Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy


Division of Respiratory Medicine, Department of Internal Medicine, Kobe University Graduate School of Medicine, 7-5-1 Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan
*
Author to whom correspondence should be addressed.
Cells 2018, 7(11), 212; https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110212
Submission received: 25 October 2018
/
Revised: 13 November 2018
/
Accepted: 15 November 2018
/
Published: 15 November 2018
(This article belongs to the Special Issue Epidermal Growth Factor Receptor Signaling)
Abstract
:Treatment with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) improves the overall survival of patients with EGFR-mutated non-small-cell lung cancer (NSCLC). First-generation EGFR-TKIs (e.g., gefitinib and erlotinib) or second-generation EGFR-TKIs (e.g., afatinib and dacomitinib) are effective for the treatment of EGFR-mutated NSCLC, especially in patients with EGFR exon 19 deletions or an exon 21 L858R mutation. However, almost all cases experience disease recurrence after 1 to 2 years due to acquired resistance. The EGFR T790M mutation in exon 20 is the most frequent alteration associated with the development of acquired resistance. Osimertinib—a third-generation EGFR-TKI—targets the T790M mutation and has demonstrated high efficacy against EGFR-mutated lung cancer. However, the development of acquired resistance to third-generation EGFR-TKI, involving the cysteine residue at codon 797 mutation, has been observed. Other mechanisms of acquired resistance include the activation of alternative pathways or downstream targets and histological transformation (i.e., epithelial–mesenchymal transition or conversion to small-cell lung cancer). Furthermore, the development of primary resistance through overexpression of the hepatocyte growth factor and suppression of Bcl-2-like protein 11 expression may lead to problems. In this report, we review these mechanisms and discuss therapeutic strategies to overcome resistance to EGFR-TKIs.
Keywords:
acquired resistance; T790M; C797S; alternative pathway; transformation; primary resistance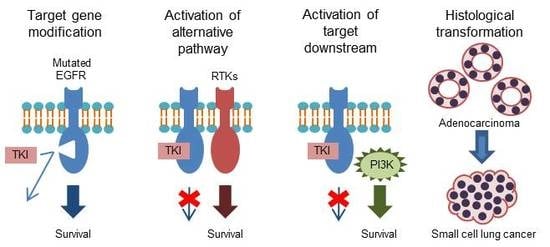
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The epidermal growth factor receptor (EGFR) is a receptor protein penetrating the cell membrane. The kinase domain is composed of the N and C lobes and an adenosine triphosphate (ATP) binding cleft between the two lobes. Following the binding of a ligand to the receptor, an asymmetric dimer is formed and the phosphate of ATP is transferred to the tyrosine residue of the regulatory domain. Various proteins are bound to this phosphorylated tyrosine and signals are transmitted downstream through the rat sarcoma (RAS)-rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB, also known as AKT) pathways. EGFR tyrosine kinase inhibitors (TKIs) competitively inhibit ATP with cleft of the kinase domain. Gefitinib, a first-generation EGFR-TKI, greatly improved the prognosis of EGFR gene mutation-positive patients with non-small-cell lung cancer (NSCLC) [1,2]. The vast majority (93%) of EGFR activation mutations (common mutations) occur in exons 19 to 21. In addition, deletion mutations in exon 19 and L858R mutations in exon 21 are particularly frequent (44.8% and 39.8%, respectively) [3]. Although EGFR-TKIs show an excellent therapeutic effect on EGFR-mutated NSCLC, most cancers develop resistance to EGFR-TKI. The T790M mutation in exon 20 is recognized in approximately half of EGFR-TKI tolerance cases [4,5]. In this review, we will summarize the mechanism of EGFR-TKI tolerance and present treatment strategies to overcome resistance. There are several outstanding reviews so far [6,7,8,9], but therapeutic drugs progress one after another and a new resistance mechanism emerges. Therefore, we would like to review focusing on the novel acquired resistance of the third-generation EGFR-TKI, osimertinib based on the latest academic information.
2. Acquired Resistance
2.1. T790M “Gatekeeper” Mutation
Despite the initial response to first-generation EGFR-TKIs, patients with NSCLCs harboring EGFR mutations acquire resistance to these agents, with a median time to disease progression of approximately 12 months [10,11,12,13,14]. The mechanisms of acquired resistance to first-generation EGFR-TKIs are summarized in Figure 1.
T790M was the first reported acquired resistance gene [4]. The T790M mutation structurally inhibits the binding of first-generation EGFR-TKIs to the ATP binding site. When the T790M mutation is added to the activation mutation of EGFR, the affinity of EGFR for ATP is increased, whereas the binding property of EGFR-TKIs is relatively decreased. Hence, the downstream signal is not inhibited and the cancer is tolerated. It has been thought that a small number of cancer cells having a secondary T790M mutation in addition to the active EGFR mutation may already be present prior to treatment with EGFR-TKIs and gradually become dominant during treatment with first-generation EGFR-TKIs (e.g., gefitinib and erlotinib). However, recent research revealed that T790M-positive cells also occur from initially T790M-negative single cells via genetic evolution [17].
Irreversible EGFR-TKIs were developed to overcome the T790M-mediated resistance. These second-generation EGFR-TKIs (e.g., afatinib and dacomitinib) irreversibly bind to the cysteine residue of the EGFR [18]. Although the second-generation EGFR-TKIs exert an effect on T790M, the half-maximal inhibitory concentration (IC50) is 30 to 100-fold higher than that observed with the activated mutation of EGFR [19]. Integrated analysis of two phase III studies (LUX-Lung 3 and 6 trials) showed that afatinib significantly prolonged overall survival compared with chemotherapy in patients with an activated EGFR mutation (hazard ratio, HR 0.81; 95% confidence interval, CI 0.66 to 0.99; p = 0.037; 27.3 vs. 24.3 months, respectively) [20,21]. Furthermore, a randomized phase IIb trial comparing afatinib and gefitinib in patients with NSCLC harboring an active EGFR mutation (LUX-Lung 7 trial) revealed superiority of afatinib regarding progression-free survival (PFS) (HR, 0.73; 95% CI, 0.57 to 0.95; p = 0.0073; 11.0 vs. 10.9 months, respectively) [22]. These results suggest that irreversible EGFR-TKIs delay the expression of T790M compared with first-generation EGFR-TKIs. However, the T790M mutation was detected in 36.4% or 47.6% of patients treated with afatinib [8,23]. In addition, the effect of afatinib was limited in patients with advanced NSCLC and T790M mutation who progressed during prior treatment with first-generation EGFR-TKIs [24].
The third-generation EGFR-TKIs (e.g., osimertinib) are pyrimidine-based irreversible EGFR-TKIs targeting the T790M mutation. Osimertinib irreversibly binds to the EGFR kinase by targeting the cysteine residue at codon 797 (C797) and has a highly selective inhibition activity [25]. It is effective for the activation of EGFR-mutated cell lines (IC50: 8–17 nmol/L in PC9 harboring an exon 19 deletion) and EGFR T790M-mutated cell lines (IC50: 5–11 nmol/L in H1975 harboring L858R/T790M). However, osimertinib is less effective in EGFR wild-type cell lines than the early-generation EGFR TKIs (IC50: 650 nmol/L in Calu3 and 461 nmol/L in H2073) [25,26,27,28]. The phase I/II clinical trials of osimertinib (AURA 1/AURA 2 trials) involving EGFR mutation-positive patients with NSCLC who became resistant to EGFR-TKI demonstrated that the median PFS was 9.6 months (95% CI, 8.3 to not reached), the overall response rate (ORR) of negative cases was 21% (95% CI, 12 to 34), and the median PFS was 2.8 months (95% CI, 2.1 to 4.3) [29]. A phase III trial confirming the effect of osimertinib in NSCLC patients with the EGFR T790M mutation (AURA3 trial) showed superiority of osimertinib over platinum therapy plus pemetrexed regarding PFS (HR, 0.30; 95% CI, 0.23 to 0.41; p < 0.001; 10.1 vs. 4.4 months, respectively) [30]. Intriguingly, another phase III trial confirming the effect of osimertinib in untreated NSCLC patients with an activated EGFR mutation (FLAURA trial) showed superiority of osimertinib over gefitinib or erlotinib regarding PFS (HR, 0.46; 95% CI, 0.37 to 0.57; p <0.001; 18.9 vs. 10.2 months, respectively) [31].
2.2. Acquired Resistance to Osimertinib
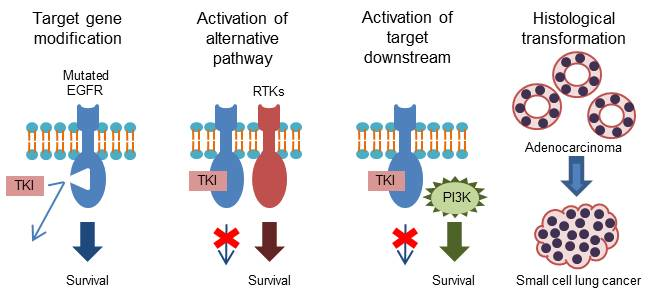
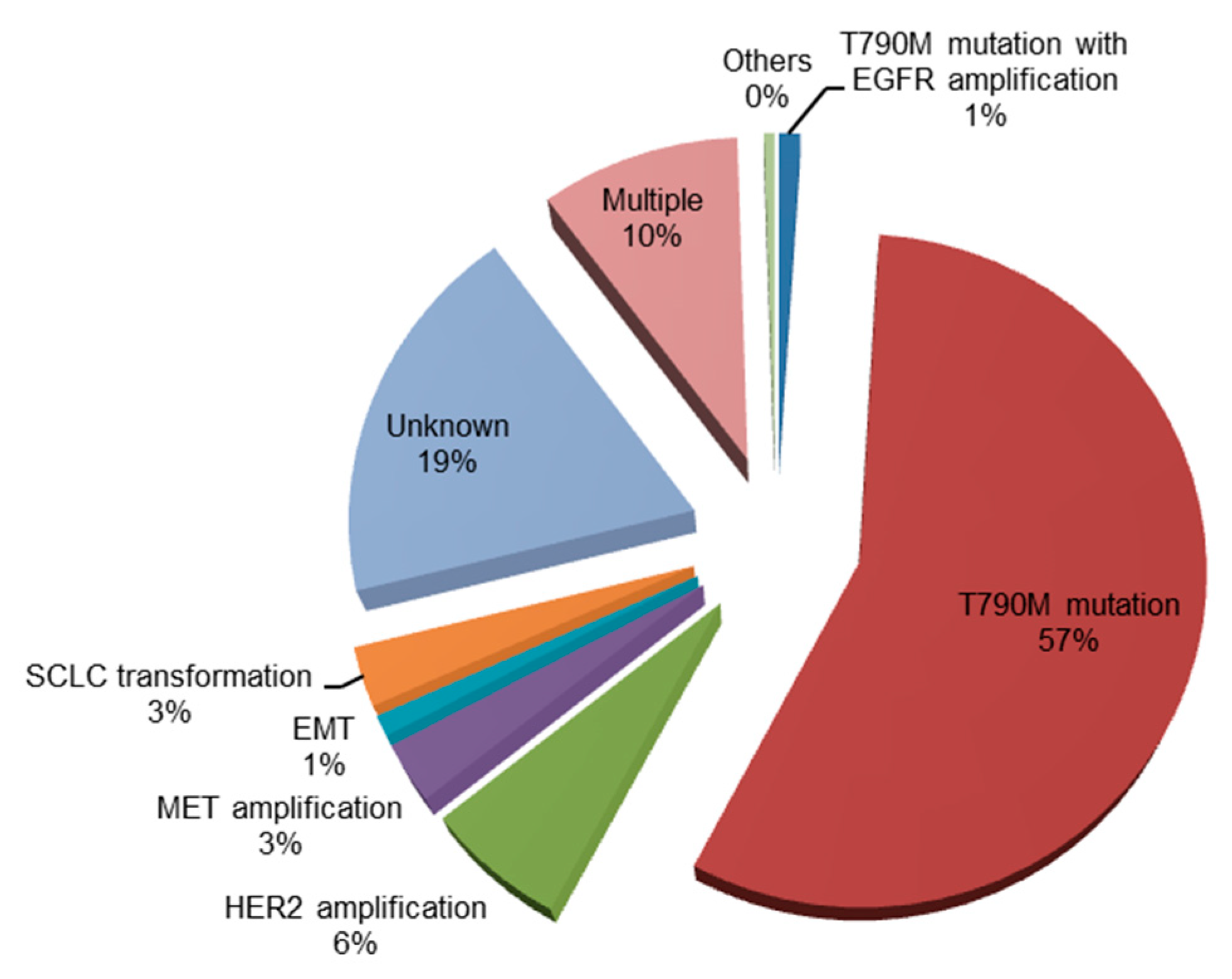
Mechanisms of acquired resistance to osimertinib are shown in Figure 2.
2.2.1. C797S Mutation
Although osimertinib shows an excellent clinical effect in the activation of both the EGFR and EGFR T790M mutations, the development of resistance is inevitable [32,37,38,39]. The C797 is present in the ATP-binding pocket in which EGFR-TKIs bind irreversibly. Therefore, the point mutation of C797S in exon 20 of EGFR—a common mechanism of acquired resistance to osimertinib—induces resistance to third-generation EGFR-TKIs [18]. Indeed, C797S-mediated resistance third-generation EGFR-TKIs develops approximately within 1 year [32,40]. Intriguingly, previous clinical trials showed that the C797S is more likely to occur in EGFR exon 19 deletion mutation [32,38,39,41,42]. The localization of T790M and C797S on the allele may be helpful in determining the treatment strategy for C797S. If C797S and T790M are in trans (i.e., on separate alleles), the resistant cells may be sensitive to the combination of first- and third-generation EGFR-TKIs [43,44]. However, if C797S and T790M are in cis (i.e., on the same allele), the cells are resistant to all EGFR-TKIs [45]. The majority of EGFR-sensitizing mutations (i.e., exon 19 deletion, exon 20 insertion, and L858R/T790M EGFR mutants), except L858R, do not require asymmetric dimerization. Hence, they do not respond to treatment with cetuximab [46]. In contrast, the L858R/T790M/C797S which partly require dimerization moderately respond to treatment with cetuximab. To overcome the EGFR/T790M/C797S mutation, use of a potent anaplastic lymphoma kinase (ALK) inhibitor (i.e., brigatinib) or mutant selective allosteric inhibitor may be an option [47,48].
2.2.2. Other EGFR Mutation
Other EGFR mutations include L792X mutation, G796S mutation, L718Q mutation and exon 20 insertion [49,50]. Exon 20 insertion inhibits the binding of EGFR-TKI to its binding site by adding residues at the N-lobe of EGFR [51]. Further studies are required for EGFR mutation, but cytotoxic anticancer agent is one of the treatment options for L718Q.
2.2.3. Oncogenic Fusion
The latest research revealed a new resistance mechanism using plasma ctDNA genomic profile. Some of these novel mechanisms are oncogenic fusions, including fibroblast growth factor receptor 3 (FGFR3)-transforming acidic coiled coil-containing protein 3 (TACC3) gene fusion, neuropathic tyrosine kinase receptor type 1 (NTRK1)-tropomyosin 3 (TPM3) gene fusion, rearranged during transfection (RET)-ELKS-Rab6-interacting protein-CAST family member 1 (ERC1) gene fusion, and spectrin beta non-erythrocytic 1 (SPTBN1)-ALK gene fusion [50,52]. The SPTBN1-ALK fusion gene was formed by the fusion of exon 7 of the SPTBN1 gene with exon 20 of the ALK gene, which was first identified in colorectal cancer [53]. SPTBN1-ALK gene fusion is a potential biomarker of cancer refractory to therapy and of a relatively poor prognosis. However, the novel SPTBN1-ALK fusion gene may become a potential target for anti-tumor therapy [54].
2.2.4. Cell Cycle Gene Alterations
2.2.5. MAPK/PI3K alterations
MAPK/PI3K alterations include v-raf murine sarcoma viral oncogene homolog B1 (BRAF)V600E mutation [55], phosphoinositide-3-kinase P110α catalytic subunit (PIK3CA)E545K mutation [50], and Kirsten rat sarcoma viral oncogene homolog (KRAS)G12D mutation [50]. These MAPK/PI3K alterations will be described in detail in Section 2.4.1, Section 2.4.2 and Section 2.4.3.
2.2.6. Others
Other mechanisms of acquired resistance to osimertinib include wild-type EGFR amplification [33], loss of T790M and other activated mutation [33], fibroblast growth factor receptor (FGFR) amplification [33], mesenchymal-epithelial transition factor (MET) amplification [56], human epidermal growth factor receptor 2 (HER2) [56], and SCLC transformation [33]. Resistance mechanisms are heterogeneous, and each number is relatively small. An in-vitro study demonstrated that the combination of rociletinib—a third-generation EGFR-TKI—or afatinib with cetuximab has an effect on wild-type EGFR amplification [57]. Loss of T790M was associated with a slightly shorter median survival. Use of cytotoxic agents is a therapeutic option in patients with loss of T790M without other mutation, and SCLC transformation.
2.3. Activation of Alternative Pathways
2.3.1. MET Gene Amplification
The most common alternative pathway is MET amplification, accounting for 5% to 10% of cases of acquired resistance to EGFR-TKIs [16,58]. The MET gene encodes the MET tyrosine kinase receptor. MET is activated by the hepatocyte growth factor (HGF) and potentiates survival through activation of ERBB3/phosphoinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling [58,59,60]. MET amplification induces autophosphorylation of MET protein and associates with ERBB3 which activates the PI3K/AKT pathway, leading to the development of resistance to EGFR-TKIs [58]. Crizotinib is a kinase inhibitor with multiple targets (including MET). Two case reports have reported that crizotinib was effective against EGFR-mutated NSCLC harboring MET amplification [61,62]. The novel potent and selective MET inhibitor capmatinib (INC280) has demonstrated preclinical activity in combination with gefitinib in EGFR-mutant, MET-amplified/overexpressing models of acquired resistance to EGFR-TKIs. A phase Ib/II study investigating the safety and efficacy of capmatinib plus gefitinib in patients with EGFR-mutated and MET-dysregulated (amplified/overexpressing) NSCLC revealed an ORR of 47% in patients with a MET gene copy number ≥6 with tolerable toxicity [63].
2.3.2. HGF Overexpression
HGF overexpression is frequently observed in 61% of patients with acquired resistance to EGFR-TKIs [64]. A high level of HGF in the serum is a poor prognostic factor in NSCLC patients treated with first- and second-generation EGFR-TKIs [65,66,67]. The HGF activates MET and induces resistance to EGFR-TKIs via the PI3K/AKT pathway [68]. In contrast to MET amplification, HGF overexpression induces resistance to EGFR-TKIs by activating the MET/PI3K/AKT pathway without the involvement of human epidermal growth factor receptor type 3 (HER3, also known as ERBB3) [68]. In a preclinical study, the combination of onartuzumab—a monovalent antibody to MET—with erlotinib blocks the HGF-induced activation of MET [69].
2.3.3. Insulin-Like Growth Factor (IGF) Upregulation
The IGF receptor 1 (IGF1R) is a transmembrane heterotetrameric protein playing a role in the promotion of oncogenic transformation, growth, and survival of cancer cells [70]. IGF1R activates two signal transduction pathways, namely the RAS/RAF/MEK/ERK and PI3K/AKT pathways. IGF1R plays a role in the development of resistance to gefitinib in the absence of the EGFR T790M mutation [71]. In addition, IGF1R is associated with the development of resistance to irreversible second-generation EGFR-TKIs in EGFR T790M-mutated lung cancer cell lines [71]. The small molecule IGF1R inhibitor AG-1024 or the monoclonal anti-IGF1/2 blocking antibody BI 836845 has been reported to restore sensitivity to treatment with third-generation EGFR-TKIs [72].
2.3.4. HER2 Amplification
HER2/ERBB2 is a member of the ERBB family and is unable to form homodimers because HER2 lack its specific ligand which has a crucial role in dimerization [73]. Therefore, HER2 is present either solely as monomer or heterodimer with other ligand-bound family members including EGFR [73,74]. HER2 and EGFR indirectly activate PI3K, and amplification of HER2 is associated with the development of acquired resistance to EGFR-TKI occurring in 12% of active EGFR-mutated NSCLC patients who develop resistance [75]. The combination of afatinib and cetuximab inhibits HER2 phosphorylation and delays the development of resistance compared with single-agent treatment (erlotinib or afatinib) in vitro and in vivo [75,76]. Trastuzumab—a human monoclonal antibody that interferes with the HER2 receptor—exerted a modest effect in lung cancer patients with HER2 amplification and HER2 overexpression [77,78]. A phase II trial involving EGFR-mutated NSCLC patients with HER2 activation showed an ORR of 41% and a median duration of response of 9 months [79].
2.3.5. Growth Arrest-Specific 6 (GAS6)/ Anexelekto (AXL) Activation
The GAS6 is a ligand of AXL which is receptor tyrosine kinase. The activation of the AXL by GAS6 leads to activation of the MEK/ERK and PI3K/AKT pathways and acquired resistance to EGFR-TKIs [80]. Furthermore, GAS6/AXL activation is often accompanied by EMT [80]. The VEGFR/MET/AXL inhibitor foretinib restored partial sensitivity of EGFR-TKI-resistant cells [27]. Furthermore, multiple AXL inhibitors such as multitarget kinase inhibitors (S49076, cabozantinib, ASLAM002, MGCD265, and MGCD516) and a specific AXL inhibitor (BGB324) are currently being developed for the treatment of solid tumors including NSCLC [81].
2.4. Activation of Downstream Targets
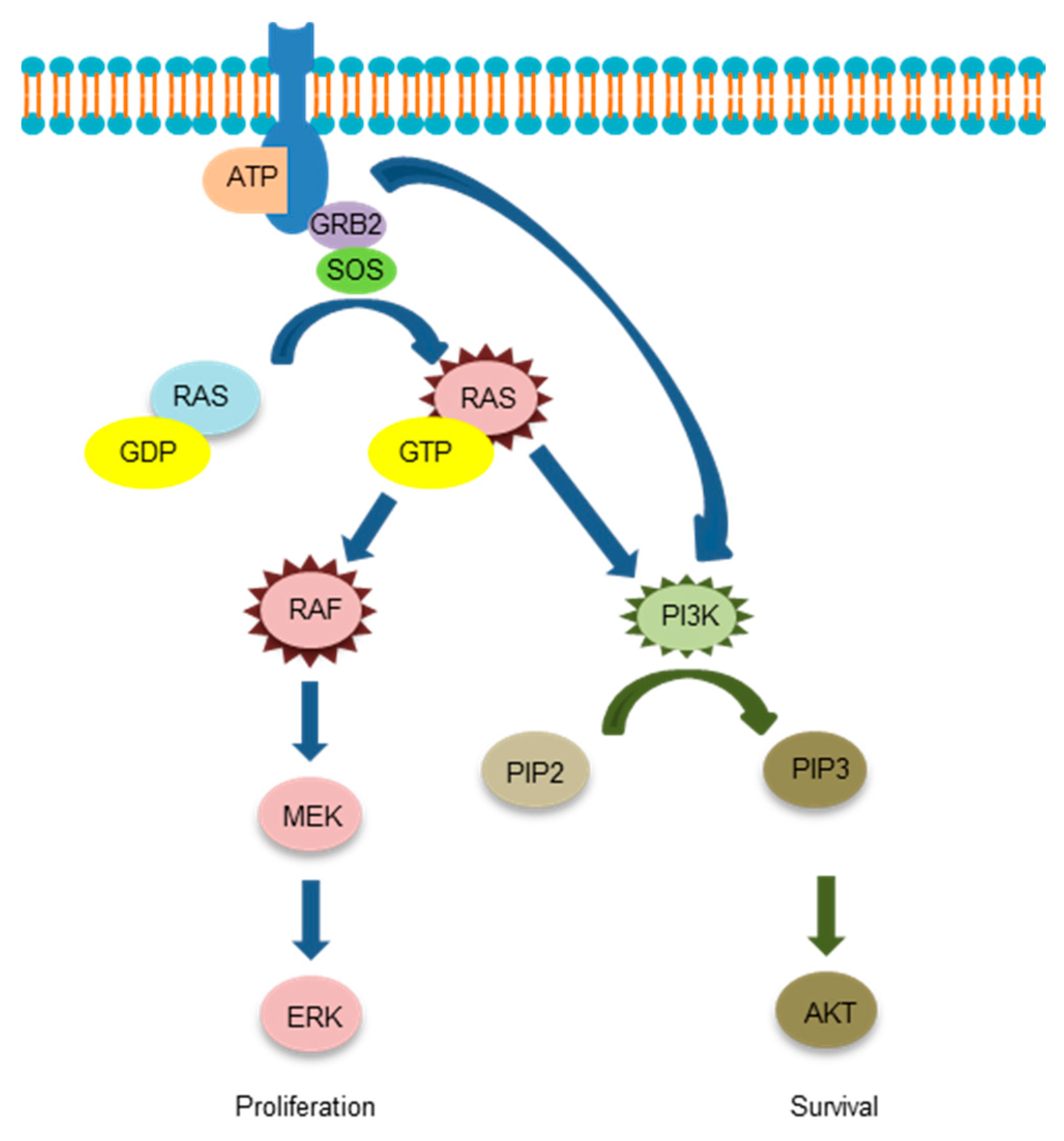
Schema of downstream signaling of EGFR is shown in Figure 3.
2.4.1. KRAS Mutation
Previously, the KRAS mutation and EGFR were considered to be exclusive genes. Indeed, KRAS and EGFR were only found in <2% of NSCLC patients [82,83,84,85]. Moreover, the KRAS mutation was observed only in a mouse model, unlike in tissue samples obtained from patients with acquired resistance to first- and second-generation EGFR-TKIs [15,23,86,87]. Recently, analysis of circulating tumor DNA revealed the emergence of distinct KRAS activating mutations (G12A, Q61H, and A146T) in three of 43 EGFR-mutant NSCLC patients with acquired activating mutations [41]. The RAS inhibitor was discovered through in-silico screening. This inhibitor suppresses the metastasis of colorectal cancer cell lines via the downregulation of lysyl oxidase [88,89]. However, the effect of RAS inhibitor on EGFR-acquired resistance remains unclear.
2.4.2. BRAF Mutation
BRAF mutations (G469A and V600E) were found in two of 195 (1%) patients with acquired resistance to EGFR-TKIs [87]. The combination of encorafenib (LGX818)—a BRAFV600E inhibitor—with osimertinib suppresses the colony formation of cells, which were collected and cultured at the time of progression in malignant pleural effusion of patients treated with osimertinib [55].
2.4.3. PIK3CA Mutation
PIK3CA encodes p110α and is one of the class I PI3K isoforms [90]. It was found in one of 37 (3%) patients with acquired resistance to EGFR-TKIs [15]. The dual inhibitor of PI3K/mTOR NVP-BEZ235 effectively inhibits the growth of gefitinib-resistant NSCLC cells by down-regulating PI3K/AKT/mTOR phosphorylation [91]. In a phase I clinical trial, the PI3K inhibitor temporarily decreased the size of the tumor in a patient with EGFR L858R/T790M/PIK3CA mutation after progression during treatment with erlotinib. However, the tumor relapsed within 1 month [92]. A phase II study showed that the combination of the AKT inhibitor MK-2206 with erlotinib in patients with advanced EGFR-mutated NSCLC who previously progressed during treatment with erlotinib showed an ORR of 9% and a PFS of 4.4 months [93].
2.4.4. Phosphatase and Tensin Homolog (PTEN) Deletion
PTEN at chromosome 10q23.3 is a tumor suppressor gene with sequence homology to protein tyrosine phosphatases. The PTEN gene is inactivated in several types of human tumors by mutation, homozygous deletion, promoter methylation, and translational modification [94]. It negatively regulates the PI3K/AKT signaling pathway [95]. The loss of PTEN function increases the level of phosphatidylinositol-(3,4,5)-triphosphate (PIP-3; a product of PI3K) and leads to AKT hyperactivation. Loss of PTEN has been shown to be involved in the development of resistance to EGFR-TKIs in certain tumor cell lines [96,97,98]. Recently, peroxisome proliferator-activated receptor gamma (PPARγ) agonist drugs such as rosiglitazone sensitize PTEN-deficient resistant human lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy [99].
2.4.5. NF-1 Deletion
NF-1 is a tumor suppressor gene encoding a neurofibromin, namely GTPase-activating protein that negatively regulates p21-RAS signaling [100]. NF-1 exerts a reverse effect on RAS by increasing the guanosine triphosphate (GTP) hydrolysis rate. Hence, its function as a tumor suppressor is believed to occur by constraining RAS activity in the normal cell. NF-1 deletion activates the MAPK pathway and was associated with the development of primary and acquired resistance of lung adenocarcinomas to EGFR TKIs in patients [101]. A MAP–ERK kinase (MEK) inhibitor restored sensitivity to erlotinib in lung cancer which was reduced NF-1 expression in vitro and in vivo [101]. Indeed, patients with plexiform neurofibromas, whose tumors harboring NF1 mutation, have shown response to this strategy of downstream inhibition [102].
2.4.6. v-crk Sarcoma Virus CT10 Oncogene Homolog (Avian)-Like (CRKL) Amplification
CRKL is an oncogene encoding an adaptor protein that participates in signal transduction of the RAS/RAF/MAPK pathway [103]. CRKL amplification was detected in EGFR-TKI-treated lung adenocarcinoma patients [103]. CRKL-specific short hairpin RNA (shRNA) suppressed the tumorigenic growth of NSCLC cells harboring the CRKL amplification [103].
2.5. Histologic Transformation
2.5.1. EMT
EMT is critical steps of morphogenesis by interconverting epithelial cell types into cells with mesenchymal attributes [104]. Molecular changes, including the loss of cell junction proteins (e.g., E-cadherin and claudins) or up-regulation of mesenchymal markers (e.g., vimentin and fibronectin), are associated with EMT [105]. Transforming growth factor (TGF)-β, Wnt, and Notch signals from the microenvironment are also associated with the induction and maintenance of EMT [104,106,107,108]. EMT was detected in 1% to 2% of patients with resistance to EGFR-TKIs [15,80,109]. Inappropriate induction of EMT in tumor cells is associated with tumor invasion, metastasis, drug resistance, and stem cell property [110,111]. Some cytotoxic agents (i.e., cisplatin, gemcitabine, etoposide, and vinorelbine) have demonstrated an antitumor effect in cells with a mesenchymal phenotype, whereas other cytotoxic agents (i.e., docetaxel and pemetrexed) are not effective in these cells [18].
2.5.2. SCLC Transformation
Transformation from EGFR-mutated adenocarcinoma to SCLC occurs in 5% of patients with acquired resistance to EGFR-TKIs [15]. Recent studies revealed that SCLC transformation is associated with inactivation of Rb and p53 [112,113]. Chemotherapy with etoposide and carboplatin was reported to be effective in two cases of SCLC transformation from EGFR-mutated adenocarcinoma [114].
3. Primary Resistance
3.1. HGF Overexpression
In a study, 29% of cases showed high resistance to HGF (primary resistance) and did not show marked response to EGFR-TKIs, despite the presence of an EGFR mutation [64]. In addition, the HGF reduced the susceptibility to irreversible EGFR-TKIs in H1975 cells harboring both the L858R activating mutation and T790M mutation in EGFR [115].
3.2. Decrease of BIM
Bcl-2-like protein 11 (BIM) is a member of the Bcl-2 protein family and a crucial mediator of apoptosis. EGFR-mutated lung cancer cells with low expression levels of BIM exhibit low sensitivity to treatment with EGFR-TKIs [116]. A decrease in BIM polymorphism, which was thought to be primary resistance to EGFR-TKI, was detected in 12% of East Asians [18]. Use of proapoptotic BH3 mimetics may be effective for the decrease of BIM.
3.3. Others
EGFR exon 20 insertions [117,118], the PIK3CA mutation [119], and PTEN deletion contribute to inhibition of cell proliferation and survival or suppression of apoptosis, which lead to primary resistance to EGFR-TKIs [120]. The majority of patients with advanced lung adenocarcinomas harboring EGFR exon 20 insertions do not respond to treatment with EGFR-TKIs. However, cytotoxic chemotherapy in these patients offers a similar overall survival to that observed in patients with a sensitizing EGFR mutation [121].
4. Summary
We reviewed the mechanisms of acquired and primary resistance to EGFR-TKIs. The development of appropriate treatment strategies according to the different mechanisms of resistance is warranted. Overcoming resistance to EGFR-TKIs may prolong patient survival. Thus, further basic research on the molecular mechanisms involved in this process and clinical research on potential therapeutic options are of crucial importance.
Author Contributions
T.N. wrote the article, T.N., M.T., and Y.N. contributed to the concept, and critically appraised and edited the article.
Funding
This research received no external funding.
Acknowledgments
We would like to express our gratitude to the members of the Division of Respiratory Medicine, Kobe University Graduate School of Medicine, for their helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Togashi, Y.; Yatabe, Y.; Mizuuchi, H.; Jangchul, P.; Kondo, C.; Shimoji, M.; Sato, K.; Suda, K.; Tomizawa, K.; et al. EGFR exon 18 mutations in lung cancer: Molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first- or third-generation TKIs. Clin. Cancer Res. 2015, 21, 5305–5313. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tam, K.Y. Combination strategies using EGFR-TKi in NSCLC therapy: Learning from the gap between pre-clinical results and clinical outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.; Gerber, D.; Gainor, J.F.; Heist, R.S.; Temel, J.S.; Shaw, A.T.; Fidias, P.; Muzikansky, A.; Engelman, J.A.; Sequist, L.V. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J. Thorac. Oncol. 2016, 11, 2022–2026. [Google Scholar] [CrossRef] [PubMed]
- Proto, C.; Lo Russo, G.; Corrao, G.; Ganzinelli, M.; Facchinetti, F.; Minari, R.; Tiseo, M.; Garassino, M.C. Treatment in EGFR-mutated non-small cell lung cancer: How to block the receptor and overcome resistance mechanisms. Tumori 2017, 103, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.; Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 2017, 8, 90557–90578. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O′Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Wu, S.G.; Liu, Y.N.; Tsai, M.F.; Chang, Y.L.; Yu, C.J.; Yang, P.C.; Yang, J.C.; Wen, Y.F.; Shih, J.Y. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016, 7, 12404–12413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. LUX-Lung 4: A phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Soejima, K.; Yasuda, H.; Hirano, T. Osimertinib for EGFR T790M mutation-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Thress, K.S.; Mooradian, M.; Heist, R.S.; Azzoli, C.G.; Temel, J.S.; Rizzo, C.; Nagy, R.J.; Lanman, R.B.; Gettinger, S.N.; et al. MET amplification (amp) as a resistance mechanism to osimertinib. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Guibert, N.M.; Paweletz, C.; Hu, Y.; Feeney, N.B.; Plagnol, V.; Poole, V.; Jones, G.; Oxnard, G.R. Early detection of competing resistance mutations using plasma next-generation sequencing (NGS) in patients (pts) with EGFR-mutant NSCLC treated with osimertinib. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Ou, Q.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Zhang, X.; Chang, Z.; Mao, Y.; Shao, Y.; Zhou, C. Investigating novel resistance mechanisms to third generation EGFR TKI osimertinib in non-small cell lung cancer patients using next generation sequencing. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Costa, D.B.; Kobayashi, S.S. Whacking a mole-cule: Clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl. Lung Cancer Res. 2015, 4, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.N.; Jung, K.S.; Yoo, K.H.; Cho, J.; Lee, J.Y.; Lim, S.H.; Kim, H.S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Acquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, e45–e47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination osimertinib and gefitinib in C797S and T790M EGFR-mutated non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC. J. Thorac. Oncol. 2016, 11, e121–e123. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Wu, Y.; Han, J.; Ahn, M.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Ying, J.; Lin, C.; Wu, J.; Guo, L.; Qiu, T.; Ling, Y.; Shan, L.; Zhou, H.; Zhao, D.; Wang, J.; et al. Anaplastic lymphoma kinase rearrangement in digestive tract cancer: Implication for targeted therapy in chinese population. PLoS ONE 2015, 10, e0144731. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.F.; Zhang, Y.; Liu, Y.Y.; Hong, X.H.; Liang, J.Y.; Tong, F.; Yang, J.S.; Liu, L. Lung adenocarcinoma harboring concomitant SPTBN1-ALK fusion, c-Met overexpression, and HER-2 amplification with inherent resistance to crizotinib, chemotherapy, and radiotherapy. J. Hematol. Oncol. 2016, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Chih-Hsin Yang, J. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Niederst, M.J.; Lennerz, J.K.; Dagogo-Jack, I.; Stevens, S.; Shaw, A.T.; Sequist, L.V.; Engelman, J.A. Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de novo MET amplification. J. Thorac. Oncol. 2016, 11, e83–e85. [Google Scholar] [CrossRef] [PubMed]
- York, E.R.; Varella-Garcia, M.; Bang, T.J.; Aisner, D.L.; Camidge, D.R. Tolerable and effective combination of full-dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR-mutant non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 2018, JCO2018777326. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kim, J.Y.; Lee, S.H.; Yoo, N.J.; Choi, B.G. Association between plasma hepatocyte growth factor and gefitinib resistance in patients with advanced non-small cell lung cancer. Lung Cancer 2011, 74, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Togashi, Y.; Fujita, S.; Sakamori, Y.; Okuda, C.; Kim, Y.H.; Mio, T.; Mishima, M. Clinical significance of serum hepatocyte growth factor and epidermal growth factor gene somatic mutations in patients with non-squamous non-small cell lung cancer receiving gefitinib or erlotinib. Med. Oncol. 2012, 29, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Cruz-Rico, G.; Soto-Perez-de-Celis, E.; Ramírez-Tirado, L.A.; Caballe-Perez, E.; Martínez-Hernández, J.N.; Martinez-Alvarez, I.; Soca-Chafre, G.; Macedo-Pérez, E.O.; Astudillo-de la Vega, H. Reduction in hepatocyte growth factor serum levels is associated with improved prognosis in advanced lung adenocarcinoma patients treated with afatinib: A phase II trial. Target. Oncol. 2016, 11, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Hashimoto, E.; Nakatani, N.; Abe, M.; Satoh, Y.; Sakata, K.; Fujii, T.; Fujimoto-Ouchi, K.; Sugimoto, M.; Nagahashi, S.; et al. Combining onartuzumab with erlotinib inhibits growth of non-small cell lung cancer with activating EGFR mutations and HGF overexpression. Mol. Cancer Ther. 2015, 14, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Tallini, G.; Finocchiaro, G.; Wilson, R.S.; Ligorio, C.; Giordano, L.; Toschi, L.; Incarbone, M.; Cavina, R.; Terracciano, L.; et al. Insulin-like growth factor receptor 1 (IGF1R) expression and survival in surgically resected non-small-cell lung cancer (NSCLC) patients. Ann. Oncol. 2010, 21, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2016, 55, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Park, S.; Kim, W.S.; Song, J.S.; Choi, C.M.; Sung, Y.H.; et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 22005–22015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.P.; Hastings, J.F.; Han, J.Z.; Croucher, D.R. The under-appreciated promiscuity of the epidermal growth factor receptor family. Front. Cell Dev. Biol. 2016, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Pirazzoli, V.; Ayeni, D.; Meador, C.B.; Sanganahalli, B.G.; Hyder, F.; de Stanchina, E.; Goldberg, S.B.; Pao, W.; Politi, K. Afatinib plus cetuximab delays resistance compared to single-agent erlotinib or afatinib in mouse models of TKI-naïve EGFR L858R-induced lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Gatzemeier, U.; Groth, G.; Butts, C.; Van Zandwijk, N.; Shepherd, F.; Ardizzoni, A.; Barton, C.; Ghahramani, P.; Hirsh, V. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann. Oncol. 2004, 15, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazières, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2016, 27, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Langen, J.D.; Kuiper, J.L.; Thunnissen, E.; Hashemi, S.M.; Monkhorst, K.; Smit, E.F. Trastuzumab and paclitaxel in patients (pts) with EGFR mutated non-small-cell lung cancer (NSCLC) that express HER2 after progression on EGFR TKI treatment. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Kim, H.R.; Cho, E.K.; Min, Y.J.; Ahn, J.S.; Ahn, M.J.; Park, K.; Cho, B.C.; Lee, J.H.; Jeong, H.C.; et al. Targeted sequencing identifies genetic alterations that confer primary resistance to EGFR tyrosine kinase inhibitor (Korean Lung Cancer Consortium). Oncotarget 2016, 7, 36311–36320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren-Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, T.; Kang, P.; Qian, K.; Deng, B.; Zhou, J.; Wang, R.; Jiang, B.; Li, K.; Liu, F.; et al. Combined analysis of rearrangement of ALK, ROS1, somatic mutation of EGFR, KRAS, BRAF, PIK3CA, and mRNA expression of ERCC1, TYMS, RRM1, TUBB3, EGFR in patients with non-small cell lung cancer and their clinical significance. Cancer Chemother. Pharmacol. 2016, 77, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, W.; He, W.; Jiang, C.; Zhou, X.; Song, X.; Zhu, J.; Fei, K.; Cao, W.; Jiang, G. Phenotype-genotype correlation in multiple primary lung cancer patients in China. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Fan, P.D.; Shen, R.; Zakowski, M.; Varmus, H. Erlotinib resistance in mouse models of epidermal growth factor receptor-induced lung adenocarcinoma. Dis. Model. Mech. 2010, 3, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, Y.; Takano, O.; Kato, I.; Takahashi, Y.; Shima, F.; Kataoka, T. Ras inhibitors display an anti-metastatic effect by downregulation of lysyl oxidase through inhibition of the Ras-PI3K-Akt-HIF-1α pathway. Cancer Lett. 2017, 410, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, Q.; Zhang, S.; Chen, J.; Huang, L.; Ren, J.; Chang, Y.; Liang, Y.; Wu, G. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. Onco Targets Ther. 2015, 8, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Lammers, P.E.; Lovly, C.M.; Horn, L. A patient with metastatic lung adenocarcinoma harboring concurrent EGFR L858R, EGFR germline T790M, and PIK3CA mutations: The challenge of interpreting results of comprehensive mutational testing in lung cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 6–11. [Google Scholar] [CrossRef]
- Lara, P.N.; Longmate, J.; Mack, P.C.; Kelly, K.; Socinski, M.A.; Salgia, R.; Gitlitz, B.; Li, T.; Koczywas, M.; Reckamp, K.L.; et al. Phase II study of the AKT inhibitor MK-2206 plus erlotinib in patients with advanced non-small cell lung cancer who previously progressed on erlotinib. Clin. Cancer Res. 2015, 21, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Yamasaki, F.; Johansen, M.J.; Zhang, D.; Krishnamurthy, S.; Felix, E.; Bartholomeusz, C.; Aguilar, R.J.; Kurisu, K.; Mills, G.B.; Hortobagyi, G.N.; et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007, 67, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Parada, L.F. Tumor microenvironment and neurofibromatosis type I: Connecting the GAPs. Oncogene 2007, 26, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yuan, C.; Cheng, C.; Zhang, Y.; Ma, Y.; Zheng, D.; Zheng, S.; Li, Y.; Jin, Y.; Sun, Y.; et al. Frequency and clinical significance of NF1 mutation in lung adenocarcinomas from East Asian patients. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Frederick, B.A.; Helfrich, B.A.; Coldren, C.D.; Zheng, D.; Chan, D.; Bunn, P.A.; Raben, D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007, 6, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009, 63, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 73, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metast. 2008, 25, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Murakami, A.; Takahashi, K. Epithelial mesenchymal transition in drug resistance and metastasis of lung cancer. Cancer Res. Treat. 2012, 44, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Lee, J.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; Kim, Y.T.; et al. Clonal history and genetic predictors of transformation into small-cell Carcinomas from lung adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.S.; Kim, S.; Kim, H.K.; Byeon, S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.L.; Han, J.; et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J. Thorac. Oncol. 2016, 11, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Matsumoto, K.; Wang, W.; Li, Q.; Nishioka, Y.; Sekido, Y.; Sone, S.; Yano, S. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin. Cancer Res. 2010, 16, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Giménez-Capitán, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, 1167–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hu, H.; Pan, Y.; Wang, R.; Li, Y.; Shen, L.; Yu, Y.; Li, H.; Cai, D.; Sun, Y.; et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE 2014, 9, e88291. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Zander, T.; Thomas, R.K.; Staratschek-Jox, A.; Claasen, J.; Wolf, J. Expression of signaling mediators downstream of EGF-receptor predict sensitivity to small molecule inhibitors directed against the EGF-receptor pathway. J. Thorac. Oncol. 2008, 3, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mechanisms of acquired resistance to first-generation tyrosine kinase inhibitors (gefitinib and erlotinib) [15,16]. EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; MET, mesenchymal–epithelial transition factor; EMT, epithelial–mesenchymal transition; SCLC, small-cell lung cancer.
Figure 1.
Mechanisms of acquired resistance to first-generation tyrosine kinase inhibitors (gefitinib and erlotinib) [15,16]. EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; MET, mesenchymal–epithelial transition factor; EMT, epithelial–mesenchymal transition; SCLC, small-cell lung cancer.

Figure 2.
Mechanisms of acquired resistance to osimertinib [32,33,34,35,36]. EGFR, epidermal growth factor receptor; MET, mesenchymal-epithelial transition factor; HER2, human epidermal growth factor receptor 2; FGFR1, fibroblast growth factor receptor 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; PIK3CA, phosphoinositide-3-kinase P110α catalytic subunit; SCLC, small-cell lung cancer.
Figure 2.
Mechanisms of acquired resistance to osimertinib [32,33,34,35,36]. EGFR, epidermal growth factor receptor; MET, mesenchymal-epithelial transition factor; HER2, human epidermal growth factor receptor 2; FGFR1, fibroblast growth factor receptor 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; PIK3CA, phosphoinositide-3-kinase P110α catalytic subunit; SCLC, small-cell lung cancer.

Figure 3.
The downstream signaling of the epidermal growth factor receptor.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110212
AMA Style
Nagano T, Tachihara M, Nishimura Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells. 2018; 7(11):212. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110212
Chicago/Turabian StyleNagano, Tatsuya, Motoko Tachihara, and Yoshihiro Nishimura. 2018. "Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy" Cells 7, no. 11: 212. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110212
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.