Dendrogenin A Synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and AML Primary Samples
2.3. Lentiviral Infection of KG-1 and HL-60 Cells
2.4. Cell Death Assay
2.5. Annexin-V/7AAD Assay
2.6. Western Blot
2.7. Autophagy
2.8. Sterol Analysis
2.9. Phosphatidyl Ethanolamine (PE) Analysis
2.10. Animals
2.11. Measure of Combination Therapy Efficacy In Vivo
2.12. Assessment of Leukemic Engraftment
2.13. Statistical Analysis
3. Results
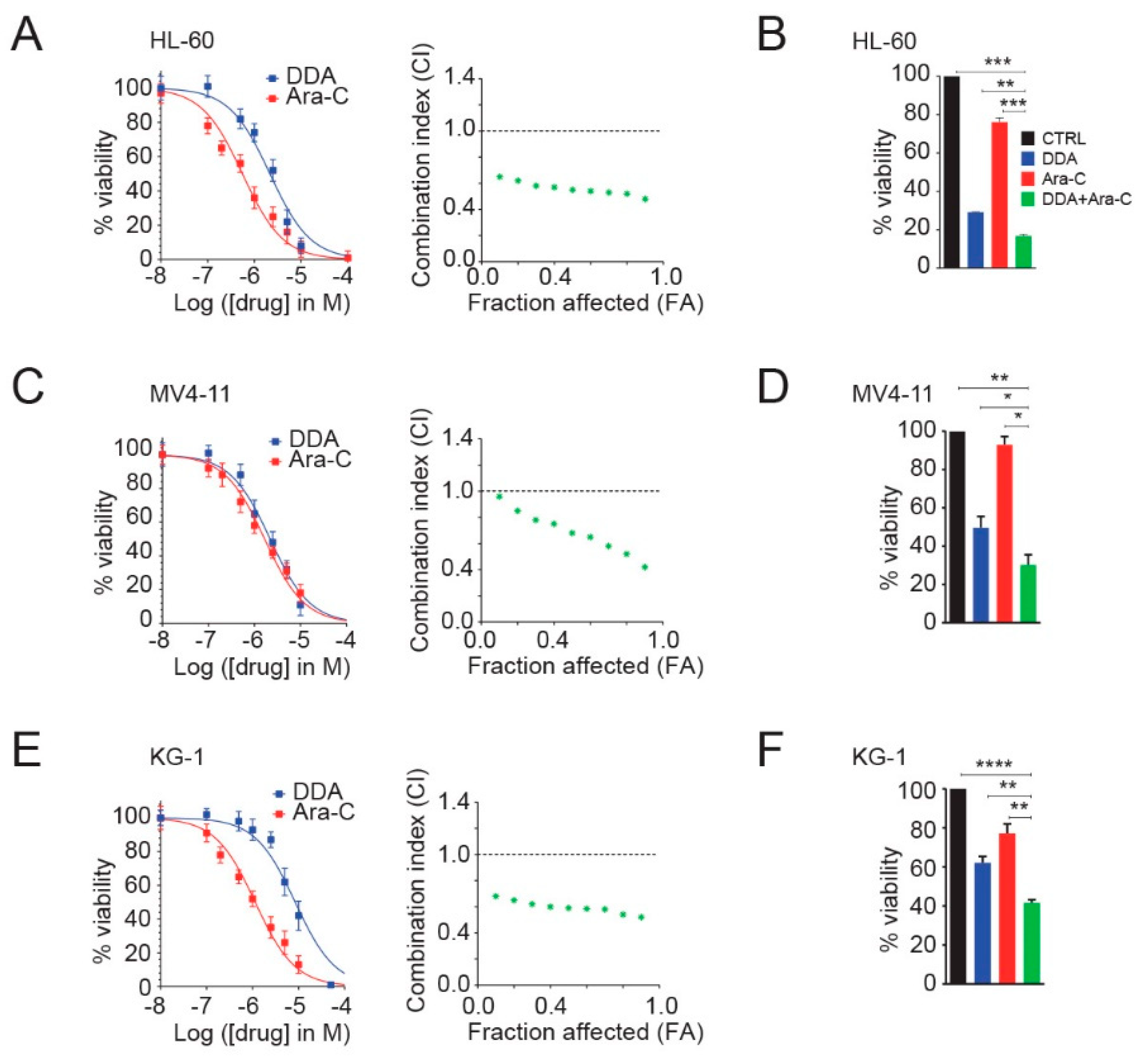
3.1. DDA Potentiates Ara-C Cytotoxicity in AML Cell Lines
3.2. Single and Combination DDA/Ara-C Treatments Induce Characteristics of Autophagy in AML Cell Lines
3.3. Autophagy Is Responsible for the Potentiation of Ara-C Cytotoxicity by DDA in AML Cell Lines
3.4. DDA Drives the Potentiation of Ara-C Cytotoxicity in AML Cell Lines in A LXRβ-Dependent Manner
3.5. DDA Potentiates Ara-C Cytotoxicity in Primary AML from Patients
3.6. DDA Potentiates the Anti-Leukemia Effects of Ara-C In Vivo in AML Cell Lines and Patient-Derived AML Xenografted in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Dendrogenin A: A Mammalian Metabolite of Cholesterol with Tumor Suppressor and Neurostimulating Properties. Curr. Med. Chem. 2015, 22, 3533–3549. [Google Scholar] [CrossRef] [PubMed]
- Silvente-Poirot, S.; Poirot, M. Cancer. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Voisin, M.; Mhamdi, L.; Dalenc, F.; Lacroix-Triki, M.; Filleron, T.; Pont, F.; Saati, T.A.; et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat. Commun. 2013, 4, 1840. [Google Scholar] [CrossRef] [Green Version]
- Silvente-Poirot, S.; Poirot, M. Cholesterol epoxide hydrolase and cancer. Curr. Opin. Pharmacol. 2012, 12, 696–703. [Google Scholar] [CrossRef]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef] [Green Version]
- Porter, F.D.; Herman, G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011, 52, 6–34. [Google Scholar] [CrossRef] [Green Version]
- Bauriaud-Mallet, M.; Vija-Racaru, L.; Brillouet, S.; Mallinger, A.; de Medina, P.; Rives, A.; Payre, B.; Poirot, M.; Courbon, F.; Silvente-Poirot, S. The cholesterol-derived metabolite dendrogenin A functionally reprograms breast adenocarcinoma and undifferentiated thyroid cancer cells. J. Steroid Biochem. Mol. Biol. 2019, 192, 105390. [Google Scholar] [CrossRef]
- Khalifa, S.A.; de Medina, P.; Erlandsson, A.; El-Seedi, H.R.; Silvente-Poirot, S.; Poirot, M. The novel steroidal alkaloids dendrogenin A and B promote proliferation of adult neural stem cells. Biochem. Biophys. Res. Commun. 2014, 446, 681–686. [Google Scholar] [CrossRef]
- de Medina, P.; Paillasse, M.R.; Payre, B.; Silvente-Poirot, S.; Poirot, M. Synthesis of new alkylaminooxysterols with potent cell differentiating activities: Identification of leads for the treatment of cancer and neurodegenerative diseases. J. Med. Chem. 2009, 52, 7765–7777. [Google Scholar] [CrossRef]
- Soules, R.; Audouard-Combe, F.; Huc-Claustre, E.; de Medina, P.; Rives, A.; Chatelut, E.; Dalenc, F.; Franchet, C.; Silvente-Poirot, S.; Poirot, M.; et al. A fast UPLC-HILIC method for an accurate quanti fi cation of dendrogenin A in human tissues. J. Steroid Biochem. Mol. Biol. 2019, 194, 105447. [Google Scholar] [CrossRef] [PubMed]
- Silvente-Poirot, S.; Segala, G.; Poirot, M.C.; Poirot, M. Ligand-dependent transcriptional induction of lethal autophagy: A new perspective for cancer treatment. Autophagy 2018, 14, 555–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvente-Poirot, S.; Dalenc, F.; Poirot, M. The Effects of Cholesterol-Derived Oncometabolites on Nuclear Receptor Function in Cancer. Cancer Res. 2018, 78, 4803–4808. [Google Scholar] [CrossRef] [Green Version]
- Poirot, M.; Silvente-Poirot, S. The tumor-suppressor cholesterol metabolite, dendrogenin A, is a new class of LXR modulator activating lethal autophagy in cancers. Biochem. Pharmacol. 2018, 153, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Freire, P.R.; Conneely, O.M. NR4A1 and NR4A3 restrict HSC proliferation via reciprocal regulation of C/EBPalpha and inflammatory signaling. Blood 2018, 131, 1081–1093. [Google Scholar] [CrossRef]
- Mullican, S.E.; Zhang, S.; Konopleva, M.; Ruvolo, V.; Andreeff, M.; Milbrandt, J.; Conneely, O.M. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat. Med. 2007, 13, 730–735. [Google Scholar] [CrossRef]
- Leignadier, J.; Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem. Pharmacol. 2017, 144, 18–28. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Tamamyan, G.; Kadia, T.; Ravandi, F.; Borthakur, G.; Cortes, J.; Jabbour, E.; Daver, N.; Ohanian, M.; Kantarjian, H.; Konopleva, M. Frontline treatment of acute myeloid leukemia in adults. Crit. Rev. Oncol. Hematol. 2017, 110, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Pettit, K.; Odenike, O. Defining and Treating Older Adults with Acute Myeloid Leukemia Who Are Ineligible for Intensive Therapies. Front. Oncol. 2015, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.S. Treating acute myeloid leukemia in older adults. Hematology 2014, 2014, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug therapy for acute myeloid leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Bhat, P.; Kriel, J.; Shubha Priya, B.; Basappa; Shivananju, N.S.; Loos, B. Modulating autophagy in cancer therapy: Advancements and challenges for cancer cell death sensitization. Biochem. Pharmacol. 2018, 147, 170–182. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Lamaziere, A.; Richard, D.; Bausero, P.; Barbe, U.; Kefi, K.; Wolf, C.; Visioli, F. Comparison of docosahexaenoic acid uptake in murine cardiomyocyte culture and tissue: Significance to physiologically relevant studies. Prostaglandins Leukot. Essent. Fatty Acids 2015, 94, 49–54. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C.; Quinn, P.J. Lipidomics in diagnosis of lipidoses. Subcell. Biochem. 2008, 49, 567–588. [Google Scholar] [CrossRef]
- Zhu, L.; Bakovic, M. Liver X Receptor Agonists Inhibit the Phospholipid Regulatory Gene CTP: Phosphoethanolamine Cytidylyltransferase-Pcyt2. Res. Lett. Biochem. 2008, 2008, 801849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh-oka, K.; Nakatogawa, H.; Ohsumi, Y. Physiological pH and acidic phospholipids contribute to substrate specificity in lipidation of Atg8. J. Biol. Chem. 2008, 283, 21847–21852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Oh-oka, K.; Ohsumi, Y. Lipidation of Atg8: How is substrate specificity determined without a canonical E3 enzyme? Autophagy 2008, 4, 911–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovic, D.; Fleer, E.A.; Breass, J.; Pfortner, J.; Schleyer, E.; Hiddemann, W. The influence of 1-beta-D-arabinofuranosylcytosine on the metabolism of phosphatidylcholine in human leukemic HL 60 and Raji cells. Leukemia 1997, 11, 2079–2086. [Google Scholar] [CrossRef]
- Kucera, G.L.; Capizzi, R.L. 1-beta-D-arabinofuranosylcytosine-diphosphate-choline is formed by the reversal of cholinephosphotransferase and not via cytidylyltransferase. Cancer Res. 1992, 52, 3886–3891. [Google Scholar]
- Daly, P.F.; Zugmaier, G.; Sandler, D.; Carpen, M.; Myers, C.E.; Cohen, J.S. Regulation of the cytidine phospholipid pathways in human cancer cells and effects of 1-beta-D-arabinofuranosylcytosine: A noninvasive 31P nuclear magnetic resonance study. Cancer Res. 1990, 50, 552–557. [Google Scholar]
- Lin, C.Y.; Gustafsson, J.A. Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 2015, 15, 216–224. [Google Scholar] [CrossRef]
- Bovenga, F.; Sabba, C.; Moschetta, A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015, 21, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Banker, D.E.; Mayer, S.J.; Li, H.Y.; Willman, C.L.; Appelbaum, F.R.; Zager, R.A. Cholesterol synthesis and import contribute to protective cholesterol increments in acute myeloid leukemia cells. Blood 2004, 104, 1816–1824. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; Appelbaum, F.R.; Willman, C.L.; Zager, R.A.; Banker, D.E. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003, 101, 3628–3634. [Google Scholar] [CrossRef] [Green Version]
- Inbar, M.; Shinitzky, M. Cholesterol as a bioregulator in the development and inhibition of leukemia. Proc. Natl. Acad. Sci. USA 1974, 71, 4229–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, P.V.; Glantz, S.T.; Scotland, S.; Kasner, M.T.; Carroll, M. Induced differentiation of acute myeloid leukemia cells by activation of retinoid X and liver X receptors. Leukemia 2014, 28, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- Calzada, E.; Onguka, O.; Claypool, S.M. Phosphatidylethanolamine Metabolism in Health and Disease. Int. Rev. Cell Mol. Biol. 2016, 321, 29–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Girardi, J.P.; Bakovic, M. Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy. Int. J. Cell Biol. 2012, 2012, 931956. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serhan, N.; Mouchel, P.-L.; de Medina, P.; Segala, G.; Mougel, A.; Saland, E.; Rives, A.; Lamaziere, A.; Despres, G.; Sarry, J.-E.; et al. Dendrogenin A Synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo. Cancers 2020, 12, 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071725
Serhan N, Mouchel P-L, de Medina P, Segala G, Mougel A, Saland E, Rives A, Lamaziere A, Despres G, Sarry J-E, et al. Dendrogenin A Synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo. Cancers. 2020; 12(7):1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071725
Chicago/Turabian StyleSerhan, Nizar, Pierre-Luc Mouchel, Philippe de Medina, Gregory Segala, Aurélie Mougel, Estelle Saland, Arnaud Rives, Antonin Lamaziere, Gaëtan Despres, Jean-Emmanuel Sarry, and et al. 2020. "Dendrogenin A Synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo" Cancers 12, no. 7: 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071725