
Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy–Lysosomal Pathway

 ,
,  , , and
, , and 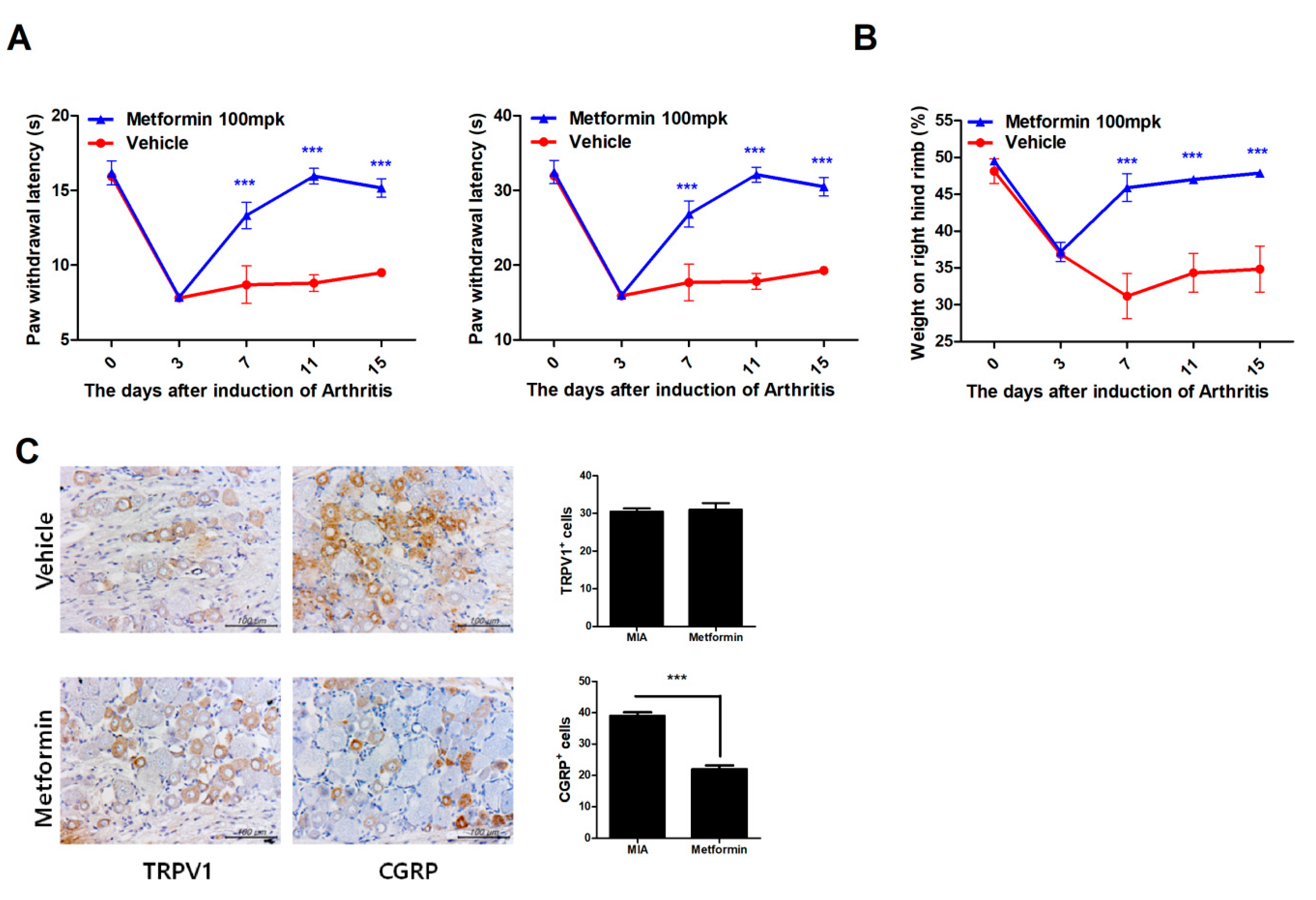{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Osteoarthritis and Treatment with Metformin
2.3. Assessment of Pain Behavior
2.4. Assessment of Weight-Bearing
2.5. Histopathological Analysis
2.6. Immunohistochemistry
2.7. Human Articular Chondrocyte Differentiation and Culture
2.8. Real-Time Polymerase Chain Reaction (PCR)
2.9. Western Blot Analysis
2.10. In Vivo Microcomputed Tomography (CT) Iimaging and Analysis
2.11. Immunofluorescence
2.12. Grading System of Kellgren and Lawrence (KL) in OA Patients
2.13. Statistical Analysis
3. Results
3.1. Metformin Reduces Pain in MIA-Induced OA Rats
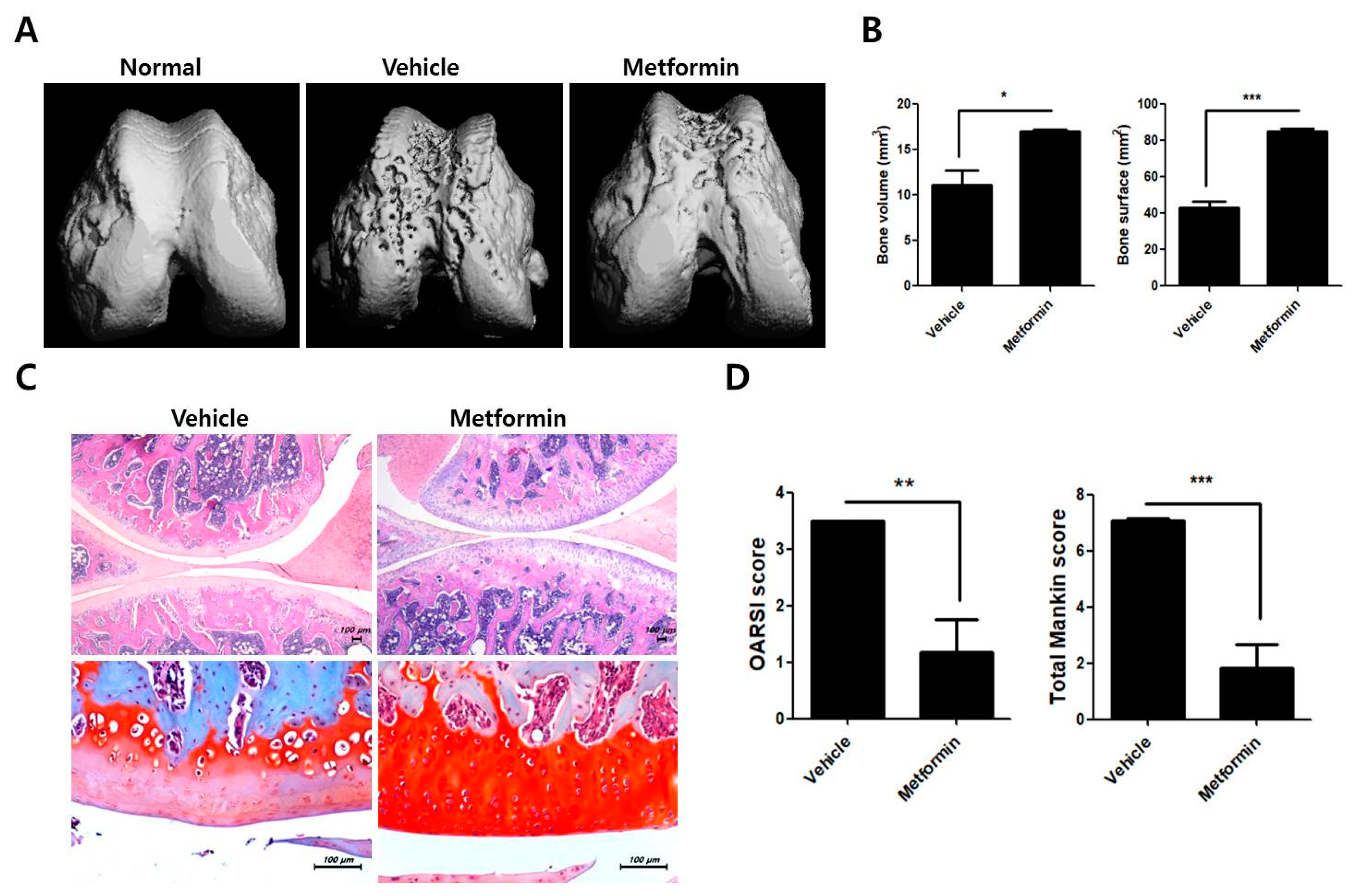
3.2. Protective Effects of Metformin in the Knee Joints of MIA-Induced OA Rats
3.3. Metformin Reduces the Levels of Inflammatory Mediators and Catabolic Factors in the Synovium of OA Rats
3.4. Metformin Decreases the Catabolic Response of Human OA Chondrocytes
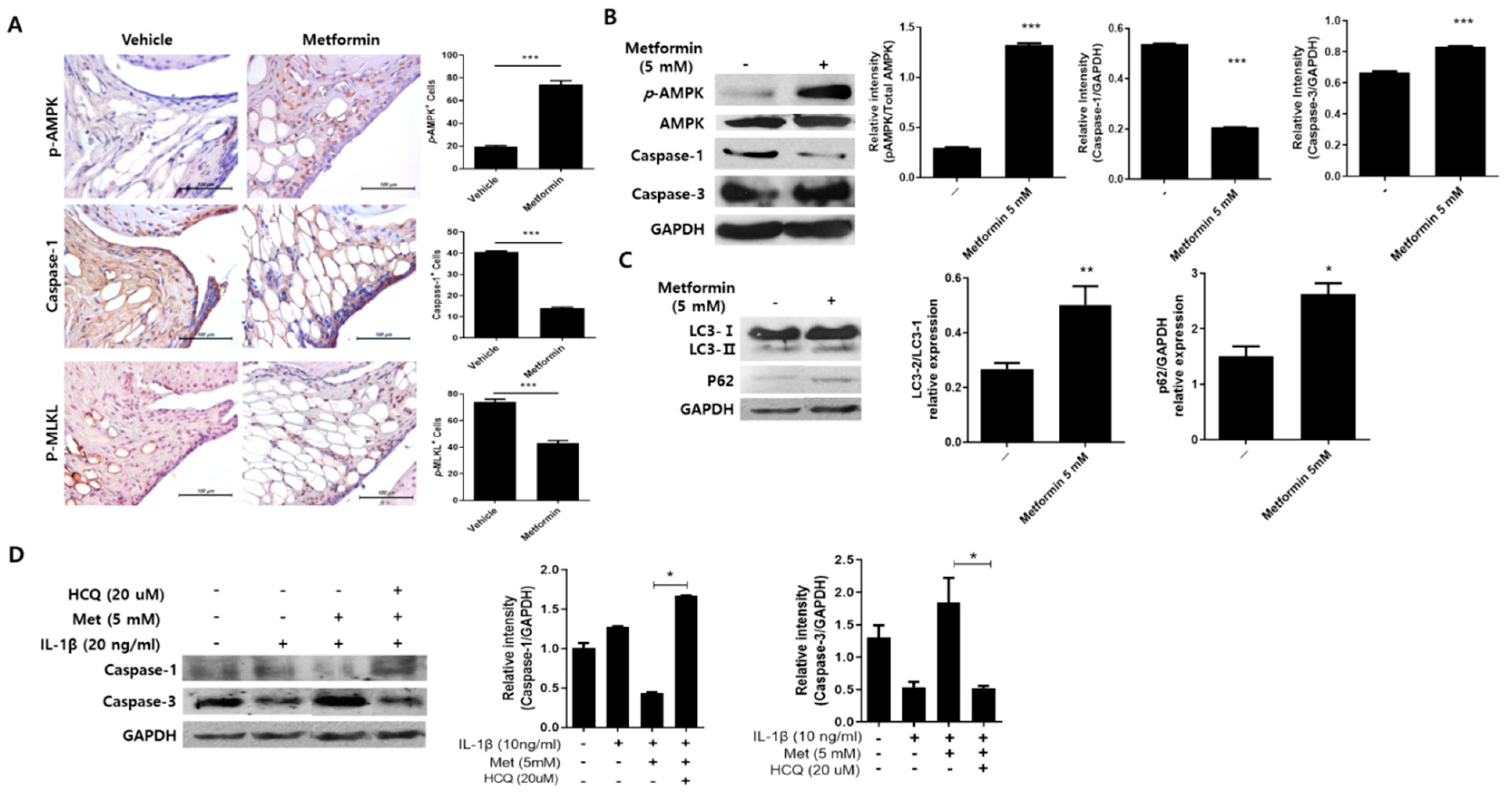
3.5. Metformin Regulates Inflammation-Induced Cell Death in the Joints of MIA-Induced OA Rats
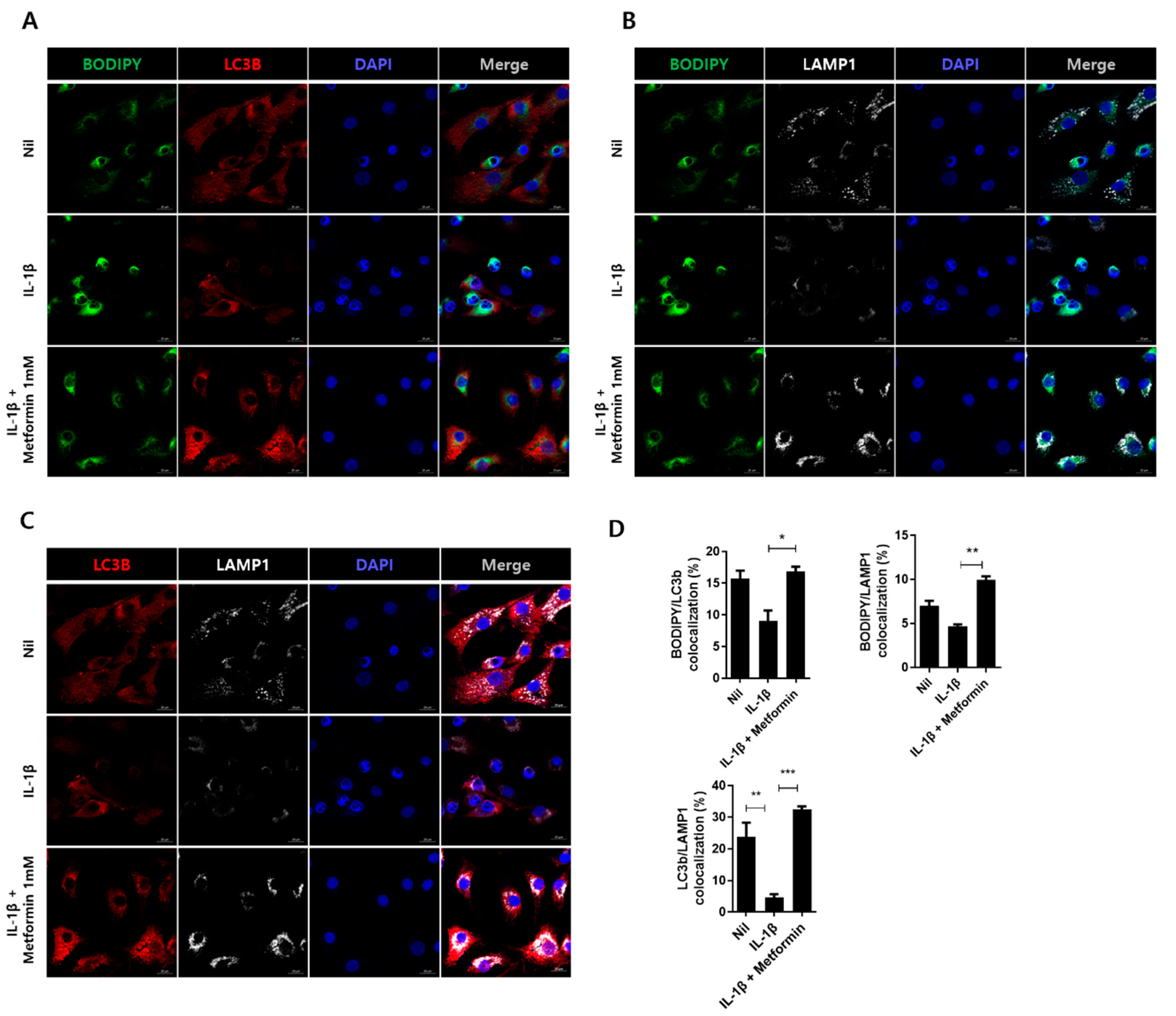
3.6. Metformin Promotes Autophagy in Human Chondrocytes
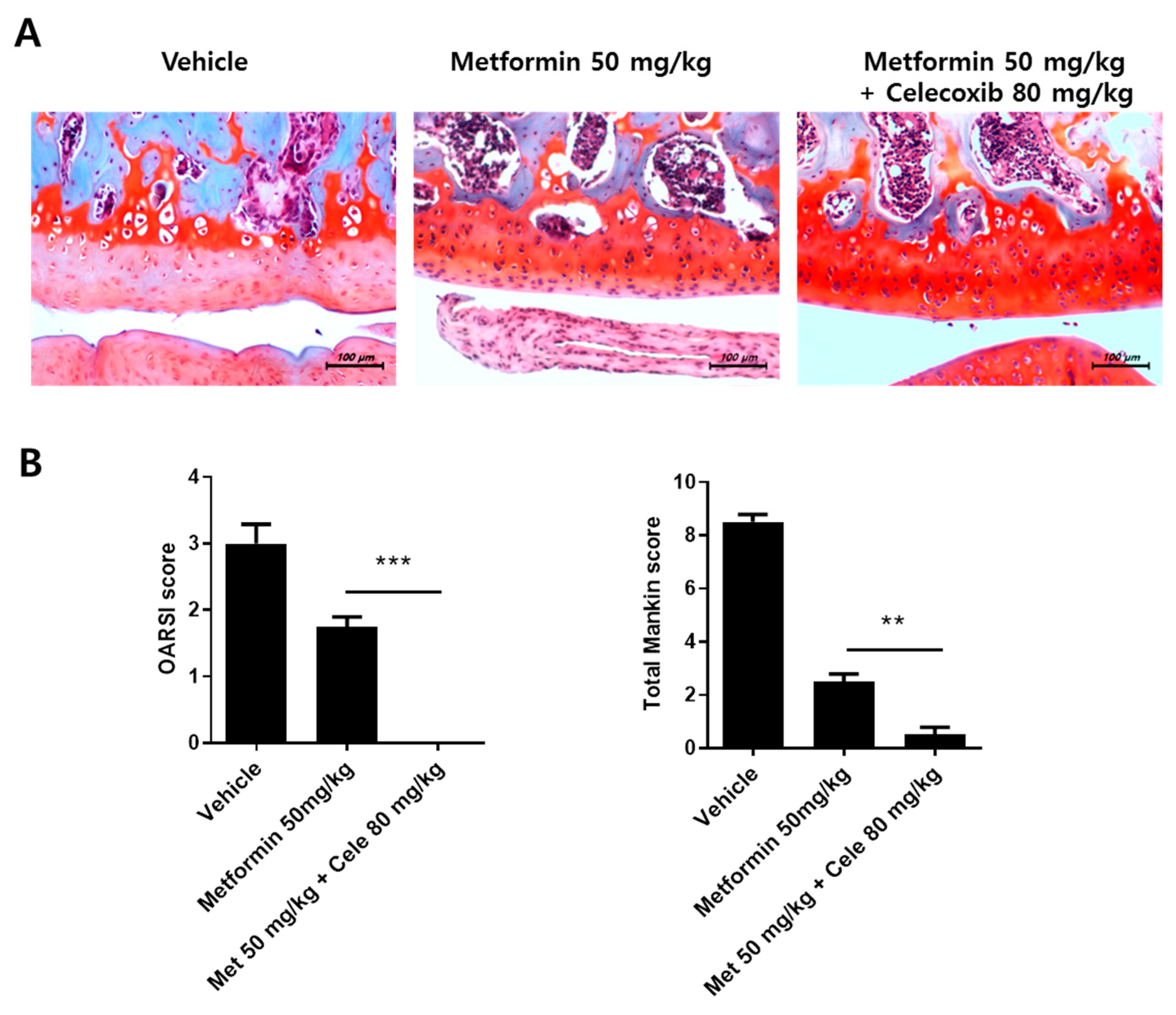
3.7. The Effect of the Combination of Metformin and Celecoxib in OA Progression
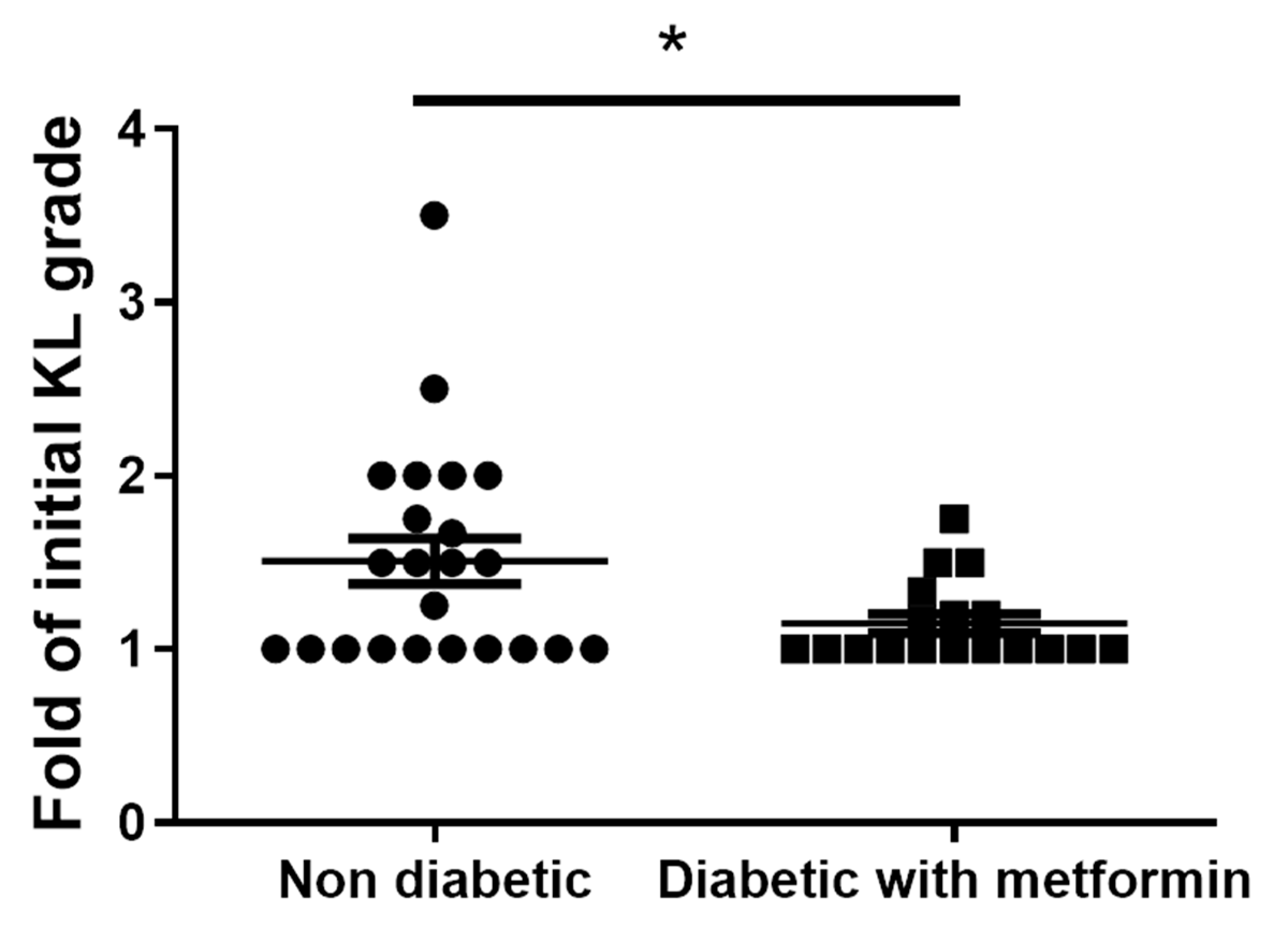
3.8. Metformin Inhibits Clinical-Grade OA and Exhibits an Anti-Inflammatory Effect in OA Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalman, D.S.; Heimer, M.; Valdeon, A.; Schwartz, H.; Sheldon, E. Effect of a natural extract of chicken combs with a high content of hyaluronic acid (Hyal-Joint) on pain relief and quality of life in subjects with knee osteoarthritis: A pilot randomized double-blind placebo-controlled trial. Nutr. J. 2008, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cart. 2014, 22, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr. Cart. 2013, 21, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Adv. Musculoskelet Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Moradi, B.; Schnatzer, P.; Hagmann, S.; Rosshirt, N.; Gotterbarm, T.; Kretzer, J.P.; Thomsen, M.; Lorenz, H.M.; Zeifang, F.; Tretter, T. CD4(+)CD25(+)/highCD127low/(-) regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints–analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res. 2014, 16, R97. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wan, J.; Anderson, W.; Sun, H.; Zhang, H.; Peng, X.; Yu, Z.; Wang, T.; Yan, X.; Smith, W. Downregulation of IL-10 secretion by Treg cells in osteoarthritis is associated with a reduction in Tim-3 expression. Biomed. Pharm. 2016, 79, 159–165. [Google Scholar] [CrossRef]
- Li, Y.S.; Luo, W.; Zhu, S.A.; Lei, G.H. T Cells in Osteoarthritis: Alterations and Beyond. Front. Immunol. 2017, 8, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Kim, E.K.; Lee, S.H.; Lee, S.Y.; Kim, J.K.; Jhun, J.Y.; Na, H.S.; Kim, S.Y.; Choi, J.Y.; Yang, C.W.; Park, S.H.; et al. Metformin ameliorates experimental-obesity-associated autoimmune arthritis by inducing FGF21 expression and brown adipocyte differentiation. Exp. Mol. Med. 2018, 50, e432. [Google Scholar] [CrossRef]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and Autoimmunity: A ‘New Deal’of An Old Drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the between Th17/Treg Balance. PLoS ONE 2015, 10, e0135858. [Google Scholar] [CrossRef] [Green Version]
- Son, H.J.; Lee, J.; Lee, S.Y.; Kim, E.K.; Park, M.J.; Kim, K.W.; Park, S.H.; Cho, M.L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014, 973986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ding, X.; Terkeltaub, R.; Lin, H.; Zhang, Y.; Zhou, B.; He, K.; Li, K.; Liu, Z.; Wei, J.; et al. Exploration of metformin as novel therapy for osteoarthritis: Preventing cartilage degeneration and reducing pain behavior. Arthritis Res. 2020, 22, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Liu, Y.; Huan, Z.; Wang, Y.; Xu, J. Metformin protects chondrocytes against IL-1β induced injury by regulation of the AMPK/NF-κ B signaling pathway. Pharmazie 2020, 75, 632–636. [Google Scholar] [CrossRef]
- Park, M.J.; Moon, S.J.; Baek, J.A.; Lee, E.J.; Jung, K.A.; Kim, E.K.; Kim, D.S.; Lee, J.H.; Kwok, S.K.; Min, J.K.; et al. Metformin Augments Anti-Inflammatory and Chondroprotective Properties of Mesenchymal Stem Cells in Experimental Osteoarthritis. J. Immunol. 2019, 203, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zweers, M.C.; de Boer, T.N.; van Roon, J.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Mastbergen, S.C. Celecoxib: Considerations regarding its potential disease-modifying properties in osteoarthritis. Arthritis Res. Ther. 2011, 13, 239. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.H.; Chung, C.H.; Lee, C.H.; Hsieh, C.H.; Hung, Y.J.; Lin, F.H.; Tsao, C.H.; Hsieh, P.S.; Chien, W.C. Combination COX-2 inhibitor and metformin attenuate rate of joint replacement in osteoarthritis with diabetes: A nationwide, retrospective, matched-cohort study in Taiwan. PLoS ONE 2018, 13, e0191242. [Google Scholar] [CrossRef] [Green Version]
- Pritzker, K.P.H.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Terkeltaub, R.; Yang, B.; Lotz, M.; Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011, 63, 1928–1937. [Google Scholar] [CrossRef]
- Moon, S.J.; Park, J.S.; Jeong, J.H.; Yang, E.J.; Park, M.K.; Kim, E.K.; Park, S.H.; Kim, H.Y.; Cho, M.L.; Min, J.K. Augmented chondroprotective effect of coadministration of celecoxib and rebamipide in the monosodium iodoacetate rat model of osteoarthritis. Arch. Pharm. Res. 2013, 36, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kohn, M.D.; Sassoon, A.A.; Fernando, N.D. Classifications in Brief: Kellgren-Lawrence Classification of Osteoarthritis. Clin. Orthop. Relat. Res. 2016, 474, 1886–1893. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, B.; Liu, W.X.; Lu, K.; Pan, H.; Wang, T.; Oh, C.D.; Yi, D.; Huang, J.; Zhao, L.; et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann. Rheum. Dis. 2020, 79, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [Green Version]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Gallelli, L.; Galasso, O.; Falcone, D.; Southworth, S.; Greco, M.; Ventura, V.; Romualdi, P.; Corigliano, A.; Terracciano, R.; Savino, R.; et al. The effects of nonsteroidal anti-inflammatory drugs on clinical outcomes, synovial fluid cytokine concentration and signal transduction pathways in knee osteoarthritis. A randomized open label trial. Osteoarthr. Cart. 2013, 21, 1400–1408. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasioli, D.J.; Kaplan, D.L. The roles of catabolic factors in the development of osteoarthritis. Tissue Eng. Part B Rev. 2014, 20, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.J.; Yu, W.B.; Luo, W.; Gao, S.G.; Li, Y.S.; Lei, G.H. Effect of osteopontin on TIMP-1 and TIMP-2 mRNA in chondrocytes of human knee osteoarthritis. Exp. Med. 2014, 8, 391–394. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, M.; Tortorella, M.; Nagase, H.; Brew, K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 2001, 276, 12501–12504. [Google Scholar] [CrossRef] [Green Version]
- Mueller, M.B.; Tuan, R.S. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: Identifying molecular targets. PMR 2011, 3, S3–S11. [Google Scholar] [CrossRef]
- Penatti, A.; Facciotti, F.; De Matteis, R.; Larghi, P.; Paroni, M.; Murgo, A.; De Lucia, O.; Pagani, M.; Pierannunzii, L.; Truzzi, M.; et al. Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res. 2017, 19, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Richter, F.; Natura, G.; Loser, S.; Schmidt, K.; Viisanen, H.; Schaible, H.G. Tumor necrosis factor causes persistent sensitization of joint nociceptors to mechanical stimuli in rats. Arthritis Rheum. 2010, 62, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Brenn, D.; Richter, F.; Schaible, H.G. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: An inflammatory mechanism of joint pain. Arthritis Rheum. 2007, 56, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Boettger, M.K.; Hensellek, S.; Richter, F.; Gajda, M.; Stockigt, R.; von Banchet, G.S.; Brauer, R.; Schaible, H.G. Antinociceptive effects of tumor necrosis factor alpha neutralization in a rat model of antigen-induced arthritis: Evidence of a neuronal target. Arthritis Rheum. 2008, 58, 2368–2378. [Google Scholar] [CrossRef]
- Del Fiacco, M.; Quartu, M.; Boi, M.; Serra, M.P.; Melis, T.; Boccaletti, R.; Shevel, E.; Cianchetti, C. TRPV1, CGRP and SP in scalp arteries of patients suffering from chronic migraine. J. Neurol. Neurosurg. Psychiatry 2015, 86, 393–397. [Google Scholar] [CrossRef]
- Riera, C.E.; Huising, M.O.; Follett, P.; Leblanc, M.; Halloran, J.; Van Andel, R.; de Magalhaes Filho, C.D.; Merkwirth, C.; Dillin, A. TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 2014, 157, 1023–1036. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Feng, L.; Feng, C.; Wang, C.X.; Xu, D.Y.; Chen, J.J.; Huang, J.F.; Tan, P.L.; Shen, J.M. Circulating microRNA let7e is decreased in knee osteoarthritis, accompanied by elevated apoptosis and reduced autophagy. Int. J. Mol. Med. 2020, 45, 1464–1476. [Google Scholar] [CrossRef]
- Wang, C.; Yao, Z.; Zhang, Y.; Yang, Y.; Liu, J.; Shi, Y.; Zhang, C. Metformin Mitigates Cartilage Degradation by Activating AMPK/SIRT1-Mediated Autophagy in a Mouse Osteoarthritis Model. Front. Pharm. 2020, 11, 1114. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Zhang, Y.; Liu, J.; Yao, Z.; Zhang, C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci. Trends 2019, 12, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.T.; Burton-Wurster, N.; Borden, C.; Hueffer, K.; Bloom, S.E.; Lust, G. Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J. Orthop. Res. 2001, 19, 703–711. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, H.S.; Kwon, J.Y.; Lee, S.-Y.; Lee, S.H.; Lee, A.R.; Woo, J.S.; Jung, K.; Cho, K.-H.; Choi, J.-W.; Lee, D.H.; et al. Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy–Lysosomal Pathway. Cells 2021, 10, 681. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030681
Na HS, Kwon JY, Lee S-Y, Lee SH, Lee AR, Woo JS, Jung K, Cho K-H, Choi J-W, Lee DH, et al. Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy–Lysosomal Pathway. Cells. 2021; 10(3):681. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030681
Chicago/Turabian StyleNa, Hyun Sik, Ji Ye Kwon, Seon-Yeong Lee, Seung Hoon Lee, A Ram Lee, Jin Seok Woo, KyungAh Jung, Keun-Hyung Cho, Jeong-Won Choi, Dong Hwan Lee, and et al. 2021. "Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy–Lysosomal Pathway" Cells 10, no. 3: 681. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030681