
SLC38A5 Modulates Ferroptosis to Overcome Gemcitabine Resistance in Pancreatic Cancer
 , and
, and
Abstract
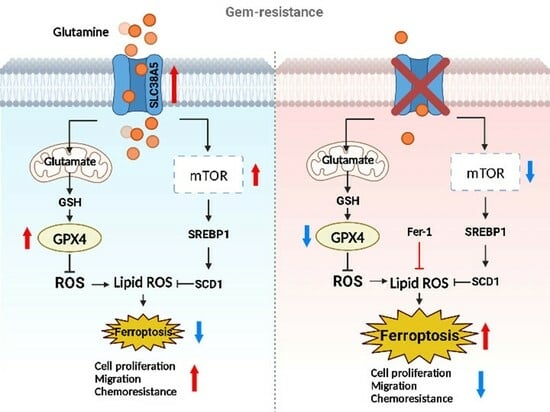
:
1. Introduction
2. Materials and Methods
2.1. The Cancer Genome Atlas Analysis
2.2. Cell Culture
2.3. Patient Tissue mRNA and Protein
2.4. Small-Interfering RNA Transfection
2.5. WST Assay
2.6. Wound Healing Assay
2.7. Invasion Assay
2.8. RNA Sequencing
2.9. Glutamine Uptake Assay
2.10. OCR and ECAR Measurement
2.11. Glutathione Level Assay
2.12. ROS and Lipid ROS Measurement
2.13. RT-PCR and qPCR
2.14. Western Blotting
2.15. Animal Studies
2.16. Statistical Analysis
3. Results
3.1. SLC38A5 Correlates with Gemcitabine-Resistant Pancreatic Cancer Patients
3.2. Deletion of SLC38A5 Downregulates Cell Viability and Migration in Gemcitabine-Resistant Pancreatic Cancer Cells
3.3. Overview of Enrichment Analyses between Gemcitabine-Resistant Cells and SLC38A5 Inhibition
3.4. SLC38A5 Modulates Lipid ROS through GSH-Mediated ROS and mTOR-SREBP1 Signaling in Gemcitabine-Resistant Pancreatic Cancer Cells
3.5. Inhibition of SLC38A5 Induces Ferroptosis in Gemcitabine-Resistant Pancreatic Cancer Cells
3.6. Suppression of SLC38A5 Inhibits Tumor Growth and Metastasis in Orthotopic Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.C.; Goggins, M.G.; Fishman, E.K. Diagnosis and detection of pancreatic cancer. Cancer J. 2017, 23, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Tingstedt, B.; Andersson, B.; Holmquist, F.; Sturesson, C.; Williamsson, C.; Sasor, A.; Borg, D.; Bauden, M.; Andersson, R. Pancreatic cancer: Yesterday, today and tomorrow. Future Oncol. 2016, 12, 1929–1946. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Setua, S.; Kumari, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Gemcitabine combination nano therapies for pancreatic cancer. Pharmaceutics 2019, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yemanyi, F.; Blomfield, A.K.; Bora, K.; Huang, S.; Liu, C.H.; Britton, W.R.; Cho, S.S.; Tomita, Y.; Fu, Z.; et al. Amino acid transporter slc38a5 regulates developmental and pathological retinal angiogenesis. eLife 2022, 11, e73105. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. Slc6a14 and slc38a5 drive the glutaminolysis and serine-glycine-one-carbon pathways in cancer. Pharmaceuticals 2021, 14, 216. [Google Scholar] [CrossRef]
- Ramachandran, S.; Sennoune, R.S.; Sharma, M.; Thangaraju, M.; Suresh, V.S.; Sneigowski, T.; Bhutia, Y.D.; Pruitt, K.; Ganapathy, V. Expression and function of slc38a5, an amino acid-coupled na+/h+ exchanger, in triple-negative breast cancer and its relevance to macropinocytosis. Biochem. J. 2021, 478, 3957–3976. [Google Scholar] [CrossRef]
- Smith, D.K.; Kates, L.; Durinck, S.; Patel, N.; Stawiski, E.W.; Kljavin, N.; Foreman, O.; Sipos, B.; Solloway, M.J.; Allan, B.B.; et al. Elevated serum amino acids induce a subpopulation of alpha cells to initiate pancreatic neuroendocrine tumor formation. Cell Rep. Med. 2020, 1, 100058. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A variant of slc1a5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020, 31, 267–283.e212. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mtor and metabolism in cancer: Lessons and innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-coa desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Xu, F.L.; Wu, X.H.; Chen, C.; Wang, K.; Huang, L.Y.; Xia, J.; Liu, Y.; Shan, X.F.; Tang, N. Slc27a5 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by downregulating glutathione reductase. Cell Death Dis. 2023, 14, 22. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes. Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Kang, H.W.; Kim, S.Y.; Kim, M.J.; Jeong, J.W.; Hong, W.C.; Fang, S.; Kim, H.S.; Lee, Y.S.; Kim, H.J.; et al. Ivermectin and gemcitabine combination treatment induces apoptosis of pancreatic cancer cells via mitochondrial dysfunction. Front. Pharmacol. 2022, 13, 934746. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, H.S.; Kim, H.J.; Kang, H.W.; Lee, D.E.; Kim, M.J.; Hong, W.C.; Kim, J.H.; Kim, M.; Cheong, J.H.; et al. The role of loxl2 induced by glucose metabolism-activated nf-κb in maintaining drug resistance through emt and cancer stemness in gemcitabine-resistant pdac. J. Mol. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.D.; Ma, L.; Flagella, M.; Geeganage, S.; Gelbert, L.M.; Slapak, C.A. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004, 64, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Z.; Li, K.; Gong, L.; Yang, Q.; Huang, X.; Hong, C.; Ding, M.; Yang, H. Linc-dync2h1-4 promotes emt and csc phenotypes by acting as a sponge of mir-145 in pancreatic cancer cells. Cell Death Dis. 2017, 8, e2924. [Google Scholar] [CrossRef] [PubMed]
- Funamizu, N.; Honjo, M.; Tamura, K.; Sakamoto, K.; Ogawa, K.; Takada, Y. Micrornas associated with gemcitabine resistance via emt, tme, and drug metabolism in pancreatic cancer. Cancers 2023, 15, 1230. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Y.; Ma, X.; Hu, H. The influence of cell cycle regulation on chemotherapy. Int. J. Mol. Sci. 2021, 22, 6923. [Google Scholar] [CrossRef]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer cell metabolism in hypoxia: Role of hif-1 as key regulator and therapeutic target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, C.; Tang, D.; Dou, Q.P.; Shen, J.; Chen, X. The role of ferroptosis in lung cancer. Biomark. Res. 2021, 9, 82. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Sappington, D.R.; Siegel, E.R.; Hiatt, G.; Desai, A.; Penney, R.B.; Jamshidi-Parsian, A.; Griffin, R.J.; Boysen, G. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of a549 and h460 lung cancer cell lines. Biochim. Biophys. Acta 2016, 1860, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. Srebp-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of pi3k-akt-mtor signaling suppresses ferroptosis via srebp-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Wattanawongdon, W.; Hahnvajanawong, C.; Namwat, N.; Kanchanawat, S.; Boonmars, T.; Jearanaikoon, P.; Leelayuwat, C.; Techasen, A.; Seubwai, W. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int. J. Oncol. 2015, 47, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes. Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Martín-Saiz, L.; Guerrero-Mauvecin, J.; Martín-Sanchez, D.; Fresnedo, O.; Gómez, M.J.; Carrasco, S.; Cannata-Ortiz, P.; Ortiz, A.; Fernandez, J.A.; Sanz, A.B. Ferrostatin-1 modulates dysregulated kidney lipids in acute kidney injury. J. Pathol. 2022, 257, 285–299. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Kim, E.S.; Koo, J.S. Amino acid transporters and glutamine metabolism in breast cancer. Int. J. Mol. Sci. 2018, 19, 907. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, K.; Huang, X.; Zhao, C.; Mei, Y.; Li, X.; Jiao, L.; Yang, H. Lncrna slc7a11-as1 promotes chemoresistance by blocking scf(β-trcp)-mediated degradation of nrf2 in pancreatic cancer. Mol. Ther. Nucleic Acids 2020, 19, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of nrf2 in ros-mediated tumor chemoresistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. Srebp activity is regulated by mtorc1 and contributes to akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Tang, D. Ferroptosis by lipid peroxidation: The tip of the iceberg? Front. Cell Dev. Biol. 2021, 9, 646890. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Sequence (5′→3′) |
|---|---|
| GAPDH F GAPDH R SLC38A5 F SLC38A5 R RRM1 F RRM1 R E-Cadherin F E-Cadhern R N-Cadhenn F N-Cadhenin R SLC7A11 F SLC7A11 R FTL F FTL R FTH F FTH R ACSL4 F ACSL4 R ALOX12 F ALOX12 R NRF2 F NRF2 R GPX4 F GPX4 R SREBP1 F SREBP1 R SCD1 F SCD1 R BAX F BAX R Caspase 3 F Caspase 3 R Caspase 9 F Caspase 9 R Mouse GAPDH F Mouse GAPDH R Mouse SLC38A5 F Mouse SLC38A5 R Mouse NRF2 F Mouse NRF2 R Mouse GPX4 F Mouse GPX4 R Mouse E-Cadhenn F Mouse E-Cadhenn R Mouse N-Cadherin F Mouse N-Cadherin R | GTCTCCTCTGACTTCAACAGCG ACCACCCTGTTGCTGTAGCCAA GTTGGGGCCATGTCCAGTTA AGTGTTTCATGAGGGCGAGG TCTCAGACGGAAACAGGCAC GCACAGGTTGCTGCATTTGA GCTCCTGAAAAGAGAGTGGAAG TGGCAGTGTCTCTCCAAATCCG CCTCCAGAGTTTACTGCCATGAC GTAGGATCTCCCCCACTGATTC TGGTCAGAAAGCCTGTTGTGT TGCTCCAATGATGGTGCCAA AAAGCTGAACCAGGCCCTTT CGAAGAGTACTCGCCCAGC AGCTCTACGCCTCCTACGTT AAGGAAGATTCGGCCACCTC TGGTTCTACTGGCCGACCTA TAGCACATGAGCCAAAGGCA GTCAACACAGGCCAGATGGA TGATGCACGTGGTCTTCACA TCCAGTCAGAAACCAGTGGAT GAATGTCTGCGCCAAAAGCTG AGAGATCAAAGAGTTCGCCGC TCTTCATCCACTTCCACAGCG TTCTCACCTCCCAGCTCTGT AGGTGAGACGTGCCAGACTT TTCGTTGCCACTTTCTTGCG CCGGGGGCTAATGTTCTTGT TCAGGATGCGTCCACCAAGAAG TGTGTCCACGGCGGCGGCAATCATC GGAAGCGAATCAATGGACTCTGG GCATCGACATCTGTACCAGACC GTTTGAGGACCTTCGACCACCT CAACGTACCAGGAGGCACTCTT TGTGAACGGATTTGGCCGTA ACTGTGCCGTTGAATTTGCC TGGCACACACTGGAGTCATC ACGGATGCCTACAACACTGG AACAGAACGGCCCTAAAGCA TGGGATTCACGCATAGGAGC CTAGTCGATCTGCATGCCCG GGCATCGTCCCCATTTACAC CAACGATCCTGACCAGCAGT TGTATTGCTGCTTGGCCTCA AGCCAACCTAACTGTCACGG ATTGCTGTTGGGGTCTGTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.J.; Kim, H.S.; Kang, H.W.; Lee, D.E.; Hong, W.C.; Kim, J.H.; Kim, M.; Cheong, J.-H.; Kim, H.J.; Park, J.S. SLC38A5 Modulates Ferroptosis to Overcome Gemcitabine Resistance in Pancreatic Cancer. Cells 2023, 12, 2509. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12202509
Kim MJ, Kim HS, Kang HW, Lee DE, Hong WC, Kim JH, Kim M, Cheong J-H, Kim HJ, Park JS. SLC38A5 Modulates Ferroptosis to Overcome Gemcitabine Resistance in Pancreatic Cancer. Cells. 2023; 12(20):2509. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12202509
Chicago/Turabian StyleKim, Myeong Jin, Hyung Sun Kim, Hyeon Woong Kang, Da Eun Lee, Woosol Chris Hong, Ju Hyun Kim, Minsoo Kim, Jae-Ho Cheong, Hyo Jung Kim, and Joon Seong Park. 2023. "SLC38A5 Modulates Ferroptosis to Overcome Gemcitabine Resistance in Pancreatic Cancer" Cells 12, no. 20: 2509. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12202509