson, M.; Stein, E.M.; Huntly, B.J.P.; Karadimitris, A.; Kamdar, M.; Fernandez de Larrea, C.; Dickinson, M.J.; Yeh, P.S.-H.; Daver, N.; Chaidos, A.; et al. A Phase I Study of GSK525762, a Selective Bromodomain (BRD) and Extra Terminal Protein (BET) Inhibitor: Results from Part 1 of Phase I/II Open Label Single Agent Study in Patients with Acute Myeloid Leukemia (AML). Blood 2017, 130 (Suppl. 1), 1377. [Google Scholar]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Wenham, R.M.; Hendrickson, A.E.W.; Armstrong, D.K.; Chan, N.; Cohn, D.E.; Lee, J.-m.; Penson, R.T.; Cristea, M.C.; et al. A phase 2 biomarker trial of combination cediranib and olaparib in relapsed platinum (plat) sensitive and plat resistant ovarian cancer (ovca). J. Clin. Oncol. 2018, 36 (Suppl. 15), 5519. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.; Dhani, N.C.; Madariaga, A.; Bonilla, L.; Lee, Y.C.; Colombo, I.; Bhat, G.; et al. Evolve: A post PARP inhibitor clinical translational phase II trial of cediranib-olaparib in ovarian cancer—A Princess Margaret Consortium—GCIG Phase II Trial. J. Clin. Oncol. 2019, 37 (Suppl. 15), 5521. [Google Scholar] [CrossRef]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Youds, J.L.; O’Neil, N.J.; Rose, A.M. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 2006, 173, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule–induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.K.; Pujade-Lauraine, E.; Aoki, D.; Mirza, M.R.; Lorusso, D.; Oza, A.M.; du Bois, A.; Vergote, I.; Reuss, A.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: Recurrent disease. Ann. Oncol. 2017, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Hanker, L.C.; Loibl, S.; Burchardi, N.; Pfisterer, J.; Meier, W.; Pujade-Lauraine, E.; Ray-Coquard, I.; Sehouli, J.; Harter, P.; du Bois, A.; et al. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann. Oncol. 2012, 23, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
DNA Double Strand Break repair by Homologous Recombination or Non-Homologous End-Joining pathway. Figure: In the absence of functional BRCA1, DNA Double strand breaks (DSBs) may be repaired by the Non-Homologous End Joining (NHEJ) pathway. Loss of 53BP1 is an acquired resistance mechanism in BRCA1 mutant tumours. 53Bp1 normally promotes DSB repair by NHEJ, by forming a 53Bp1-RIF1 complex that protects DSB ends from endonuclease processing. Loss of 53BP1 prohibits a recruitment of Shieldin (SHLD) complex to DSB, allowing 5′ end resection, and reactivation of HR in a BRCA1 independent manner.
Figure 1.
DNA Double Strand Break repair by Homologous Recombination or Non-Homologous End-Joining pathway. Figure: In the absence of functional BRCA1, DNA Double strand breaks (DSBs) may be repaired by the Non-Homologous End Joining (NHEJ) pathway. Loss of 53BP1 is an acquired resistance mechanism in BRCA1 mutant tumours. 53Bp1 normally promotes DSB repair by NHEJ, by forming a 53Bp1-RIF1 complex that protects DSB ends from endonuclease processing. Loss of 53BP1 prohibits a recruitment of Shieldin (SHLD) complex to DSB, allowing 5′ end resection, and reactivation of HR in a BRCA1 independent manner.

Figure 2.
Overlapping mechanisms of resistance to Platinum and PARP-inhibitor treatment. Platinum and PARPi demonstrate distinct and overlapping mechanisms of acquired treatment resistance. Whilst both demonstrate reactivation of HRR and loss of 53BP1, and replication fork protection promoting progression through the cell cycle, platinum resistance is additionally associated with defects in the NER pathway, including increased ERCC1 activity. Reduced intracellular drug accumulation is achieved via copper transporters for platinum, and MDR1 drug efflux pump for PARPi. In both instances, an immunosuppressive microenvironment and desmoplastic stroma likely contribute to tumour progression. Abbreviations; HRR, homologous recombination repair, MDR1, Multidrug Resistance Protein 1, ERCC1, DNA excision repair protein ERCC-1, PARPi, PARP inhibitor.
Figure 2.
Overlapping mechanisms of resistance to Platinum and PARP-inhibitor treatment. Platinum and PARPi demonstrate distinct and overlapping mechanisms of acquired treatment resistance. Whilst both demonstrate reactivation of HRR and loss of 53BP1, and replication fork protection promoting progression through the cell cycle, platinum resistance is additionally associated with defects in the NER pathway, including increased ERCC1 activity. Reduced intracellular drug accumulation is achieved via copper transporters for platinum, and MDR1 drug efflux pump for PARPi. In both instances, an immunosuppressive microenvironment and desmoplastic stroma likely contribute to tumour progression. Abbreviations; HRR, homologous recombination repair, MDR1, Multidrug Resistance Protein 1, ERCC1, DNA excision repair protein ERCC-1, PARPi, PARP inhibitor.


{kind=link}
{kind=link}
Table 1.
Examples of current active trials that target DNA Damage Repair pathways to overcome resistance to PARPi and Platinum.
Table 1.
Examples of current active trials that target DNA Damage Repair pathways to overcome resistance to PARPi and Platinum.
| Study Name/NCT | Target | Study Treatment | Study Population | Study Phase |
|---|---|---|---|---|
| DUETTE NCT04239014 | ATR/PARP | Ceralasertib (AZD6738) + Olaparib, or Olaparib monotherapy, or Placebo | Relapsed platinum-sensitive OC, who have acquired resistance from prior PARPi treatment | II, RCT |
| NCT02723864 | ATR/PARP | VX-970 + Veliparib and Cisplatin | Advanced refractory solid tumours | I |
| NCT02901899 | DNMT/PD-1 | Guadecitabine + pembrolizumab | Recurrent Platinum Resistant OC | II, open-label |
| NCT03924245 | HDAC/PARP | Entinostat + Olaparib | Recurrent platinum refractory and resistant EOC | I/II |
| NCT02915523 | PD-L1/HDAC | Avelumab ± Entinostat | Advanced OC Which Has Progressed or Recurred After First-line Platinum-based Chemotherapy and at Least Two Subsequent Lines of Treatment | Ib/II, RCT |
| NCT02797977 | CHK1 | SRA737 + gemcitabine + cisplatin, or gemcitabine monotherapy | Advanced solid tumours, including HGSOC which is BRCA1 and BRCA2 wild type. | I/II, non-randomised |
| NCT03057145 | CHK1/PARP | (Prexasertib) LY2606368 + Olaparib | Advanced solid tumours | I |
| NCT03579316 | WEE-1/PARP | Adavosertib (AZD1775) + Olaparib, or adavosertib monotherapy | Recurrent OC with progression on prior PARPi therapy | II, RCT |
| NCT02502266 | Angiogenesis/PARP | Cediranib + Olaparib, or chemotherapy | Platinum resistant or Refractory OC | III, RCT |
Abbreviations; ATR, ataxia telangiectasia and Rad3-related protein; PARP, Poly (ADP-ribose) polymerase; DNMT, DNA methyltransferase, PD-1, Programmed cell death protein 1; PD-L1, Programmed death-ligand 1; HDAC, Histone deacetylases; CHK1, Checkpoint kinase 1; RCT, Randomised Control Trial.
Table 2.
Contribution of DNA Damage Repair pathway alterations to Platinum and PARPi resistance.
| DNA Damage Repair Pathway | Key Pathway Functions | Key Genes | Effect of Pathway Alterations on Therapeutic Resistance |
|---|---|---|---|
| Homologous Recombination (HR) | Repair of DSBs or stalled replication forks during S and G2 phases of cell cycle | BRCA1, BRCA2, RAD51, HSP90 | Reactivation of HR pathway enables repair of DSBs and resolves replisome blocks, promoting cancer cell progression through the cell cycle despite the presence of cytotoxic DNA damage. |
| Non-Homologous End Joining (NHEJ) | Repair of DSBs during interphase | 53BP1 | Loss of 53BP1 re-wires NHEJ pathway, reactivating HR independent of BRCA1 |
| Base Excision Repair (BER) | Repair of SSBs and DNA base lesions | PARP-1, XRCC1, Pol β | Functional BER pathway leads to loss of synthetic lethality and PARPi resistance |
| Nucleotide Excision Repair (NER) | Removes “bulky lesions” which distort the DNA double helix, including intra-strand crosslinks formed by platinum adducts. | ERCC1, XPF | Upregulation of ERCC1 and XPF potentially restores NER function. NER pathway alteration potentially confers sensitivity to platinum, and not PARPi. |
| Fanconi Anemia (FA) | Removes intra-strand DNA crosslinks, coordinates DNA replication by fine-tuning mitotic checkpoints and replication fork stabilisation | FANCC, FANCD2, FANCA | Mutations in FA pathway genes may have a similar effect to BRCA1 and BRCA2 mutation, in promoting progression of cancer cell through the cell cycle, even in setting of DNA damage and replication stress |
| Mismatch Repair (MMR) Deficiency | Recognise, excise and resynthesise mismatched or unmatched DNA base pairs or insertion-deletion loops. | MLH1, MSH2 | MMR deficiency results in microsatellite instability, interfering with detection of cytotoxic DNA damage, allowing cancer cells to proliferate despite DNA damage. |
DSBs; Double Strand DNA breaks, SSBs; Single Strand DNA breaks, PARPi; PARP inhibitor.
Table 3.
Mechanisms of reactivation of Homologous Recombination repair.
| Resistance Mechanism | Function |
|---|---|
| BRCA (or HR gene) reversion mutation | Restores open reading frame of gene, resulting in functional protein expression |
| Loss of BRCA1 promoter methylation | Restores BRCA1 function |
| Upregulated HSP90 | Promotes BRCA-independent RAD51 loading onto damaged DNA |
| BRCA1 C-terminal domain mutation | Upregulation of BRCA1, in absence of BRCA1 reversion mutation |
| Loss 53BP1 | Recruits Shieldin complex to inhibit DNA resection, initiating HR in a BRCA-independent manner. |
Abbreviations; HR, Homologous Recombination; HSP90, Heat Shock Protein 90.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McMullen, M.; Karakasis, K.; Madariaga, A.; Oza, A.M. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers 2020, 12, 1607. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061607
AMA Style
McMullen M, Karakasis K, Madariaga A, Oza AM. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers. 2020; 12(6):1607. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061607
Chicago/Turabian StyleMcMullen, Michelle, Katherine Karakasis, Ainhoa Madariaga, and Amit M. Oza. 2020. "Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer" Cancers 12, no. 6: 1607. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061607
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.