Immunotherapy for Hepatocellular Carcinoma: A 2021 Update

 ,
,  , , ,
, , ,  , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma
2.1. Nivolumab
2.2. Pembrolizumab
2.3. Atezolizumab
2.4. Tremelimumab
3. Vaccine Therapy in Hepatocellular Carcinoma
3.1. Alpha-Fetoprotein (AFP) Peptide
3.2. Glypican-3 (GPC3)
3.3. Multidrug Resistance-Associated Protein 3 (MRP3)
3.4. NY-ESO-1 and MAGE-A
3.5. Dendritic Cell Vaccine
3.6. Oncolytic Viruses
4. Adoptive Cell Transfer in Hepatocellular Carcinoma
4.1. CIK Cells
4.2. Chimeric Antigen Receptor T Cells (CAR-T)
5. Combinations Strategies of Immunotherapies
6. Hepatitis Infection and Immunotherapy
7. Predictive Biomarkers in HCC Immunotherapy
8. Conclusions
Funding
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Chen, Q.; Chhatwal, J.; Hoshida, Y. Changing epidemiology of hepatocellular carcinoma and role of surveillance. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Humana Press Cham: Totowa, NJ, USA, 2019; pp. 53–67. [Google Scholar] [CrossRef]
- World Health Organization. Projections of Mortality and Causes of Death, 2016 to 2060. Available online: http://www.who.int/healthinfo/global_burden_disease/projections/en/ (accessed on 9 November 2018).
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Muralidharan, P.; Raj, J.P. Update in global trends and aetiology of hepatocellular carcinoma. Współczesna Onkologia 2018, 22, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Buonaguro, L.; Mauriello, A.; Cavalluzzo, B.; Petrizzo, A.; Tagliamonte, M. Immunotherapy in hepatocellular carcinoma. Ann. Hepatol. 2019, 18, 291–297. [Google Scholar] [CrossRef]
- Schildberg, F.A.; Hegenbarth, S.I.; Schumak, B.; Scholz, K.; Limmer, A.; Knolle, P.A. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur. J. Immunol. 2008, 38, 957–967. [Google Scholar] [CrossRef]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef] [Green Version]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular carcinomas originate predominantly from hepatocytes and benign lesions from hepatic progenitor cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-D.; Song, G.-W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association between expression level of PD1 by tumor-infiltrating CD8+ T cells and features of hepatocellular carcinoma. Gastroenterology 2018, 155, 1936–1950.e1917. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; De Moura, M.C.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.K.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.I.; Jeong, D.; Ji, S.; Ahn, T.S.; Bae, S.H.; Chin, S.; Chung, J.C.; Kim, H.C.; Lee, M.S.; Baek, M.-J. Overexpression of PD-L1 and PD-L2 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. Treat. 2017, 49, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 2018, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- De Galarreta, M.R.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehí, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Sachdeva, M.; Arora, S.K. Prognostic role of immune cells in hepatocellular carcinoma. EXCLI J. 2020, 19, 718–733. [Google Scholar] [CrossRef]
- Kapanadze, T.; Gamrekelashvili, J.; Ma, C.; Chan, C.; Zhao, F.; Hewitt, S.; Zender, L.; Kapoor, V.; Felsher, D.W.; Manns, M.P.; et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J. Hepatol. 2013, 59, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2017, 67, 931–944. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J. The role of MDSCs in hepatocellular carcinoma—In vivo veritas? J. Hepatol. 2013, 59, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Z.; Wang, L.; Tian, G.; Tian, J.; Yang, Z.; Cao, G.; Zhou, H.; Zhao, L.; Wu, Z.; et al. Critical role of myeloid-derived suppressor cells in tumor-induced liver immune suppression through inhibition of NKT cell function. Front. Immunol. 2017, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.-Y.; Li, L.; Zheng, L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.-H.; Levy, L.; Zolotriov, L.; Granot, Z.; Fridlender, Z.G. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, e1356965. [Google Scholar] [CrossRef]
- Sharma, S.; Khosla, R.; David, P.; Rastogi, A.; Vyas, A.; Singh, D.; Bhardwaj, A.; Sahney, A.; Maiwall, R.; Sarin, S.K.; et al. CD4+CD25+CD127low regulatory T cells play predominant anti-tumor suppressive role in hepatitis B virus-associated hepatocellular carcinoma. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Arii, S.; Yamaoka, Y.; Futagawa, S.; Inoue, K.; Kobayashi, K.; Kojiro, M.; Makuuchi, M.; Nakamura, Y.; Okita, K.; Yamada, R. Results of surgical and nonsurgical treatment for small-sized hepatocellular carcinomas: A retrospective and nationwide survey in Japan. Hepatology 2000, 32, 1224–1229. [Google Scholar] [CrossRef]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Golabi, P.; Fazel, S.; Otgonsuren, M.; Sayiner, M.; Locklear, C.T.; Younossi, Z.M. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine 2017, 96, e5904. [Google Scholar] [CrossRef] [PubMed]
- Büttner, N.; Schmidt, N.; Thimme, R. Perspectives of immunotherapy in hepatocellular carcinoma (HCC). Zeitschrift für Gastroenterologie 2016, 54, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Mylonas, K.S.; Katsaros, I.; Zhao, M.; A Ajani, J.; Psyrri, A.; Karamouzis, M.V.; Liakakos, T. Immunotherapy for esophageal cancer: A 2019 update. Immunotherapy 2020, 12, 203–218. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Herzberg, B.; Campo, M.J.; Gainor, J. Immune checkpoint inhibitors in non-small cell lung cancer. Oncology 2017, 22, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41, S3–S13. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Wang, B.-J.; Bao, J.-J.; Wang, J.-Z.; Wang, Y.; Jiang, M.; Xing, M.-Y.; Zhang, W.-G.; Qi, J.-Y.; Roggendorf, M.; Lu, M.-J.; et al. Immunostaining of PD-1/PD-Ls in liver tissues of patients with hepatitis and hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Decade in review-cancer immunotherapy: Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol. 2014, 11, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Brower, V. Checkpoint blockade immunotherapy for cancer comes of age. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, F.; Vacchelli, E.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide vaccines in cancer therapy. Oncoimmunology 2013, 2, e26621. [Google Scholar] [CrossRef] [Green Version]
- Vacchelli, E.; Martins, I.; Eggermont, A.; Fridman, W.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide vaccines in cancer therapy. Oncoimmunology 2012, 1, 1557–1576. [Google Scholar] [CrossRef] [Green Version]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The potential of CAR T cell therapy in pancreatic cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y.-H. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2017, 32, 317–326. [Google Scholar] [CrossRef]
- Greten, T.F.; Sangro, B. Targets for immunotherapy of liver cancer. J. Hepatol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Xing, N.; Luan, L.; Xu, H.; Sharma, R.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.-C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.; Finn, R.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs. sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Sharp, M.; Corp., D. Study of Pembrolizumab (MK-3475) vs. Best Supportive Care in Participants with Previously Systemically Treated Advanced Hepatocellular Carcinoma (MK-3475-240/KEYNOTE-240). 2016. Available online: https://clinicaltrials.gov/show/NCT02702401 (accessed on 11 June 2020).
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Randomised efficacy and safety results for atezolizumab (Atezo) + bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, v875. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; De La Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.-I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; Elgindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Tsoris, A.; Marlar, C.A. Use of the Child Pugh Score in Liver Disease; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- The Clatterbridge Cancer Centre NHS Foundation Trust. Nivolumab in Combination with TACE/TAE for Patients with Intermediate Stage HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT04268888 (accessed on 16 July 2020).
- Eli Lilly and Company; Squibb, B.-M. A Study of Galunisertib (LY2157299) in Combination with Nivolumab in Advanced Refractory Solid Tumors and in Recurrent or Refractory NSCLC, or Hepatocellular Carcinoma. 2015. Available online: https://clinicaltrials.gov/show/NCT02423343 (accessed on 5 August 2020).
- Suzhou Kintor Pharmaceutical Inc. Combination of GT90001 and Nivolumab in Patients with Metastatic Hepatocellular Carcinoma (HCC). 2019. Available online: https://clinicaltrials.gov/show/NCT03893695 (accessed on 14 September 2020).
- SignalRX Pharmaceuticals, Inc.; University of California, San Diego. Phase 1 Study of SF1126 in Combination with Nivolumab in Patients with Advanced Hepatocellular Carcinoma. 2017. Available online: https://clinicaltrials.gov/show/NCT03059147 (accessed on 14 August 2019).
- Bristol-Myers Squibb; Ono Pharmaceutical Co. Ltd. A Study of Nivolumab in Participants with Hepatocellular Carcinoma Who Are at High Risk of Recurrence after Curative Hepatic Resection or Ablation. 2017. Available online: https://clinicaltrials.gov/show/NCT03383458 (accessed on 28 August 2020).
- AIO-Studien-gGmbH; Bristol-Myers Squibb. Transarterial Chemoembolization in Combination with Nivolumab Performed for Intermediate Stage Hepatocellular Carcinoma. 2018. Available online: https://clinicaltrials.gov/show/NCT03572582 (accessed on 14 July 2020).
- Merck Sharp & Dohme Corp. Study of Pembrolizumab (MK-3475) or Placebo Given With Best Supportive Care in Asian Participants with Previously Treated Advanced Hepatocellular Carcinoma (MK-3475-394/KEYNOTE-394). 2017. Available online: https://clinicaltrials.gov/show/NCT03062358 (accessed on 27 January 2020).
- Roswell Park Cancer Institute; National Cancer Institute (NCI); Merck Sharp & Dohme Corp. Sorafenib Tosylate and Pembrolizumab in Treating Patients with Advanced or Metastatic Liver Cancer. 2017. Available online: https://clinicaltrials.gov/show/NCT03211416 (accessed on 3 September 2020).
- Merck Sharp & Dohme Corp.; Eisai Inc. Safety and Efficacy of Lenvatinib (E7080/MK-7902) in Combination with Pembrolizumab (MK-3475) Versus Lenvatinib as First-Line Therapy in Participants with Advanced Hepatocellular Carcinoma (MK-7902-002/E7080-G000-311/LEAP-002). 2018. Available online: https://clinicaltrials.gov/show/NCT03713593 (accessed on 10 April 2020).
- Institut für Klinische Krebsforschung IKF GmbH at Krankenhaus Nordwest. IMMULAB-Immunotherapy with Pembrolizumab in Combination with Local Ablation in Hepatocellular Carcinoma (HCC). 2019. Available online: https://clinicaltrials.gov/show/NCT03753659 (accessed on 28 May 2020).
- University Health Network, Toronto. Study of Pembrolizumab and Radiotherapy in Liver Cancer. 2018. Available online: https://clinicaltrials.gov/show/NCT03316872 (accessed on 17 December 2019).
- Autumn McRee, M.D.; Merck Sharp & Dohme Corp.; Hoosier Cancer Research Network. Pembrolizumab Plus Y90 Radioembolization in HCC Subjects. 2017. Available online: https://clinicaltrials.gov/show/NCT03099564 (accessed on 15 October 2019).
- Sun Yat-sen University. Anti-PD-1therapy Combined with Thermal Ablation for Advanced HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT03939975 (accessed on 19 August 2019).
- Kelley, R.K.; Oliver, J.W.; Hazra, S.; Benzaghou, F.; Yau, T.; Cheng, A.-L.; Rimassa, L. Cabozantinib in combination with atezolizumab versus sorafenib in treatment-naive advanced hepatocellular carcinoma: COSMIC-312 Phase III study design. Future Oncol. 2020, 16, 1525–1536. [Google Scholar] [CrossRef]
- Exelixis. Study of Cabozantinib in Combination with Atezolizumab versus Sorafenib in Subjects with Advanced HCC Who Have Not Received Previous Systemic Anticancer Therapy. 2018. Available online: https://clinicaltrials.gov/show/NCT03755791 (accessed on 25 September 2020).
- Roche, H.-L. A Study of Atezolizumab Plus Bevacizumab Versus Active Surveillance as Adjuvant Therapy in Patients with Hepatocellular Carcinoma at High Risk of Recurrence After Surgical Resection or Ablation. 2019. Available online: https://clinicaltrials.gov/show/NCT04102098 (accessed on 29 September 2020).
- Ludwig-Maximilians-University of Munich. Atezolizumab/Bevacizumab Followed by On-Demand TACE or Initial Synchronous Treatment with TACE and Atezolizumab/Bevacizumab. 2020. Available online: https://clinicaltrials.gov/show/NCT04224636 (accessed on 8 April 2020).
- AVEO Pharmaceuticals, Inc.; AstraZeneca. A Study of Tivozanib in Combination with Durvalumab in Subjects with Untreated Advanced Hepatocellular Carcinoma. 2019. Available online: https://clinicaltrials.gov/show/NCT03970616 (accessed on 14 September 2020).
- BeiGene. Phase 3 Study of Tislelizumab Versus Sorafenib in Participants With Unresectable HCC. 2017. Available online: https://clinicaltrials.gov/show/NCT03412773 (accessed on 29 June 2020).
- Jiangsu HengRui Medicine Co., Ltd. A Study to Evaluate SHR-1210 in Subjects with Advanced HCC. 2016. Available online: https://clinicaltrials.gov/show/NCT02989922 (accessed on 27 December 2017).
- AstraZeneca. Assess Efficacy and Safety of Durvalumab Alone or Combined with Bevacizumab in High Risk of Recurrence HCC Patients after Curative Treatment. 2019. Available online: https://clinicaltrials.gov/show/NCT03847428 (accessed on 25 September 2020).
- Seoul National University Hospital. Safety and Efficacy Study of Radioembolization in Combination with Durvalumab in Locally Advanced and Unresectable HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT04124991 (accessed on 18 October 2019).
- The First Affiliated Hospital with Nanjing Medical University. SHR-1210 Combined with Apatinib Mesylate in the Perioperative Treatment of Hepatocellular Carcinoma. 2019. Available online: https://clinicaltrials.gov/show/NCT04297202 (accessed on 27 August 2020).
- Institut Bergonié; Bayer; Merck KGaA. A Phase I/II Study of Regorafenib Plus Avelumab in Solid Tumors. 2018. Available online: https://clinicaltrials.gov/show/NCT03475953 (accessed on 31 July 2020).
- Sun Yat-sen University; Innovent Biologics, Inc. TAI Combined with PD-1 Inhibitor in Locally Advanced, Potentially Resectable HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT03869034 (accessed on 27 August 2020).
- Asan Medical Center; Samsung Medical Center; Bundang CHA Hospital. Combination of Regorafenib and Nivolumab in Unresectable Hepatocellular Carcinoma. 2020. Available online: https://clinicaltrials.gov/show/NCT04310709 (accessed on 18 June 2020).
- Tianjin Medical University Cancer Institute and Hospital. PD-1 Monoclonal Antibody, Lenvatinib and TACE in the Treatment of HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT04273100 (accessed on 17 February 2020).
- Shanghai Zhongshan Hospital. Stereotactic Body Radiation Therapy Combined with Anti-PD-1 Antibody in Patients with Hepatocellular Carcinoma. 2019. Available online: https://clinicaltrials.gov/show/NCT03857815 (accessed on 19 March 2019).
- Tremelimumab. Drugs R D 2010, 10, 123–132. [CrossRef]
- Kumar, J.; Habib, N.A.; Huang, K.-W.; Podda, M.; Warwick, J.; Arasaradnam, R. Immunological basis of genesis of hepatocellular carcinoma: Unique challenges and potential opportunities through immunomodulation. Vaccines 2020, 8, 247. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: Immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, Y.; Yoshikawa, T.; Ofuji, K.; Yoshimura, M.; Tsuchiya, N.; Takahashi, M.; Nobuoka, D.; Gotohda, N.; Takahashi, S.; Kato, Y.; et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology 2016, 5, e1129483. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Nakagawa, H.; Kitahara, M.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Iida, N.; Fushimi, K.; Kaneko, S. Phase I trial of multidrug resistance-associated protein 3-derived peptide in patients with hepatocellular carcinoma. Cancer Lett. 2015, 369, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.H.; Midgley, R.S.; Mirza, N.; Torr, E.E.; Ahmed, F.; Steele, J.C.; Steven, N.M.; Kerr, D.J.; Young, L.S.; Adams, D.H. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 2009, 49, 124–132. [Google Scholar] [CrossRef]
- Rizell, M.; Sternby Eilard, M.; Andersson, M.; Andersson, B.; Karlsson-Parra, A.; Suenaert, P. Phase 1 trial with the cell-based immune primer ilixadencel, alone, and combined with sorafenib, in advanced hepatocellular carcinoma. Front. Oncol. 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-based oncolytic immunotherapy pexastimogene devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: A randomized multicenter Phase IIb trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.J. Role of alpha-fetoprotein in the diagnosis and management of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1999, 14, S32–S36. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Meng, W.S.; Koh, A.; Vollmer, C.M.; Ribas, A.; Dissette, V.B.; Faull, K.; Glaspy, J.A.; McBride, W.H.; Economou, J.S. T cell responses to HLA-A*0201-restricted peptides derived from human alpha fetoprotein. J. Immunol. 2001, 166, 5300–5308. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, L.H.; Koh, A.; Meng, W.; Vollmer, C.M.; Ribas, A.; Dissette, V.; Lee, E.; Glaspy, J.A.; McBride, W.H.; Economou, J.S. Generation of human T-cell responses to an HLA-A2.1-restricted peptide epitope derived from alpha-fetoprotein. Cancer Res. 1999, 59, 3134–3142. [Google Scholar] [PubMed]
- Butterfield, L.H.; Economou, J.S.; Gamblin, T.C.; Geller, D.A. Alpha fetoprotein DNA prime and adenovirus boost immunization of two hepatocellular cancer patients. J. Transl. Med. 2014, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A new target for diagnosis and treatment of hepatocellular carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, Y.; Suzuki, H.; Hirohashi, T.; Tyson, C.A.; Sugiyama, Y. cDNA cloning and inducible expression of human multidrug resistance associated protein 3 (MRP3). FEBS Lett. 1998, 433, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Mizukoshi, E.; Honda, M.; Arai, K.; Yamashita, T.; Nakamoto, Y.; Kaneko, S. Expression of multidrug resistance-associated protein 3 and cytotoxic T cell responses in patients with hepatocellular carcinoma. J. Hepatol. 2008, 49, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T.; Tanaka, H.; Fujimoto, S.; Kimura, T.; Okamoto, K.; Miyamoto, H.; Muguruma, N.; et al. MRP3 as a novel resistance factor for sorafenib in hepatocellular carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Terashima, T.; Yamashita, T.; Mizukoshi, E.; Sakai, A.; Nakamoto, Y.; Honda, M.; et al. Randomized, phase II study comparing interferon combined with hepatic arterial infusion of fluorouracil plus cisplatin and fluorouracil alone in patients with advanced hepatocellular carcinoma. Oncology 2011, 81, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Kerkar, S.P.; Wang, Z.F.; Lasota, J.; Park, T.; Patel, K.; Groh, E.; Rosenberg, S.A.; Miettinen, M.M. MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J. Immunother. 2016, 39, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Roch, N.; Kutup, A.; Vashist, Y.; Yekebas, E.; Kalinin, V.; Izbicki, J.R. Coexpression of MAGE-A peptides and HLA class I molecules in hepatocellular carcinoma. Anticancer Res. 2010, 30, 1617–1623. [Google Scholar]
- Zerbini, A.; Pilli, M.; Soliani, P.; Ziegler, S.; Pelosi, G.; Orlandini, A.; Cavallo, C.; Uggeri, J.; Scandroglio, R.; Crafa, P.; et al. Ex vivo characterization of tumor-derived melanoma antigen encoding gene-specific CD8+cells in patients with hepatocellular carcinoma. J. Hepatol. 2004, 40, 102–109. [Google Scholar] [CrossRef]
- Palucka, K.; Ueno, H.; Fay, J.; Banchereau, J. Dendritic cells and immunity against cancer. J. Intern. Med. 2011, 269, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Luan, W.; Warren, L.; Kadri, H.; Kim, K.W.; Goz, V.; Blank, S.; Isabel Fiel, M.; Hiotis, S.P. Autologous tumor cell lysate-loaded dendritic cell vaccine inhibited tumor progression in an orthotopic murine model for hepatocellular carcinoma. Ann. Surg. Oncol. 2016, 23, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J. Hepatol. 2017, 67, 739–748. [Google Scholar] [CrossRef]
- Iwashita, Y.; Tahara, K.; Goto, S.; Sasaki, A.; Kai, S.; Seike, M.; Chen, C.L.; Kawano, K.; Kitano, S. A phase I study of autologous dendritic cell-based immunotherapy for patients with unresectable primary liver cancer. Cancer Immunol. Immunother. 2003, 52, 155–161. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Nakamoto, Y.; Arai, K.; Yamashita, T.; Mukaida, N.; Matsushima, K.; Matsui, O.; Kaneko, S. Enhancement of tumor-specific T-cell responses by transcatheter arterial embolization with dendritic cell infusion for hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2164–2174. [Google Scholar] [CrossRef]
- FullHope Biomedical Co., Ltd.; Taipei Veterans General Hospital, Taiwan. Dendritic Killer Cell-Based Immunotherapy for Solid Tumors. 2014. Available online: https://clinicaltrials.gov/show/NCT02882659 (accessed on 30 August 2016).
- Guangxi Medical University. A Study of DC-CIK to Treat Hepatocellular Carcinoma. 2013. Available online: https://clinicaltrials.gov/show/NCT01821482 (accessed on 12 April 2013).
- Second Military Medical University. Immunotherapy Using Precision T Cells Specific to Multiple Common Tumor-Associated Antigen Combined with Transcatheter Arterial Chemoembolization for the Treatment of Advanced Hepatocellular Carcinoma. 2015. Available online: https://clinicaltrials.gov/show/NCT02638857 (accessed on 1 January 2016).
- Chinese PLA General Hospital; Likang Life Sciences Holdings Limited. A Study Combining Personalized Neoantigen-Based Dendritic Cell Vaccine with Microwave Ablation for the Treatment of Hepatocellular Carcinoma. 2018. Available online: https://clinicaltrials.gov/show/NCT03674073 (accessed on 18 October 2018).
- Kirn, D.; Martuza, R.L.; Zwiebel, J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001, 7, 781–787. [Google Scholar] [CrossRef]
- Liu, T.C.; Galanis, E.; Kirn, D. Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat. Clin. Pract. Oncol. 2007, 4, 101–117. [Google Scholar] [CrossRef]
- SillaJen, Inc. Hepatocellular Carcinoma Study Comparing Vaccinia Virus Based Immunotherapy Plus Sorafenib vs. Sorafenib Alone. 2015. Available online: https://clinicaltrials.gov/show/NCT02562755 (accessed on 12 August 2019).
- Li, S.; Yang, F.; Ren, X. Immunotherapy for hepatocellular carcinoma. Drug Discov. Ther. 2015, 9, 363–371. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, C.; Lu, B. Cytokine-induced killer cells promote antitumor immunity. J. Transl. Med. 2013, 11, 83. [Google Scholar] [CrossRef] [Green Version]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Hiramatsu, N.; Sakamori, R.; Yamaguchi, S.; Hayashi, N. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J. Hepatol. 2005, 43, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, A.; Pilli, M.; Laccabue, D.; Pelosi, G.; Molinari, A.; Negri, E.; Cerioni, S.; Fagnoni, F.; Soliani, P.; Ferrari, C.; et al. Radiofrequency thermal ablation for hepatocellular carcinoma stimulates autologous NK-cell response. Gastroenterology 2010, 138, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Seigo Nishida; Florida Department of Health; Seigo Nishida, University of Miami. Safety Study of Liver Natural Killer Cell Therapy for Hepatoma Liver Transplantation. 2010. Available online: https://clinicaltrials.gov/show/NCT01147380 (accessed on 10 March 2016).
- Samsung Medical Center. To Evaluate the Efficacy and Safety of MG4101 (Ex Vivo Expanded Allogeneic NK Cell). 2014. Available online: https://clinicaltrials.gov/show/NCT02008929 (accessed on 3 December 2015).
- Jiang, S.S.; Tang, Y.; Zhang, Y.J.; Weng, D.S.; Zhou, Z.G.; Pan, K.; Pan, Q.Z.; Wang, Q.J.; Liu, Q.; He, J.; et al. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget 2015, 6, 41339–41349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, D.; Qiang, L.; Jian, W.; Ti, Z.; Da-Lu, K. A randomized, controlled trial of postoperative adjuvant cytokine-induced killer cells immunotherapy after radical resection of hepatocellular carcinoma. Dig. Liver Dis. 2009, 41, 36–41. [Google Scholar] [CrossRef]
- Pan, K.; Li, Y.Q.; Wang, W.; Xu, L.; Zhang, Y.J.; Zheng, H.X.; Zhao, J.J.; Qiu, H.J.; Weng, D.S.; Li, J.J.; et al. The efficacy of cytokine-induced killer cell infusion as an adjuvant therapy for postoperative hepatocellular carcinoma patients. Ann. Surg. Oncol. 2013, 20, 4305–4311. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Kim, Y.; Shuang, Z.Y.; Zhang, Y.J.; Lao, X.M.; Li, Y.Q.; Chen, M.S.; Pawlik, T.M.; Xia, J.C.; et al. A randomized controlled trial on patients with or without adjuvant autologous cytokine-induced killer cells after curative resection for hepatocellular carcinoma. Oncoimmunology 2016, 5, e1083671. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.S.; Liu, M.X.; Zhang, B.; Shi, M.; Lei, Z.Y.; Sun, W.B.; Du, Q.Y.; Chen, J.M. Antitumor activities of human autologous cytokine-induced killer (CIK) cells against hepatocellular carcinoma cells in vitro and in vivo. World J. Gastroenterol. 2002, 8, 464–468. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391.e1386. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Xu, Y.C.; Tang, L.; Zhang, Z.; Wang, J.; Wang, H.X. Cytokine-induced killer (CIK) cell therapy for patients with hepatocellular carcinoma: Efficacy and safety. Exp. Hematol. Oncol. 2012, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.M.; Li, W.; Li, S.; Gao, F.; Zhou, Q.M.; Wu, F.M.; He, N.; Pan, C.C.; Xia, J.C.; Wu, P.H.; et al. Cytokine-induced killer cells in combination with transcatheter arterial chemoembolization and radiofrequency ablation for hepatocellular carcinoma patients. J. Immunother. 2013, 36, 287–293. [Google Scholar] [CrossRef]
- Weng, D.S.; Zhou, J.; Zhou, Q.M.; Zhao, M.; Wang, Q.J.; Huang, L.X.; Li, Y.Q.; Chen, S.P.; Wu, P.H.; Xia, J.C. Minimally invasive treatment combined with cytokine-induced killer cells therapy lower the short-term recurrence rates of hepatocellular carcinomas. J. Immunother. 2008, 31, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Song, B.G.; Lee, J.-H.; Lee, H.Y.; Kim, S.W.; Chang, Y.; Lee, Y.B.; Cho, E.J.; Yu, S.J.; Sinn, D.H.; et al. Adjuvant cytokine-induced killer cell immunotherapy for hepatocellular carcinoma: A propensity score-matched analysis of real-world data. BMC Cancer 2019, 19, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wen, J.; Yi, H.; Hou, X.; Yin, Y.; Ye, G.; Wu, X.; Jiang, X. Split chimeric antigen receptor-modified T cells targeting glypican-3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qin, W.; Liu, T.; Jiang, D.; Cui, L.; Liu, X.; Fang, Y.; Tang, X.; Jin, H.; Qian, Q. PiggyBac-engineered T cells expressing a glypican-3-specific chimeric antigen receptor show potent activities against hepatocellular carcinoma. Immunobiology 2020, 225. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Luo, H.; Shi, B.; Di, S.; Sun, R.; Su, J.; Liu, Y.; Li, H.; Jiang, H.; Li, Z. Combined antitumor effects of sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol. Ther. 2019, 27, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Carsgen Therapeutics, Ltd.; NanJing PLA 81 Hospital; First Affiliated Hospital of Zhejiang University; RenJi Hospital. Chimeric Antigen Receptor T Cells Targeting Glypican-3. 2019. Available online: https://clinicaltrials.gov/show/NCT03884751 (accessed on 15 October 2019).
- Baylor College of Medicine; Center for Cell and Gene Therapy, Baylor College of Medicine; The Methodist Hospital System. Glypican 3-Specific Chimeric Antigen Receptor Expressing T Cells for Hepatocellular Carcinoma (GLYCAR). 2019. Available online: https://clinicaltrials.gov/show/NCT02905188 (accessed on 10 July 2020).
- The Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School. GPC3-targeted CAR-T Cell for Treating GPC3 Positive Advanced HCC. 2019. Available online: https://clinicaltrials.gov/show/NCT04121273 (accessed on 11 October 2019).
- Zhejiang University; Carsgen Therapeutics, Ltd. 4th Generation Chimeric Antigen Receptor T Cells Targeting Glypican-3. 2019. Available online: https://clinicaltrials.gov/show/NCT03980288 (accessed on 15 October 2019).
- Second Affiliated Hospital of Guangzhou Medical University; Hunan Zhaotai Yongren Medical Innovation Co. Ltd.; Guangdong Zhaotai InVivo Biomedicine Co. Ltd.; First Affiliated Hospital, Sun Yat-Sen University. GPC3-T2-CAR-T Cells for Immunotherapy of Cancer With GPC3 Expression. 2017. Available online: https://clinicaltrials.gov/show/NCT03198546 (accessed on 26 November 2019).
- Baylor College of Medicine; The Methodist Hospital System. T Cells Co-Expressing a Second Generation Glypican 3-Specific Chimeric Antigen Receptor with Cytokines Interleukin-21 and 15 as Immunotherapy for Patients with Liver Cancer (TEGAR). 2020. Available online: https://clinicaltrials.gov/show/NCT04093648 (accessed on 20 May 2020).
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric antigen receptor-glypican-3 T-Cell therapy for advanced hepatocellular carcinoma: Results of phase 1 trials. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- RenJi Hospital. Anti-GPC3 CAR T for Treating Patients with Advanced HCC. 2015. Available online: https://clinicaltrials.gov/show/NCT02395250 (accessed on 28 August 2019).
- Fuda Cancer Hospital, Guangzhou. CAR-T Cell Immunotherapy for HCC Targeting GPC3. 2015. Available online: https://clinicaltrials.gov/show/NCT02723942 (accessed on 16 July 2020).
- PersonGen BioTherapeutics (Suzhou) Co., Ltd.; The First People’s Hospital of Hefei; Hefei Binhu Hospital. Phase I/II Study of Anti-Mucin1 (MUC1) CAR T Cells for Patients With MUC1+ Advanced Refractory Solid Tumor. 2015. Available online: https://clinicaltrials.gov/show/NCT02587689 (accessed on 5 December 2016).
- First Affiliated Hospital of Chengdu Medical College. A Clinical Research of CAR T Cells Targeting EpCAM Positive Cancer. 2017. Available online: https://clinicaltrials.gov/show/NCT03013712 (accessed on 6 January 2017).
- Takayama, T.; Sekine, T.; Makuuchi, M.; Yamasaki, S.; Kosuge, T.; Yamamoto, J.; Shimada, K.; Sakamoto, M.; Hirohashi, S.; Ohashi, Y.; et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: A randomised trial. Lancet 2000, 356, 802–807. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Gracián, A.C.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb; Ono Pharmaceutical Co. Ltd. An Immuno-Therapy Study to Evaluate the Effectiveness, Safety and Tolerability of Nivolumab or Nivolumab in Combination with Other Agents in Patients with Advanced Liver Cancer. 2012. Available online: https://clinicaltrials.gov/show/NCT01658878 (accessed on 30 June 2020).
- Imperial College London; Bristol-Myers Squibb. Safety and Bioactivity of Ipilimumab and Nivolumab Combination Prior to Liver Resection in Hepatocellular Carcinoma. 2019. Available online: https://clinicaltrials.gov/show/NCT03682276 (accessed on 11 June 2019).
- Bristol-Myers Squibb. A Study of Nivolumab in Combination with Ipilimumab in Participants with Advanced Hepatocellular Carcinoma. 2019. Available online: https://clinicaltrials.gov/show/NCT04039607 (accessed on 23 September 2020).
- National Health Research Institutes, Taiwan; National Taiwan University Hospital; Taipei Veterans General Hospital, Taiwan; National Cheng-Kung University Hospital; China Medical University Hospital; Chang Gung Memorial Hospital. Nivolumab Plus Ipilimumab as Neoadjuvant Therapy for Hepatocellular Carcinoma (HCC). 2018. Available online: https://clinicaltrials.gov/show/NCT03510871 (accessed on 27 April 2018).
- M.D. Anderson Cancer Center; National Cancer Institute (NCI). Nivolumab with or without Ipilimumab in Treating Patients with Resectable Liver Cancer. 2017. Available online: https://clinicaltrials.gov/show/NCT03222076 (accessed on 19 August 2020).
- Transgene. A Trial to Evaluate the Safety and Efficacy of the Combination of the Oncolytic Immunotherapy Pexa-Vec with the PD-1 Receptor Blocking Antibody Nivolumab in the First-Line Treatment of Advanced Hepatocellular Carcinoma (HCC). 2017. Available online: https://clinicaltrials.gov/show/NCT03071094 (accessed on 12 March 2020).
- City of Hope Medical Center; National Cancer Institute (NCI). Vaccine Therapy and Pembrolizumab in Treating Patients with Solid Tumors That Have Failed Prior Therapy. 2015. Available online: https://clinicaltrials.gov/show/NCT02432963 (accessed on 30 April 2019).
- Geneos Therapeutics. GNOS-PV02 Personalized Neoantigen Vaccine, INO-9012 and Pembrolizumab in Subjects With Advanced HCC. 2020. Available online: https://clinicaltrials.gov/show/NCT04251117 (accessed on 19 May 2020).
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; Bristol-Myers Squibb. DNAJB1-PRKACA Fusion Kinase Peptide Vaccine Combined with Nivolumab and Ipilimumab for Patients With Fibrolamellar Hepatocellular Carcinoma. 2020. Available online: https://clinicaltrials.gov/show/NCT04248569 (accessed on 13 April 2020).
- AstraZeneca. Study of Durvalumab and Tremelimumab as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma. 2017. Available online: https://clinicaltrials.gov/show/NCT03298451 (accessed on 27 August 2020).
- National Cancer Institute (NCI); National Institutes of Health Clinical Center (CC). A Pilot Study of Combined Immune Checkpoint Inhibition in Combination with Ablative Therapies in Subjects with Hepatocellular Carcinoma (HCC) or Biliary Tract Carcinomas (BTC). 2016. Available online: https://clinicaltrials.gov/show/NCT02821754 (accessed on 13 May 2020).
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Shimizu, K.; Kotera, Y.; Aruga, A.; Takeshita, N.; Katagiri, S.; Ariizumi, S.; Takahashi, Y.; Yoshitoshi, K.; Takasaki, K.; Yamamoto, M. Postoperative dendritic cell vaccine plus activated T-cell transfer improves the survival of patients with invasive hepatocellular carcinoma. Hum. Vaccin. Immunother. 2014, 10, 970–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.L.; Ji, Q.H.; An, J.J.; Masuda, S.; Tsuneyama, K. Clinicopathological analysis of CD8-positive lymphocytes in the tumor parenchyma and stroma of hepatocellular carcinoma. Oncol. Lett. 2014, 8, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.J.; Zhang, Y.; Jin, G.X.; Yao, L.; Wu, D.Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef]
- Sun, D.W.; An, L.; Huang, H.Y.; Sun, X.D.; Lv, G.Y. Establishing peripheral PD-L1 as a prognostic marker in hepatocellular carcinoma patients: How long will it come true? Clin. Transl. Oncol. 2020. [Google Scholar] [CrossRef]
- Li, B.; Yan, C.; Zhu, J.; Chen, X.; Fu, Q.; Zhang, H.; Tong, Z.; Liu, L.; Zheng, Y.; Zhao, P.; et al. Anti-PD-1/PD-L1 blockade immunotherapy employed in treating hepatitis B virus infection-related advanced hepatocellular carcinoma: A literature review. Front. Immunol. 2020, 11, 1037. [Google Scholar] [CrossRef]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef]
- Veigl, M.L.; Kasturi, L.; Olechnowicz, J.; Ma, A.H.; Lutterbaugh, J.D.; Periyasamy, S.; Li, G.M.; Drummond, J.; Modrich, P.L.; Sedwick, W.D.; et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 8698–8702. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Okamoto, W.; Shitara, K.; Kojima, T.; Morizane, C.; Naito, Y.; Yuki, S.; Kagawa, Y.; Narita, Y.; Nakashima, Y.; et al. Large-scale analyses of tumor mutation burdens (TMBs) across various advanced gastrointestinal (GI) malignancies in the nationwide cancer genome screening project, SCRUM-Japan GI-SCREEN. J. Clin. Oncol. 2018, 36, 12094. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazachkov, Y.; Yoffe, B.; Khaoustov, V.I.; Solomon, H.; Klintmalm, G.B.; Tabor, E. Microsatellite instability in human hepatocellular carcinoma: Relationship to p53 abnormalities. Liver 1998, 18, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Togni, R.; Bagla, N.; Muiesan, P.; Miquel, R.; O’Grady, J.; Heaton, N.; Knisely, A.S.; Portmann, B.; Quaglia, A. Microsatellite instability in hepatocellular carcinoma in non-cirrhotic liver in patients older than 60 years. Hepatol. Res. 2009, 39, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Goumard, C.; Desbois-Mouthon, C.; Wendum, D.; Calmel, C.; Merabtene, F.; Scatton, O.; Praz, F. Low levels of microsatellite instability at simple repeated sequences commonly occur in human hepatocellular carcinoma. Cancer Genomics Proteomics 2017, 14, 329–339. [Google Scholar] [CrossRef]
- Kawaoka, T.; Ando, Y.; Yamauchi, M.; Suehiro, Y.; Yamaoka, K.; Kosaka, Y.; Fuji, Y.; Uchikawa, S.; Morio, K.; Fujino, H.; et al. Incidence of microsatellite instability-high hepatocellular carcinoma among Japanese patients and response to pembrolizumab. Hepatol. Res. 2020. [Google Scholar] [CrossRef]
- Dominguez, D.A.; Wang, X.W. Impact of next-generation sequencing on outcomes in hepatocellular carcinoma: How precise are we really? J. Hepatocell Carcinoma 2020, 7, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Kim, E.; Lisby, A.; Ma, C.; Lo, N.; Ehmer, U.; Hayer, K.E.; Furth, E.E.; Viatour, P. Promotion of growth factor signaling as a critical function of beta-catenin during HCC progression. Nat. Commun. 2019, 10, 1909. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, C.; Ren, J.; Ma, Z.M.; Xie, Y.H.; Wen, Y.M. Gene-expression profiles of a hepatitis B small surface antigen-secreting cell line reveal upregulation of lymphoid enhancer-binding factor 1. J. Gen. Virol. 2007, 88, 2966–2976. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, J.; Ma, Z.M.; Zhao, C.; Wan, D.F.; Wen, Y.M. Role of hepatitis B surface antigen in the development of hepatocellular carcinoma: Regulation of lymphoid enhancer-binding factor 1. J. Exp. Clin. Cancer Res. 2009, 28, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]


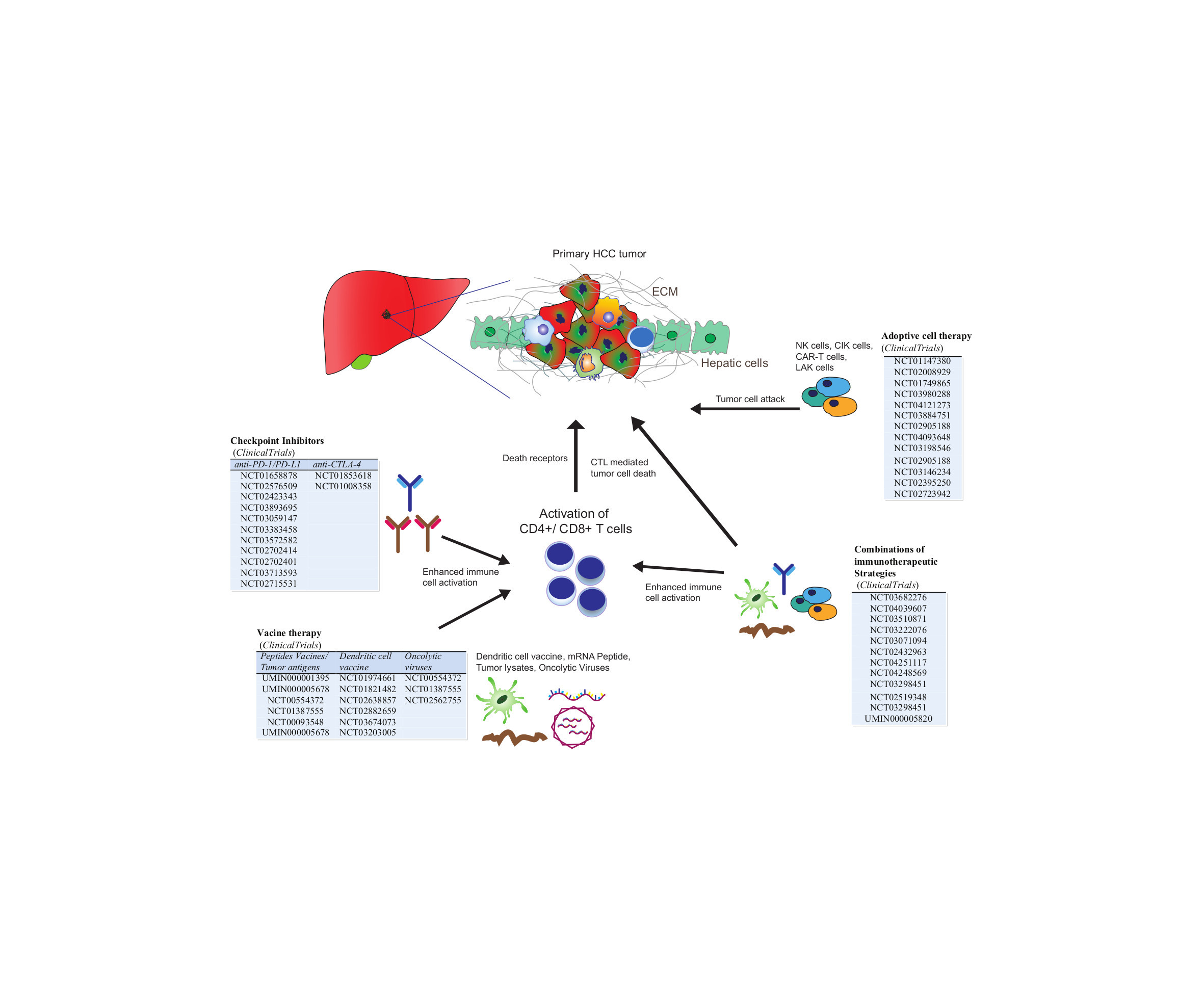{kind=link}
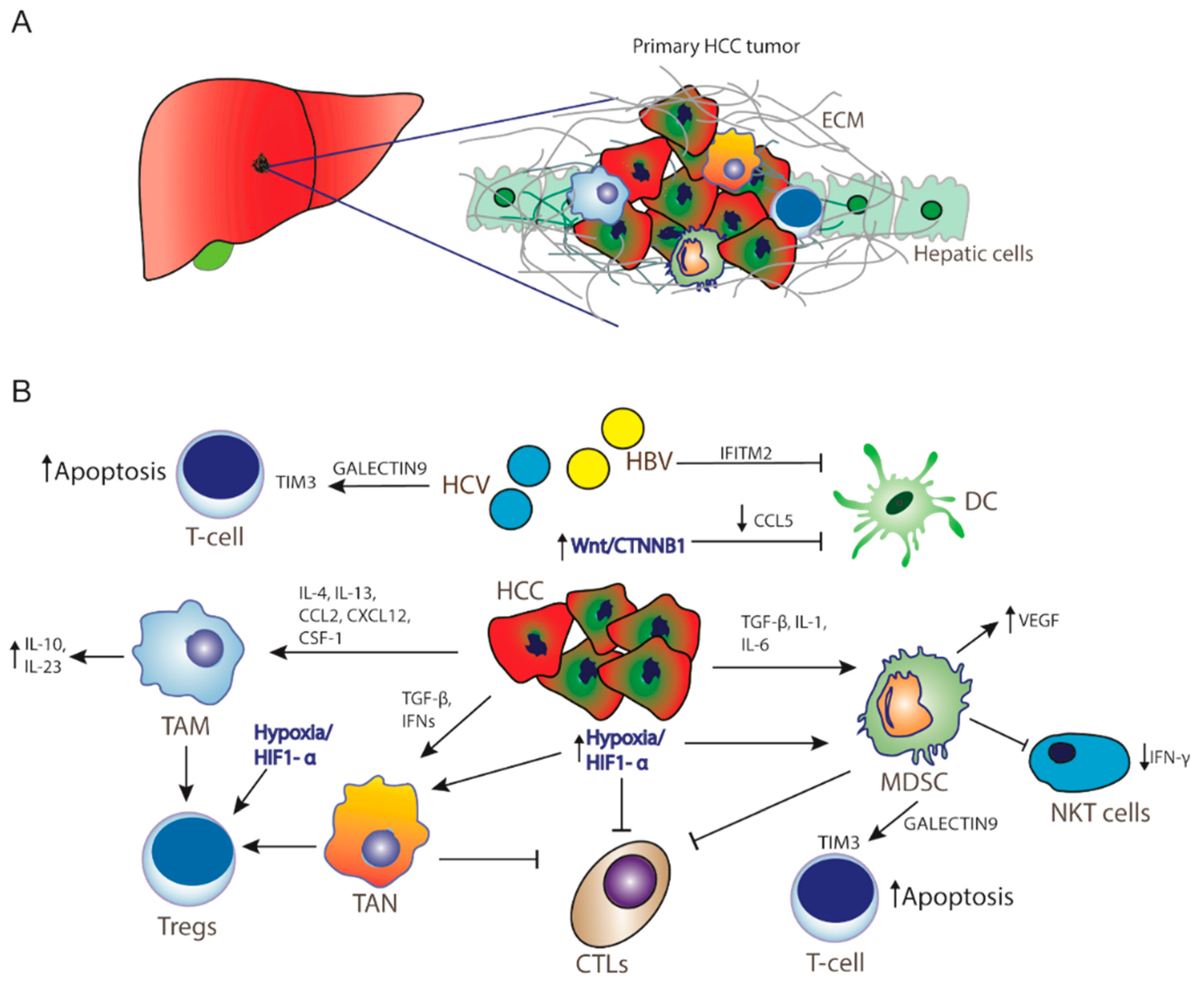{kind=link}
| Intervention | Cancer Stage | Clinical Phase/Identifier | Progression Free Survival (PFS) (Months, 95% CI) | Median Overall Survival (OS) (Months, 95% CI) | Response Rates (%, 95% CI) | Bibliography |
|---|---|---|---|---|---|---|
| Nivolumab | Advanced HCC | Phase I/II, NCT01658878 | 3.4 (1.6–6.9), for DS 4.1 (3.7–5.5), for EX | 15.0 (9.6–20.2), for DS NR, for EX | 15% (6–28), for DS 20% (15–26), for EX | [52] |
| Nivolumab | Advanced HCC | Phase III, NCT02576509 | 3.7 (3.1–3.9) | 16.4 (13.9–18.4) | 15% | [53] |
| Sorafenib | 3.8 (3.7–4.5) | 14.7 (11.9–17.2) (HR 0.84, p = 0.0419) | 7% | |||
| Pembrolizumab, sorafenib | Advanced HCC | Phase II, NCT02702414 | 4.8 (3.4–6.6) | 12,9 (9,7–15,5) | 17% (11–26) | [54] |
| Pembrolizumab | Second-line, Advanced HCC | Phase III NCT02702401 | 3.0 (2.8–4.1) | 13.9 (11.6–16.0) | 18.3 (14.0–23.4) | [55] |
| placebo | 2.8 (2.5–4.1) | 10.6 (8.3–13.5) (HR 0.781, p = 0.023) | 4.4 (1.6–9.4) | |||
| Pembrolizumab, Lenvatinib | Unresectable HCC | Phase Ib | 9.3 per mRECIST 8.6 per RECIST v1.1. | 22.0 | 46.0% (36.0–56.3) per mRECIST 36.0% (26.6%–46.2) per RECIST v1.1 | [56] |
| Atezolizumab, Bevacizumab | Unresectable HCC | Phase Ib NCT02715531 | 5.6 (3.6–7.4) | [57] | ||
| Atezolizumab | 3.4 (1.9–5.2) (HR 0.55, p = 0.0108) | |||||
| Atezolizumab/Bevacizumab | Unresectable HCC | Phase III NCT03434379 | 6.8 (5.7–8.3) | 67.2% (61.3–73.1) | [58] | |
| Sorafenib | 4.3 (4.0–5.6) (HR 0.59, p < 0.001) | 54.6% (45.2–64.0) 12 months response | ||||
| Tremelimumab | HCC | Phase II NCT01008358 | 6.48 (3.95–9.14) | 17.6% | [59] | |
| Tremelimumab, RFA or TACE | Advanced HCC | Phase I/II NCT01853618 | 7.5 (5.6–9.3) | 8.4 (6.5–10.3) | [60] |
| Intervention | Cancer Stage | Clinical Phase/Identifier | Progression Free Survival (PFS) (Months, 95% CI) | Median Overall Survival (OS) (Months, 95% CI) | Response Rates (%, 95% CI) | Bibliography | |
|---|---|---|---|---|---|---|---|
| GPC3-vaccine | Advanced HCC | Phase I, UMIN000001395 | 3.4 (2.1–4.6) | 9.0 (8.0–10.0) | 91% | [93] | |
| GPC3-vaccine, Surgery and RFA | Adjuvant therapy | Phase II | 20.1 (14.7–25.5) | 1 year at 24%, 2 years at 52.4% | [94] | ||
| MRP3 | HLA-A24-positive | Phase I UMIN000005678 | 14.0 (9.6–18.5) | 72.7% | [95] | ||
| DCs | HCC patients | Phase II | 6 months at 33%, 1 year at 11% | 5.5 | [96] | ||
| Ilixadencel | HCC patients | Phase I NCT01974661 | 5.5 | 7.4, for 1 0 × 106 cells 11.8, for 20 × 106 cells | 73% | [97] | |
| JX-594 | Advanced HCC | Phase II NCT00554372 | 14.1, for high-dose 6.7, for low-dose | 57%, for high-dose 67%, for low-dose | [98] | ||
| JX-594 | Advanced HCC, previously treated with sorafenib | Phase IIb NCT01387555 | 1.8 (1.5–2.8) | 4.2 | [99] | ||
| BSC | 2.8 (1.5–NA) | 4.4 (HR, 1.19, p = 0.428) | |||||
| Intervention | Cancer Stage | Clinical Phase/Identifier | Progression free survival (PFS) (Months, 95% CI) | Median Overall Survival (OS) (Months, 95% CI) | Response Rates (%, 95% CI) | Bibliography | |
|---|---|---|---|---|---|---|---|
| CIK, TACE, and RFA | Advance HCC | 17 (10.96–23.04) | 56 (38.09–73.91) | [138] | |||
| TACE, RFA | 10 (8.57–11.44) | 31 (24.53–37.47) | |||||
| CAR-T cells, cyclophosphamide, and fludarabine | Advanced GPC3+ HCC (Child–Pugh A) | Phase I NCT02905188 NCT03146234 | 3.2 and 3.6 (for two patients) | 9.1 (1.5–20) | Two partial responses | [151] | |
| Adjuvant-adoptive immunotherapy | Adjuvant treatment, Resected HCC | 48% (37–59) | [156] | ||||
| control | 33% (22–43) | ||||||
| Intervention | Cancer Stage | Clinical Phase/Identifier | Progression Free Survival (PFS) (Months, 95% CI) | Median Overall Survival (OS) (Months, 95% CI) | Response Rates (%, 95% CI) | Bibliography |
|---|---|---|---|---|---|---|
| NIVO (1mg/kg), IPI (3mg/kg) | Sorafenib-treated advanced hepatocellular carcinoma patients | NCT01658878 | 54.0 (39.0–68.0) | 23.0 (9.0–NA) | 32% | [157] |
| NIVO (3mg/kg), IPI (1mg/kg) | 43.0 (29.0–58.0) | 12.0 (8.0–15.0) | 31% | |||
| NIVO (3mg/kg), IPI (1mg/kg) | 49.0 (34.0–64.0) | 13.0 (7.0–33.0) | 31% | |||
| NIVO, CABO | Sorafenib or experienced advanced hepatocellular carcinoma patients | NCT01658878 | 5.5 | Not reached | 81% | [158] |
| NIVO, IPI, CABO | 6.8 | Not reached | 83% | |||
| ATVAC | Resected, invasive HCC | UMIN000005820 | 24.5 (7.8–41.2) | 97.7 (48.6–146.7) | [171] | |
| Surgery alone | 12.6 (6.9–18.3) | 41.0 (16.3–65.8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kole, C.; Charalampakis, N.; Tsakatikas, S.; Vailas, M.; Moris, D.; Gkotsis, E.; Kykalos, S.; Karamouzis, M.V.; Schizas, D. Immunotherapy for Hepatocellular Carcinoma: A 2021 Update. Cancers 2020, 12, 2859. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102859
Kole C, Charalampakis N, Tsakatikas S, Vailas M, Moris D, Gkotsis E, Kykalos S, Karamouzis MV, Schizas D. Immunotherapy for Hepatocellular Carcinoma: A 2021 Update. Cancers. 2020; 12(10):2859. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102859
Chicago/Turabian StyleKole, Christo, Nikolaos Charalampakis, Sergios Tsakatikas, Michail Vailas, Dimitrios Moris, Efthymios Gkotsis, Stylianos Kykalos, Michalis V. Karamouzis, and Dimitrios Schizas. 2020. "Immunotherapy for Hepatocellular Carcinoma: A 2021 Update" Cancers 12, no. 10: 2859. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102859