
NAFLD-Related Hepatocarcinoma: The Malignant Side of Metabolic Syndrome

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
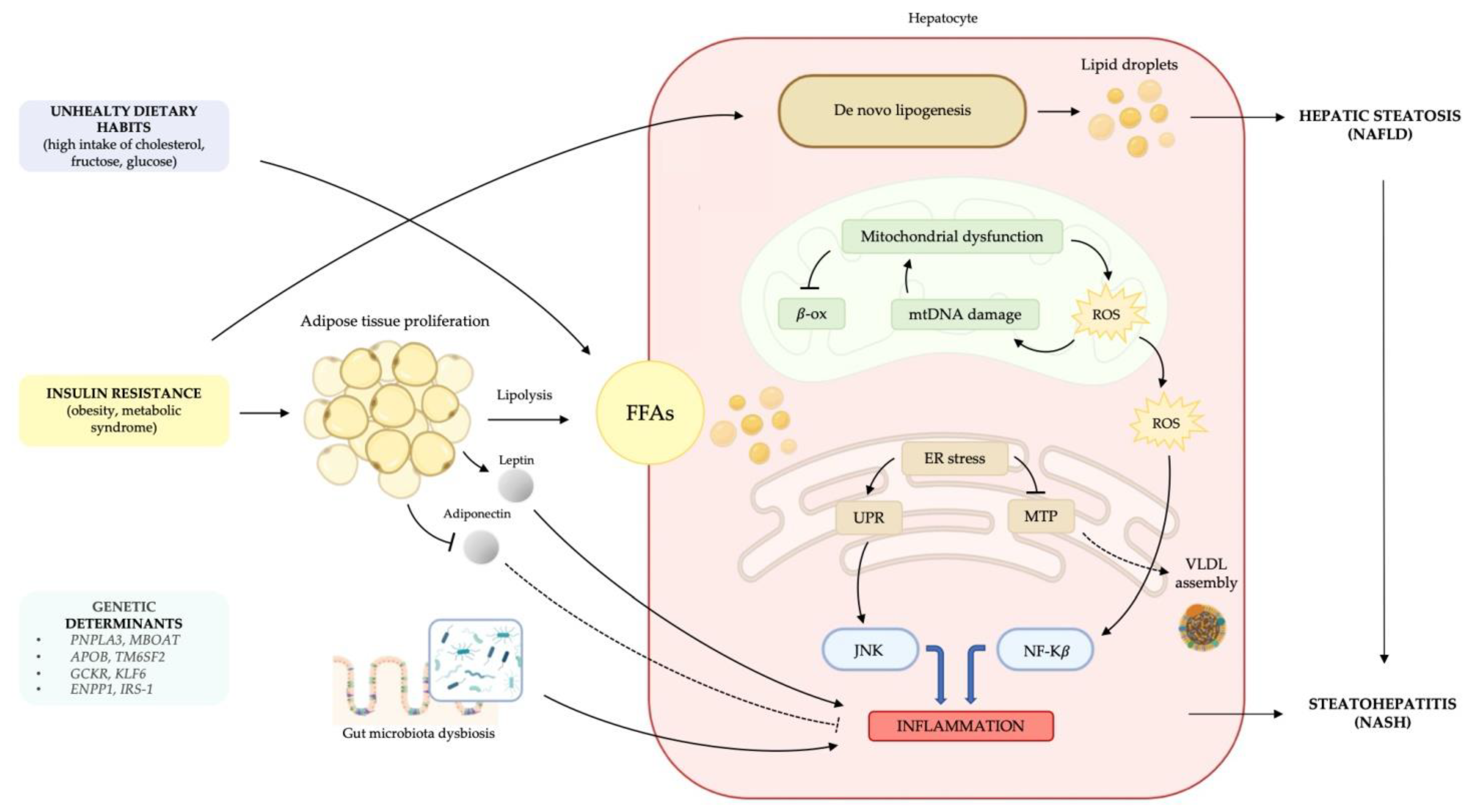
2. Understanding Molecular Pathways Underlying NAFLD
2.1. Metabolic Dysfunction: Lipid Accumulation, Lipotoxicity and Insulin Resistance
2.2. The Onset of a Chronic Inflammatory State
2.3. Genetic Predisposition
3. From NAFLD to HCC, a Roadmap Not So Winding
4. Clinical Impact of Different Pathogenetic Drivers in the Management of HCC
NAFLD and HCC Treatment
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- IARC. Working group on the evaluation of carcinogenic risks to humans. Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–441. [Google Scholar]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global epidemiology of hepatocellular carcinoma: An Emphasis on Demographic and Regional Variability. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- De Mitri, M.S.; Poussin, K.; Baccarini, P.; Pontisso, P.; D’Errico, A.; Simon, N.; Grigioni, W.; Alberti, A.; Beaugrand, M.; Pisi, E. HCV-associated liver cancer without cirrhosis. Lancet Lond. Engl. 1995, 345, 413–415. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Flisiak-Jackiewicz, M.; Bobrus-Chociej, A.; Wasilewska, N.; Lebensztejn, D.M. From nonalcoholic fatty liver disease (NAFLD) to metabolic dysfunction-associated fatty liver disease (MAFLD)—New terminology in pediatric patients as a step in good scientific direction? J. Clin. Med. 2021, 10, 924. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef]
- Masuoka, H.C.; Chalasani, N. Nonalcoholic Fatty Liver Disease: An Emerging Threat to Obese and Diabetic Individuals. Ann. New York Acad. Sci. 2013, 1281, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Tsuneto, A.; Hida, A.; Sera, N.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Maemura, K.; Akahoshi, M. Fatty liver incidence and predictive variables. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2010, 33, 638–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of Hepatic Steatosis in an Urban Population in the United States: Impact of Ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.-C.; Roudot-Thoraval, F. The Burden of Liver Disease in Europe: A Review of Available Epidemiological Data. J. Hepatol. 2013, 58, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Linee Guida Epatocarcinoma. Available online: https://www.aiom.it/linee-guida-aiom-2020-epatocarcinoma/ (accessed on 15 June 2021).
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [Green Version]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Alexander, M.; Loomis, A.K.; Van Der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef]
- Lee, S.S.; Park, S.H. Radiologic evaluation of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7392–7402. [Google Scholar] [CrossRef]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and its management. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2020, 35, 72–84. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH Pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Brozinick, J.T.; Wang, L.-P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.; Dhaliwal, J.; Mouzaki, M. Lean non-alcoholic fatty liver disease. Clin. Nutr. Edinb. Scotl. 2019, 38, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Hui, T.Y.; Young, S.G.; Davis, R.A. Blocking microsomal triglyceride transfer protein interferes with ApoB secretion without causing retention or stress in the ER. J. Lipid Res. 2003, 44, 978–985. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Liddle, C. Nonalcoholic fatty liver disease: Pathogenesis and potential for nuclear receptors as therapeutic targets. Mol. Pharm. 2008, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Pacini, G.; Musso, G.; Gambino, R.; Mecca, F.; Depetris, N.; Cassader, M.; David, E.; Cavallo-Perin, P.; Rizzetto, M. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: Further evidence for an etiologic association. Hepatology 2002, 35, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Durazzo, M.; Pagano, G.; Cassader, M. Adiponectin gene polymorphisms modulate acute adiponectin response to sietary fat: Possible pathogenetic role in NASH. Hepatology 2008, 47, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic Fatty Liver Disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Roiban, A.L.; Both, E.; Huţanu, A. Serum leptin and leptin resistance correlations with NAFLD in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2018, 34, e3050. [Google Scholar] [CrossRef]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of Ob/Ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Xu, C.-F.; Yu, C.-H.; Chen, W.-X.; Li, Y.-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef]
- Duvnjak, M.; Barsić, N.; Tomasić, V.; Lerotić, I. Genetic polymorphisms in non-alcoholic fatty liver disease: Clues to pathogenesis and disease progression. World J. Gastroenterol. 2009, 15, 6023–6027. [Google Scholar] [CrossRef] [PubMed]
- Miyaaki, H.; Nakao, K. Significance of genetic polymorphisms in patients with nonalcoholic fatty liver disease. Clin. J. Gastroenterol. 2017, 10, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomba, R.; Schork, N.; Chen, C.-H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant Rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Gastaldelli, A.; Vetter, D.; Patman, G.L.; Pascoe, L.; Hannivoort, R.A.; Lee, U.E.; Fiel, I.; Muñoz, U.; Ciociaro, D.; et al. Glucokinase links krüppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology 2012, 55, 1083–1093. [Google Scholar] [CrossRef]
- Gambino, R.; Cassader, M.; Pagano, G.; Durazzo, M.; Musso, G. Polymorphism in microsomal triglyceride transfer protein: A link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology 2007, 45, 1097–1107. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, J.; Zhao, Z.; Zhang, J.; Dong, M.; Ma, X.; Liu, S.; Xin, Y. The Association between SNPs Rs1800591 and Rs3816873 of the MTTP gene and nonalcoholic fatty liver disease: A meta-analysis. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2020. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects from Hepatocellular Carcinoma Development in Alcoholic Liver Disease—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30908678/ (accessed on 15 June 2021).
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef]
- Margini, C.; Dufour, J.F. The Story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016, 36, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugianesi, E.; Leone, N.; Vanni, E.; Marchesini, G.; Brunello, F.; Carucci, P.; Musso, A.; De Paolis, P.; Capussotti, L.; Salizzoni, M.; et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002, 123, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Jeong, S.-H.; Byoun, Y.-S.; Chung, S.M.; Seong, M.H.; Sohn, H.R.; Min, B.; Jang, E.S.; Kim, J.-W.; Park, G.J.; et al. Clinical features and outcome of cryptogenic hepatocellular carcinoma compared to those of viral and alcoholic hepatocellular carcinoma. BMC Cancer 2013, 13, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 8. [Google Scholar] [CrossRef]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular mechanisms: Connections between nonalcoholic fatty liver disease, steatohepatitis and hepatocellular carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron metabolism in nonalcoholic fatty liver disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Chettouh, H.; Lequoy, M.; Fartoux, L.; Vigouroux, C.; Desbois-Mouthon, C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 2015, 35, 2203–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the P53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Enhanced Preneoplastic Liver Lesion Development under ‘Selection Pressure’ Conditions after Administration of Deoxycholic or Lithocholic Acid in the Initiation Phase in Rats. Carcinogenesis. Oxford Academic. Available online: https://0-academic-oup-com.brum.beds.ac.uk/carcin/article-abstract/11/8/1323/316006?redirectedFrom=fulltext (accessed on 15 June 2021).
- Schnabl, B. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003, 37, 653–664. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Nakano association between appendectomy and fibrosis progression in nonalcoholic fatty liver disease. Gastroenterol. Res. 2013. [CrossRef] [Green Version]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 Rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Scragg, J.; Avery, L.; Cassidy, S.; Taylor, G.; Haigh, L.; Boyle, M.; Trenell, M.I.; Anstee, Q.M.; McPherson, S.; Hallsworth, K. Feasibility of a very low calorie diet to achieve a sustainable 10% weight loss in patients with nonalcoholic fatty liver disease. Clin. Transl. Gastroenterol. 2020, 11. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Pellegrini, E.; Lugari, S.; Mondelli, A.; Bursi, S.; Onfiani, G.; Carubbi, F.; Lonardo, A. Statins and nonalcoholic fatty liver disease in the era of precision medicine: More Friends than Foes. Atherosclerosis 2019, 284, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Eslami, L.; Merat, S.; Malekzadeh, R.; Nasseri-Moghaddam, S.; Aramin, H. Statins for non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Lonardo, A.; Loria, P. Potential for statins in the chemoprevention and management of hepatocellular carcinoma: Statins and hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2012, 27, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase lb study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. Late-Breaking Abstract-3: CheckMate 459: Long-Term Efficacy Outcomes with Nivolumab Versus Sorafenib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma. Oncology PRO. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-world-gi-2020-virtual/late-breaking-abstract-3-checkmate-459-long-term-efficacy-outcomes-with-nivolumab-versus-sorafenib-as-first-line-treatment-in-patients-with-advan (accessed on 9 June 2021).
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. of Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Updated efficacy and safety of KEYNOTE-224: A phase II study of pembrolizumab (pembro) in patients with advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2020, 38, 518. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Tokushige, K.; Hashimoto, E.; Horie, Y.; Taniai, M.; Higuchi, S. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease, alcoholic liver disease, and chronic liver disease of unknown etiology: Report of the nationwide survey. J. Gastroenterol. 2011, 46, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Trials | Outcomes (Experimental Arm vs. Control Arm) | Non-Viral Etiology (Experimental Arm vs. Control Arm) |
|---|---|---|
| SHARP: sorafenib vs. placebo (1st line) NCT00105443 | OS: 10.7 vs. 7.9 months; HR 0.69, 95% CI, 0.55–0.87, p < 0.001 PR: 2% vs. 1% SD: 71% vs. 67% 1-yr survival rate: 44% vs. 33% | Alcoholic 26% vs. 26% Other 9% vs. 10% Unknown 16% vs. 19% |
| REFLECT: lenvatinib vs sorafenib (1st line) a NCT01761266 | OS: 13.6 vs. 12.3 months; HR 0.92, 95% CI 0.79–1.06, p not significant PFS: 7.4 vs. 3.7 months; HR 0.66, 95% CI 0.57−0.77, p < 0.0001 ORR: 24.1% vs. 9.2%; OR 3.13, 95% CI 2.15–4.56, p < 0.0001 | Alcoholic 8% vs. 4% Other 8% vs. 7% Unknown 13% vs. 14% |
| IMBRAVE 150: atezolizumab-bevacizumab vs sorafenib (1st line) b NCT03434379 | OS at 6 months: 84.8% (95% CI 80.9–88.7) vs. 72.2% (95% CI 65.1–79.4) OS at 12 months: 67.2% (95% CI 61.3–73.1) vs. 54.6% (95% CI, 45.2–64.0) PFS: 6.8 vs. 4.3 months; HR 0.59, 95% CI, 0.47–0.76, p < 0.001 ORR: 27.3% vs. 11.9%, p < 0.001 | Non-viral 30% vs. 32% |
| KEYNOTE 524: lenvatinib-pembrolizumab (1st line) a NCT03006926 | OS: 22.0 months (95% CI, 20.4 months-NE) PFS: 8.2 months (95% CI, 7.4–9.7 months) ORR: 46.0% (95% CI, 36.0–56.3%) | Alcoholic 28% Other 22% |
| CHECKMATE 459: nivolumab-sorafenib (1st line) b NCT02576509 | OS: 16.4 vs. 14.8 months; HR 0.85, 95% CI 0.72–1.00, p = 0.0522 | Non-viral 45% vs. 45% |
| RESORCE: regorafenib vs placebo (2nd line) a NCT01774344 | OS: 10.6 vs. 7.8 months; HR 0.63, 95% CI 0.50–0.79, p < 0.0001 PFS: 3.1 vs. 1.5 months; HR 0.46, 95% CI 0.37–0.56), p < 0.0001 ORR: 11% vs. 4%, p = 0.0047 | Alcoholic 24% vs. 28% NASH 7% vs. 7% Other 7% vs. 5% Unknown 17% vs. 16% |
| CELESTIAL: cabozantinib vs. placebo (2nd line) b NCT01908426 | OS: 10.2 vs. 8.0 months; HR 0.76, 95% CI 0.63–0.92, p = 0.005 PFS: 5.2 vs. 1.9 months; HR 0.44, 95% CI 0.36–0.52, p < 0.001 ORR: 4% vs. <1%, p = 0.009 | Alcoholic 24% vs. 16% NASH 9% vs. 10% Other 5% vs. 7% Unknown 16% vs. 20% |
| KEYNOTE 240: pembrolizumab vs. placebo (2nd line) b NCT02702401 | OS: 13.9 vs. 10.6 months; HR 0.78, 95% CI 0.611–0.998; p = 0.0238 PFS: 3.0 vs. 2.8 months; HR 0.718, 95% CI 0.570–0.904, p = 0.0022 ORR: 18.3% vs. 4.4%, p = 0.00007 | Non-viral 58.6% vs. 63.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelotti, A.; de Scordilli, M.; Palmero, L.; Guardascione, M.; Masala, M.; Roncato, R.; Foltran, L.; Ongaro, E.; Puglisi, F. NAFLD-Related Hepatocarcinoma: The Malignant Side of Metabolic Syndrome. Cells 2021, 10, 2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082034
Michelotti A, de Scordilli M, Palmero L, Guardascione M, Masala M, Roncato R, Foltran L, Ongaro E, Puglisi F. NAFLD-Related Hepatocarcinoma: The Malignant Side of Metabolic Syndrome. Cells. 2021; 10(8):2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082034
Chicago/Turabian StyleMichelotti, Anna, Marco de Scordilli, Lorenza Palmero, Michela Guardascione, Mario Masala, Rossana Roncato, Luisa Foltran, Elena Ongaro, and Fabio Puglisi. 2021. "NAFLD-Related Hepatocarcinoma: The Malignant Side of Metabolic Syndrome" Cells 10, no. 8: 2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082034