Mesenchymal Stem Cells (MSCs) Coculture Protects [Ca2+]i Orchestrated Oxidant Mediated Damage in Differentiated Neurons In Vitro

 , , and
, , and
Abstract
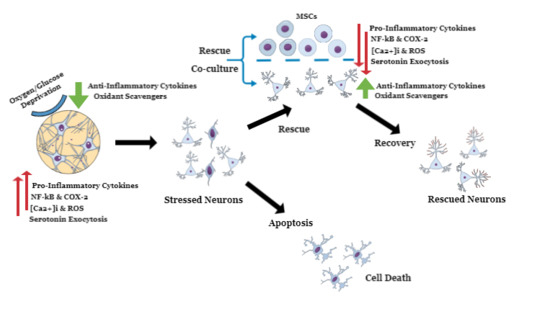
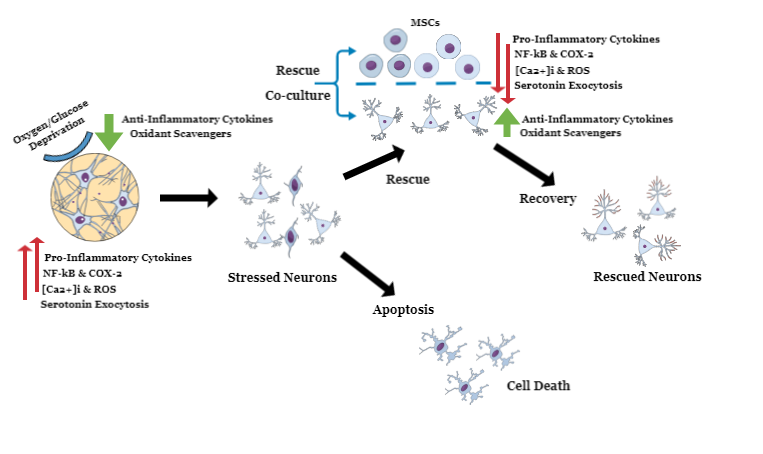
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Design of the Study
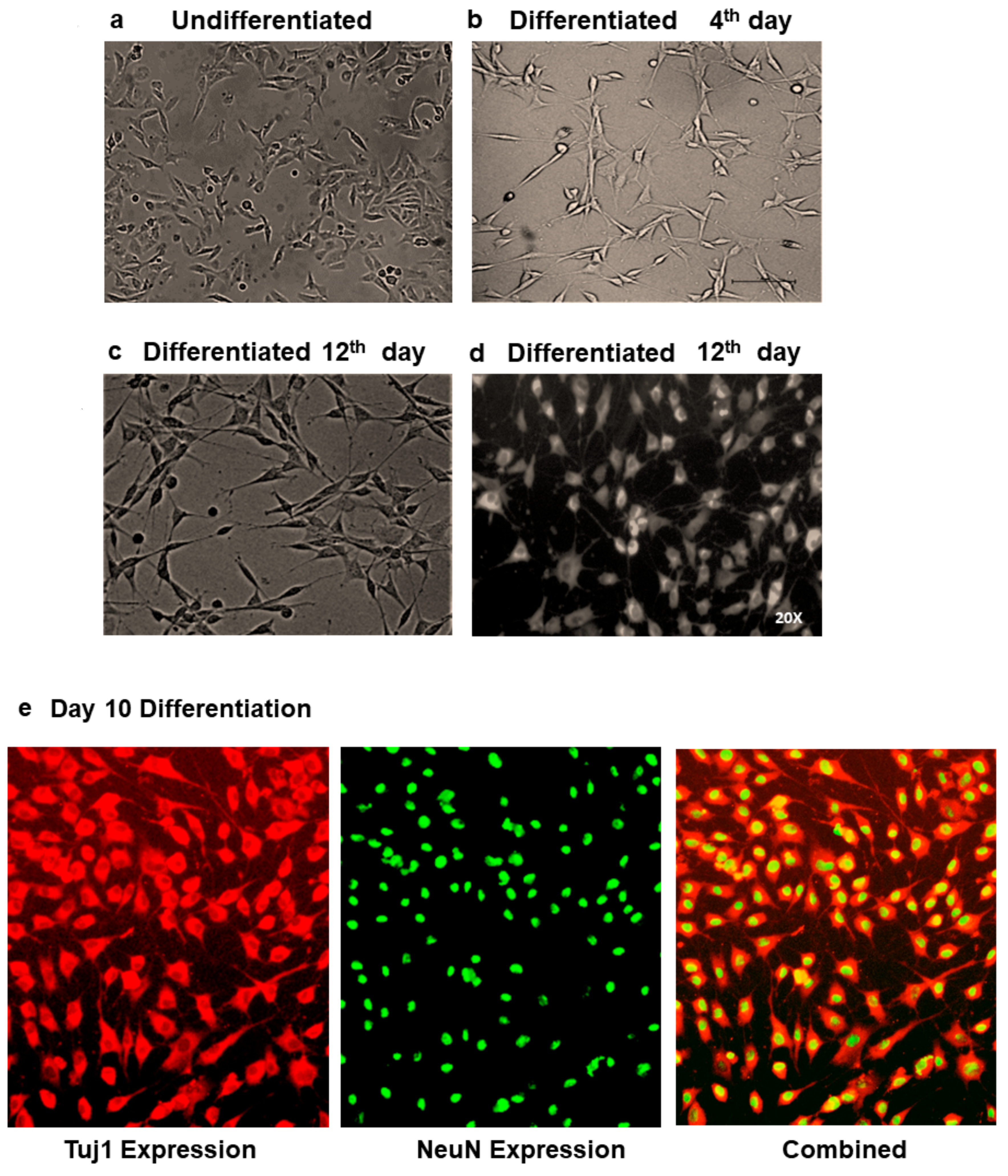
2.2. Neuronal Differentiation of SH-SY5Y Neuroblastoma Cells
2.3. Scratch Assay
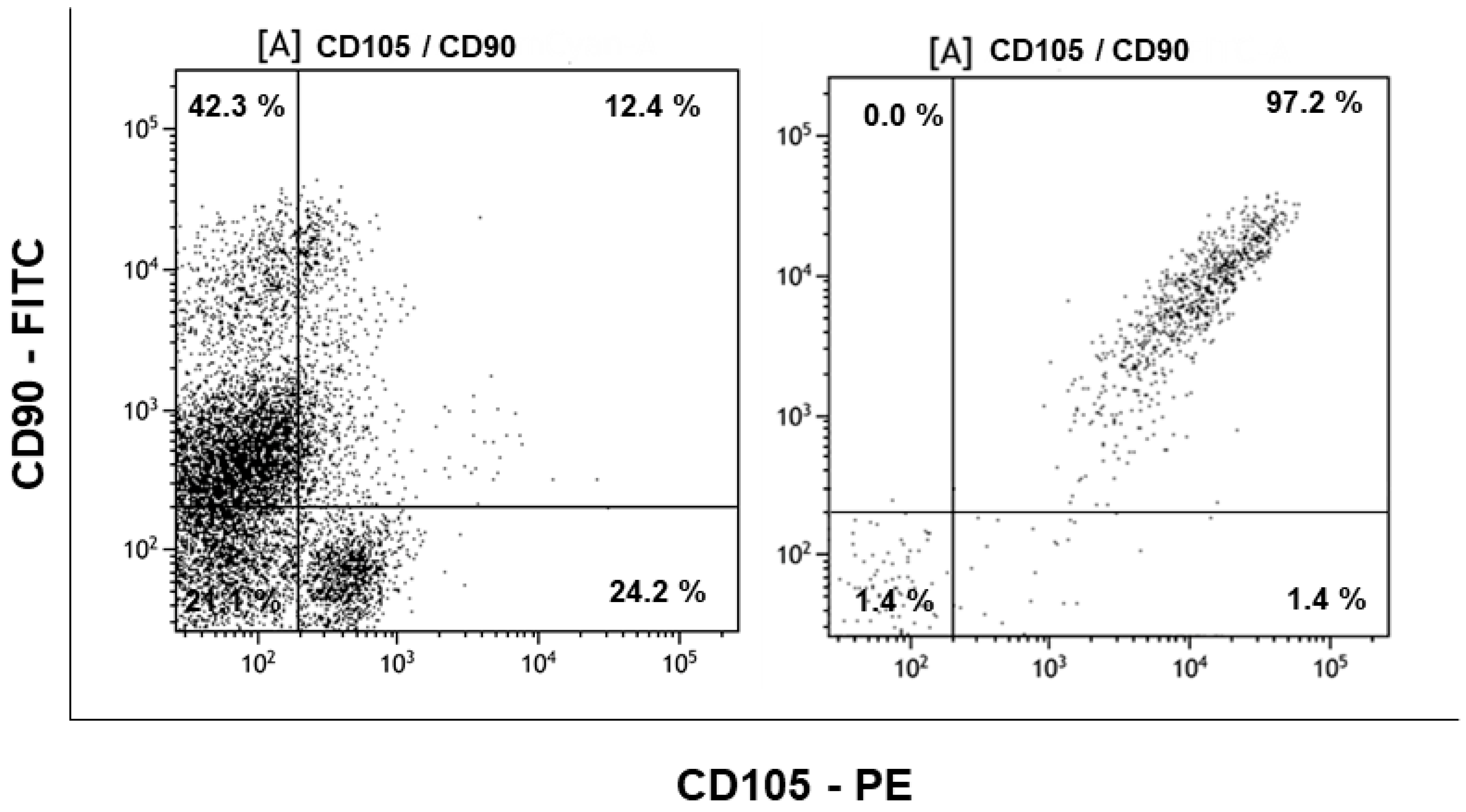
2.4. Human Umbilical-Cord Blood Mesenchymal Stem Cells (UCBMSCs)
2.5. In Vitro Cerebral Ischemia
2.6. In Vitro Differentiated Neuronal Cells and MSCs Coculture Experiments
2.7. Enzyme Linked Immunosorbent Assay (ELISA)
2.8. Cell-Death Assay
2.9. Real-Time PCR
2.10. Annexin V Assay
2.11. [Ca2+]ic Assay
2.12. ROS Assay
2.13. Western Blot
2.14. Statistical Analysis
3. Results
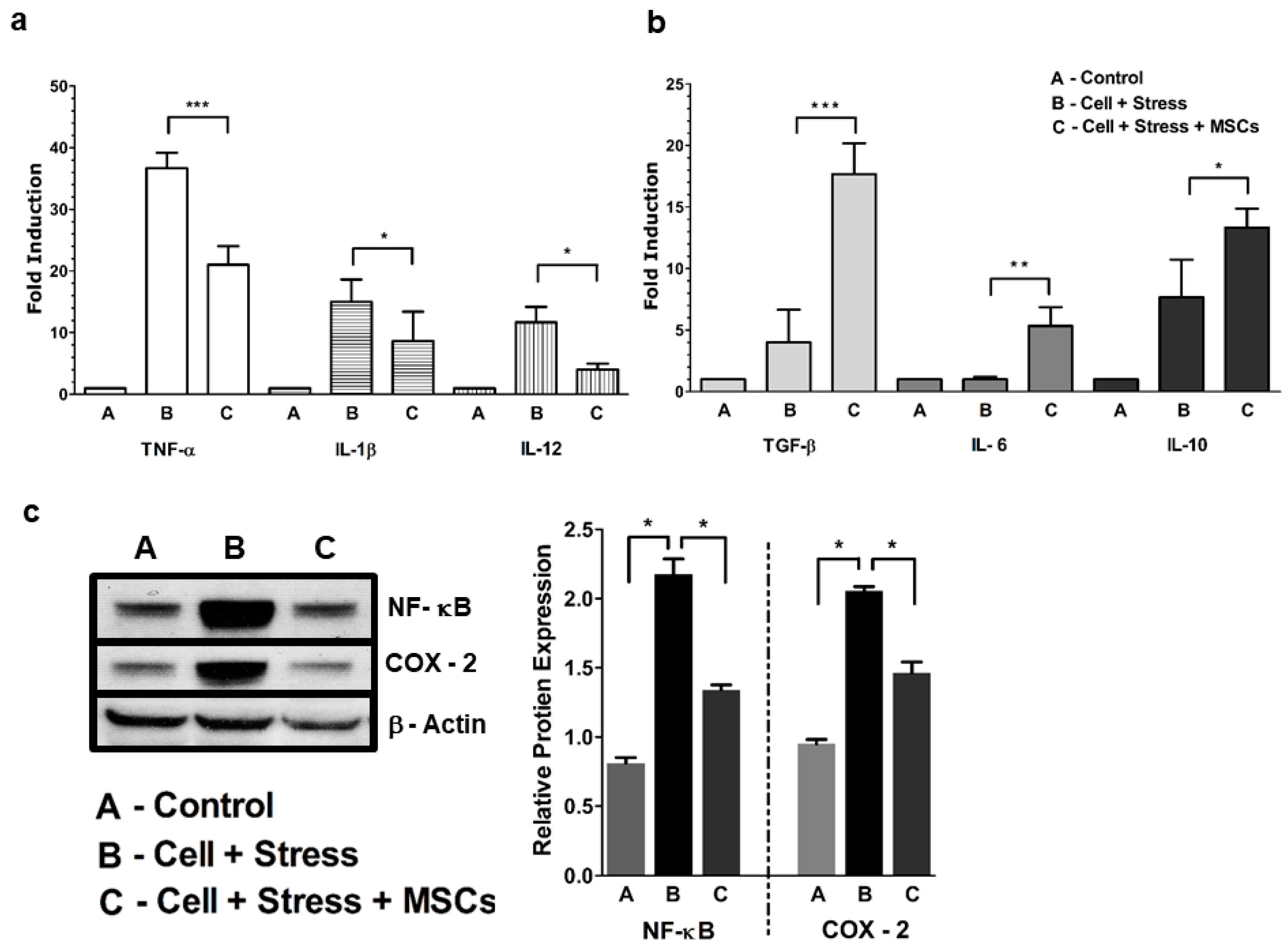
3.1. MSCs Coculture Alleviates Neuronal Ischemia Characterized by NF-κB-Mediated Proinflammatory Cytokines
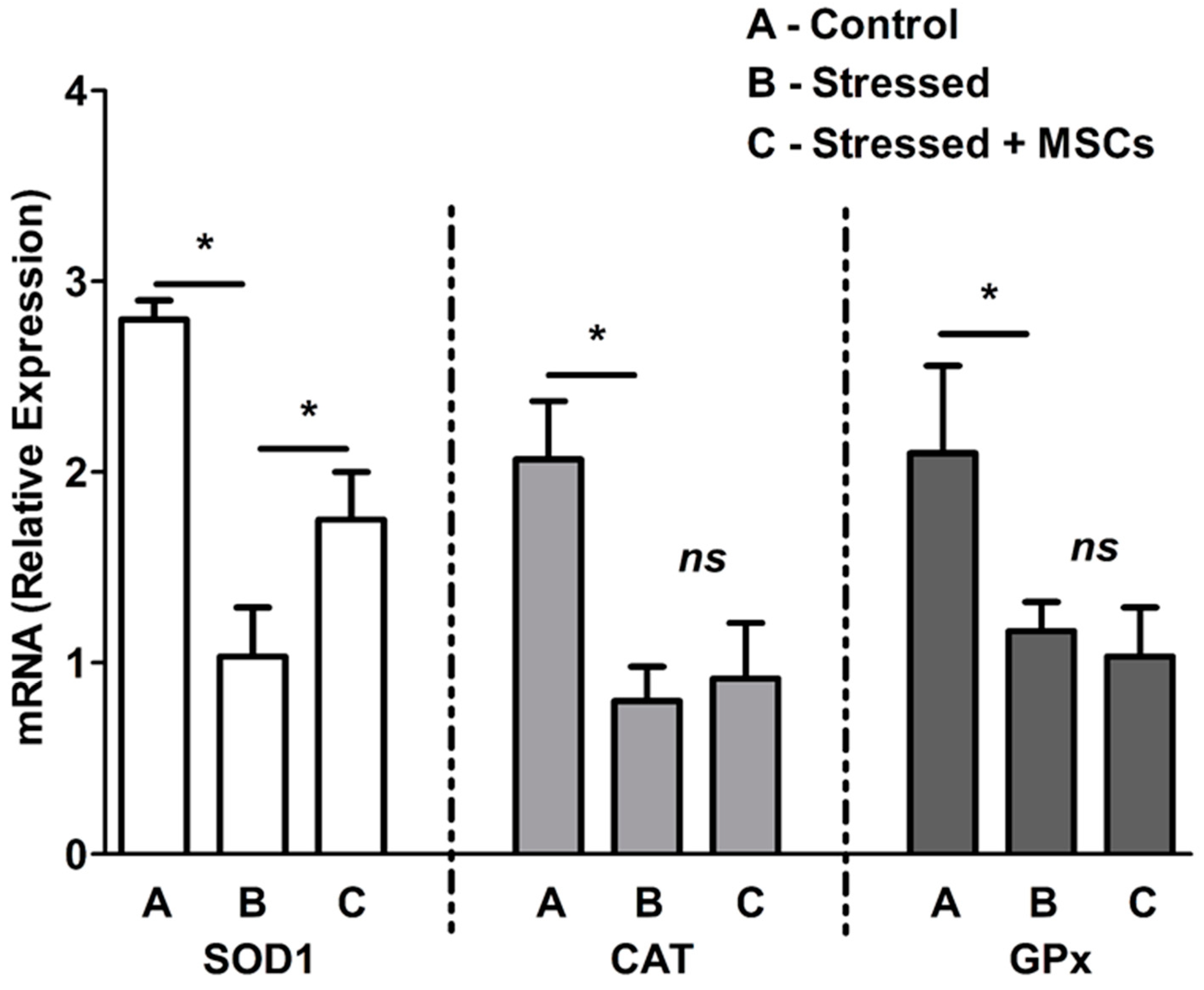
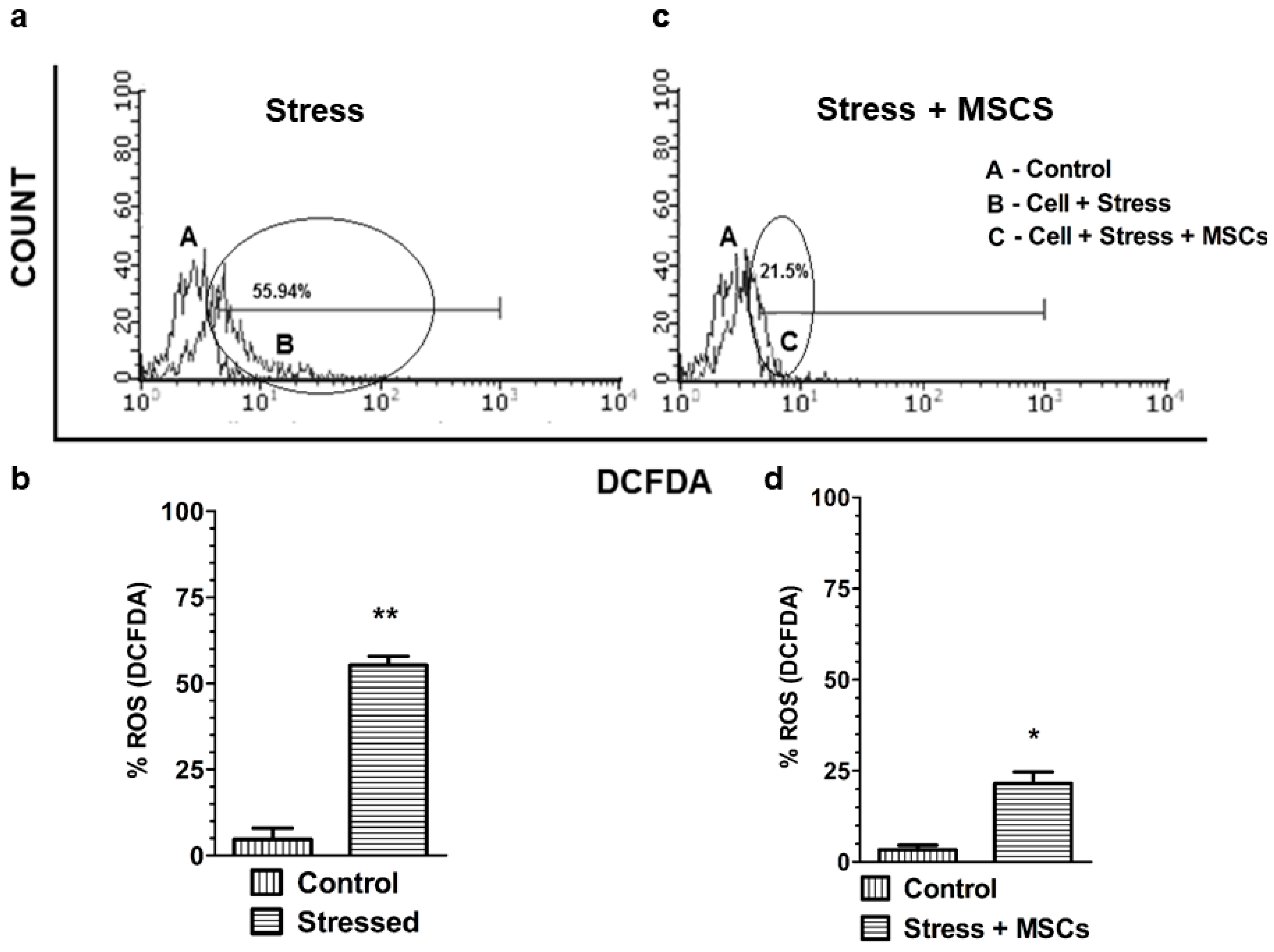
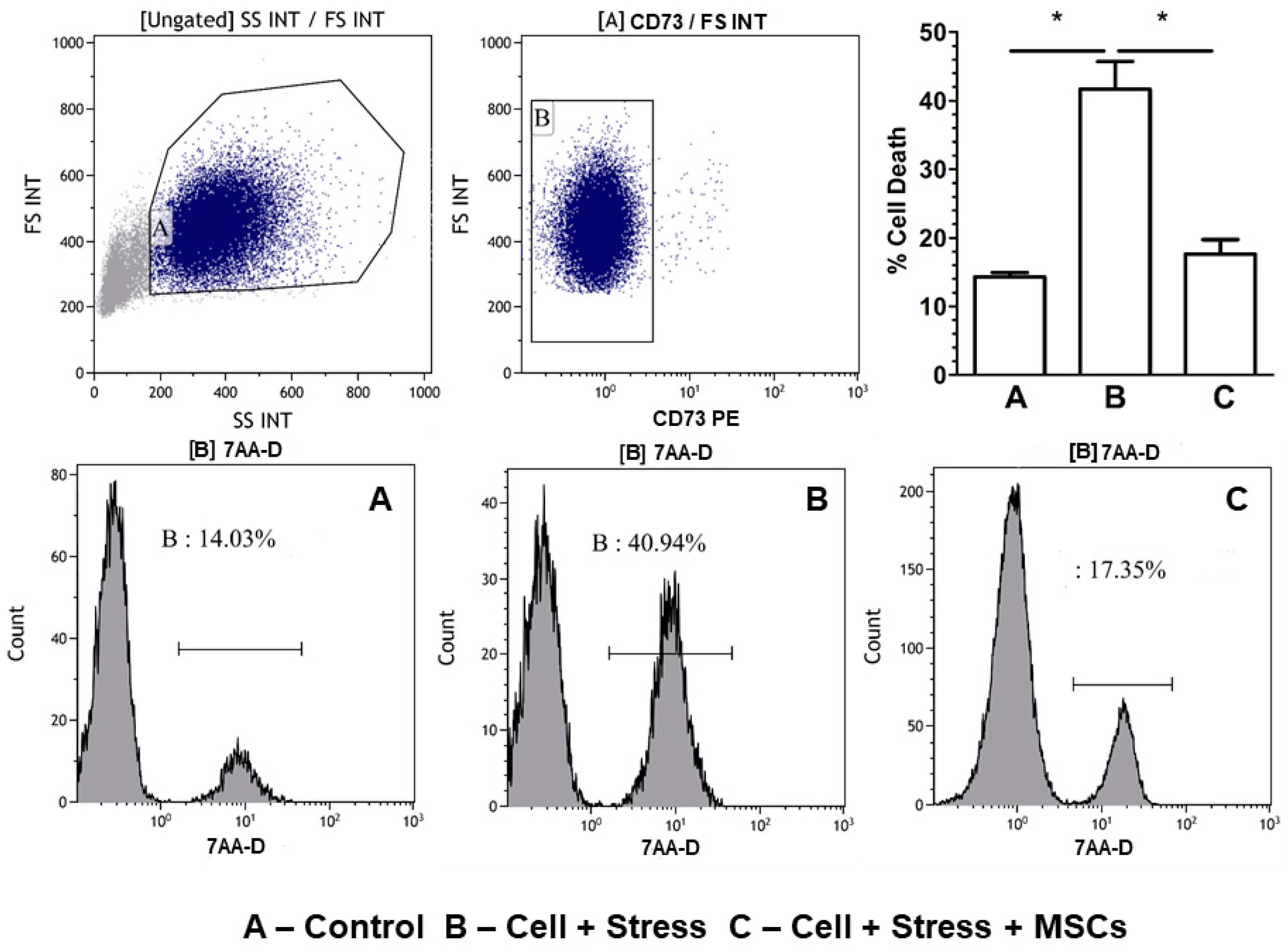
3.2. Inflammatory Neuronal Cell Death Characterized by Low Antioxidant Enzymes and Free-Radical Increase are Rescued by MSCs Coculture
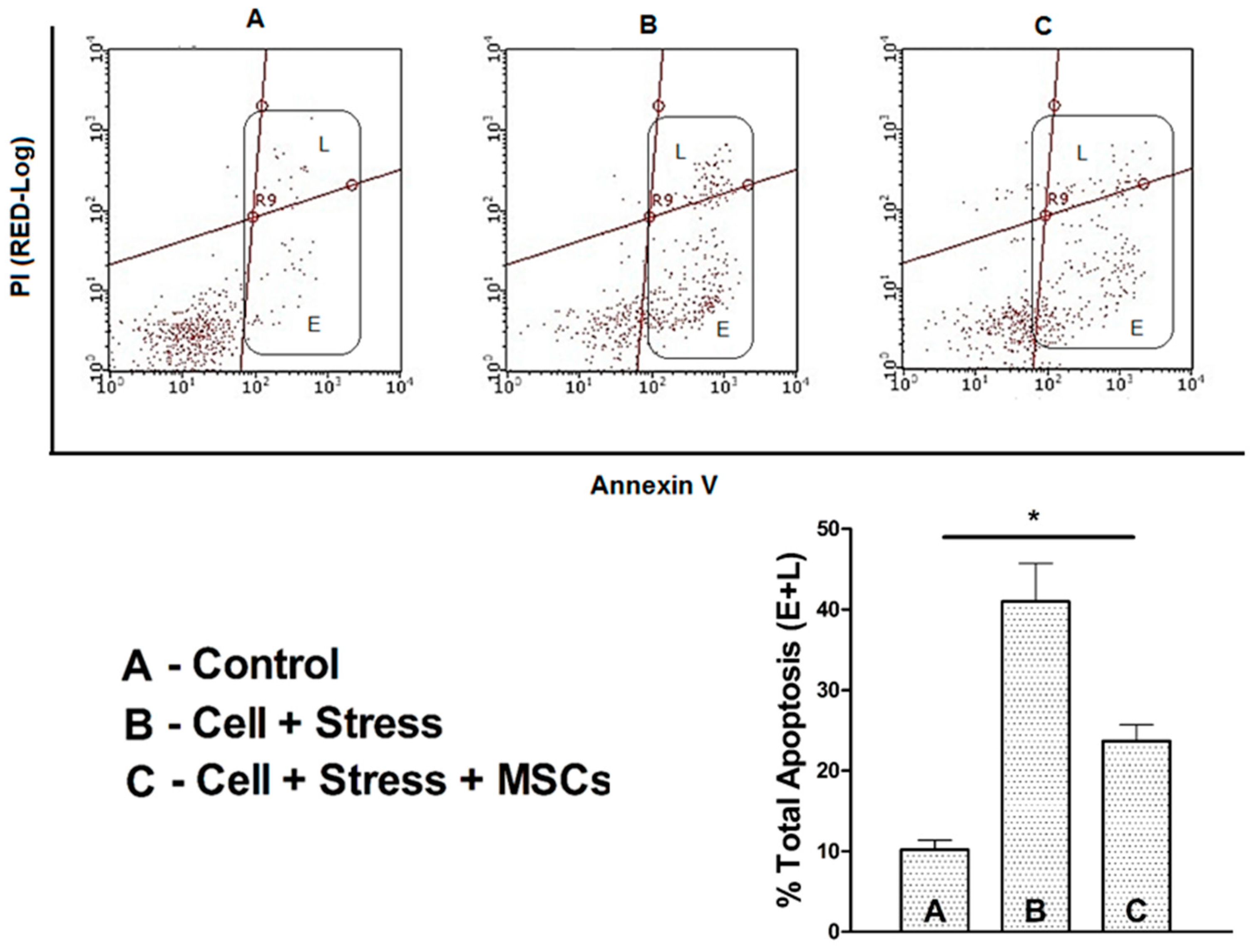
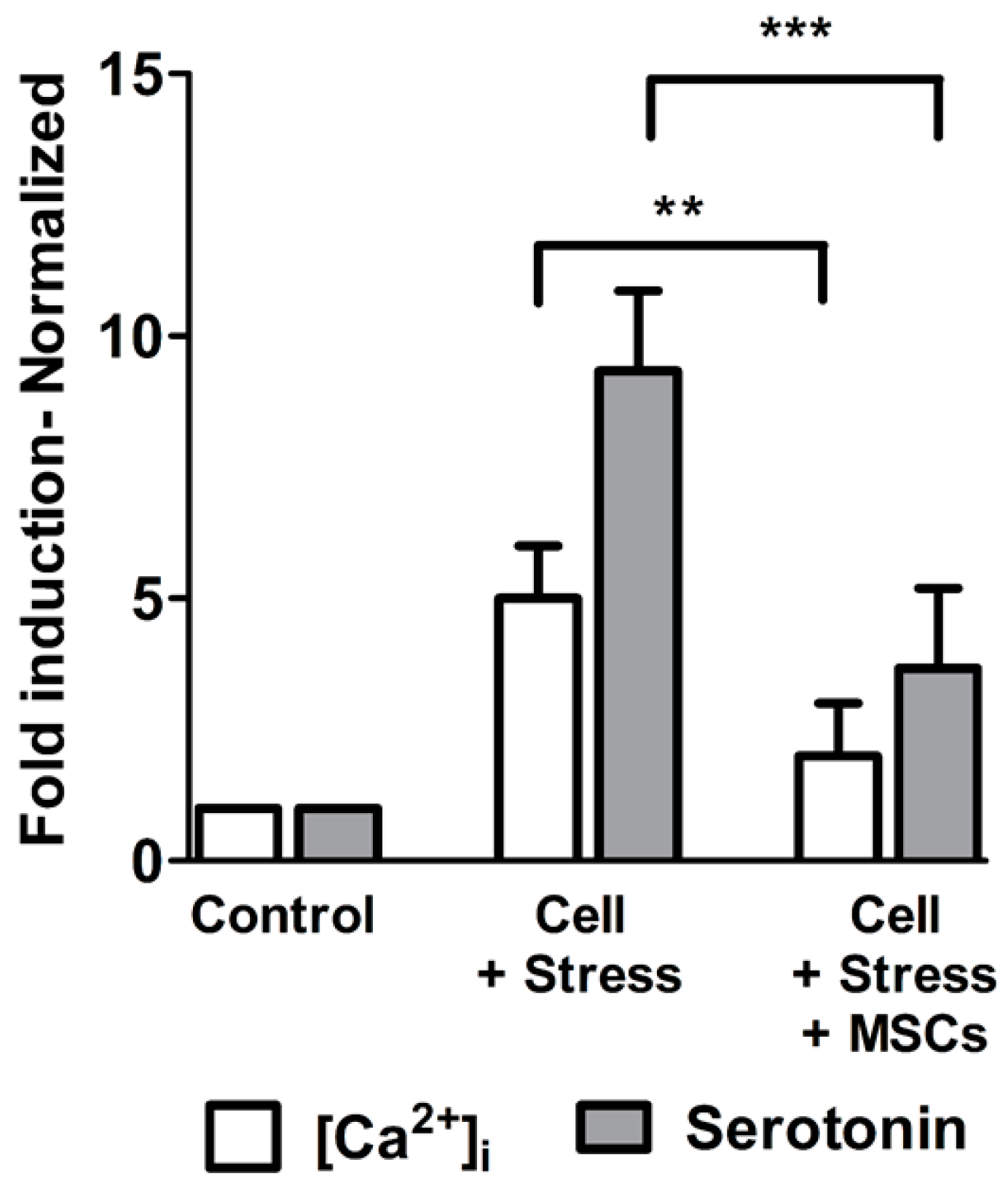
3.3. Elevated Serotonin Release as Altered Neuronal-Cell function is Resuced Upon [Ca2+]i Regulation by MSCs Coculture
4. Discussion
4.1. Glucose-Oxygen Deprivation Model Induced NF-kB-Mediated Inflammation in Neuronal Cells
4.2. High ROS Levels Characterized by Elevated [Ca2+]i Skew Neuronal Cells to Apoptosis
4.3. MSCs Coculture Rescues Stressed Neuronal Cells from Apoptosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, W.; Hsuan, Y.C.; Lin, M.T.; Kuo, T.W.; Lin, C.H.; Su, Y.C.; Niu, K.C.; Chang, C.P.; Lin, H.J. Human Umbilical Cord Mesenchymal Stem Cells Preserve Adult Newborn Neurons and Reduce Neurological Injury after Cerebral Ischemia by Reducing the Number of Hypertrophic Microglia/Macrophages. Cell Transplant. 2017, 26, 1798–1810. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Kaindl, A.M. Neuronal Death and Oxidative Stress in the Developing Brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J. Transl. Med. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med. 2010, 5, 121–143. [Google Scholar] [CrossRef] [Green Version]
- Bergstrom, T.; Forsberg-Nilsson, K. Neural stem cells: Brain building blocks and beyond. Ups. J. Med. Sci. 2012, 117, 132–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cardenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014, 28, 4936–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; Madesh, M. Regulation of the mitochondrial Ca(2+) uniporter by MICU1 and MICU2. Biochem. Biophys. Res. Commun. 2014, 449, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Invest. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and Inflammation in Viral Central Nervous System Infections. Mediators Inflamm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zundorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, F.; Klein, O.; Klose, J.; Priller, J. Mesenchymal stromal cells rescue cortical neurons from apoptotic cell death in an in vitro model of cerebral ischemia. Cell. Mol. Neurobiol. 2012, 32, 567–576. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Ex. 2016. [Google Scholar] [CrossRef] [Green Version]
- Messi, E.; Florian, M.C.; Caccia, C.; Zanisi, M.; Maggi, R. Retinoic acid reduces human neuroblastoma cell migration and invasiveness: Effects on DCX, LIS1, neurofilaments-68 and vimentin expression. BMC Cancer 2008, 8. [Google Scholar] [CrossRef]
- Chandramoorthy, H.C.; Bin-Jaliah, I.; Karari, H.; Rajagopalan, P.; Ahmed Shariff, M.E.; Al-Hakami, A.; Al-Humayad, S.M.; Baptain, F.A.; Ahmed, H.S.; Yassin, H.Z.; et al. MSCs ameliorates DPN induced cellular pathology via [Ca(2+)]i homeostasis and scavenging the pro-inflammatory cytokines. J. Cell. Physiol. 2018, 233, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, Y.; Cheng, W.; Chen, R.; Liu, T.; Chen, Z.; Tan, S. High glucose induces apoptosis and suppresses proliferation of adult rat neural stem cells following in vitro ischemia. BMC Neurosci. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol. Cell. Biol. 2011, 31, 3745–3758. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, K.; Wieltsch, C.; Blumer, N.; Mehlich, M.; Pathak, H.; Khan, A.Q.; Hildebrandt, H.; Frieling, H. A cell culture model for investigation of synapse influenceability: Epigenetics, expression and function of gene targets important for synapse formation and preservation in SH-SY5Y neuroblastoma cells differentiated by retinoic acid. J. Neural Transm. 2017, 124, 1341–1367. [Google Scholar] [CrossRef]
- Lopes, F.M.; da Motta, L.L.; De Bastiani, M.A.; Pfaffenseller, B.; Aguiar, B.W.; de Souza, L.F.; Zanatta, G.; Vargas, D.M.; Schönhofen, P.; Londero, G.F.; et al. RA Differentiation Enhances Dopaminergic Features, Changes Redox Parameters, and Increases Dopamine Transporter Dependency in 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Cells. Neurotox. Res. 2017, 31, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Y.T.; Lau, W.K.; Yu, M.S.; Lai, C.S.; Yeung, S.C.; So, K.F.; Chang, R.C.C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef]
- Korecka, J.A.; van Kesteren, R.E.; Blaas, E.; Spitzer, S.O.; Kamstra, J.H.; Smit, A.B.; Swaab, D.F.; Verhaagen, J.; Bossers, K. Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Neirinckx, V.; Coste, C.; Rogister, B.; Wislet-Gendebien, S. Concise review: Adult mesenchymal stem cells, adult neural crest stem cells, and therapy of neurological pathologies: A state of play. Stem Cells Transl. Med. 2013, 2, 284–296. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Widera, D.; Kaltschmidt, C. Signaling via NF-κB in the nervous system. BBA Mol. Cell Res. 2005, 1745, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immun. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Russo, S.J.; Ferguson, D.; Nestler, E.J.; Duman, R.S. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. USA 2010, 107, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Sarnico, I.; Ingrassia, R.; Boroni, F.; Branca, C.; Benarese, M.; Faraco, G.; Blasi, F.; Chiarugi, A.; Spano, P.; et al. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis. 2010, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef]
- Planas, A.M.; Soriano, M.A.; Rodriguez-Farre, E.; Ferrer, I. Induction of cyclooxygenase-2 mRNA and protein following transient focal ischemia in the rat brain. Neurosci. Lett. 1995, 200, 187–190. [Google Scholar] [CrossRef]
- Krause, D.L.; Muller, N. Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, 2010. [Google Scholar] [CrossRef]
- Hirst, W.D.; Young, K.A.; Newton, R.; Allport, V.C.; Marriott, D.R.; Wilkin, G.P. Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Mol. Cell. Neurosci. 1999, 13, 57–68. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, F.; Dawson, V.L.; Dawson, T.M. Free radicals as mediators of neuronal injury. Cell. Mol. Neurobiol. 1998, 18, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidara, M.A.; Assiri, A.S.; Youssef, M.A.; Mahmoud, M.M.; Ahmed, M.S.E.; Al-Hakami, A.; Chandramoorthy, H.C. Differentiated mesenchymal stem cells ameliorate cardiovascular complications in diabetic rats. Cell Tissue Res. 2015, 359, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirley, R.; Ord, E.N.; Work, L.M. Oxidative Stress and the Use of Antioxidants in Stroke. Antioxid. 2014, 3, 472–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Fattman, C.L.; Schaefer, L.M.; Oury, T.D. Extracellular superoxide dismutase in biology and medicine. Free Radic. Biol. Med. 2003, 35, 236–256. [Google Scholar] [CrossRef]
- Yagi, H.; Soto-Gutierrez, A.; Parekkadan, B.; Kitagawa, Y.; Tompkins, R.G.; Kobayashi, N.; Yarmush, M.L. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010, 19, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Lee, R.H.; Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res. Ther. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Palagano, E.; Villa, A.; Menale, C. Soluble Factors on Stage to Direct Mesenchymal Stem Cells Fate. Front. Bioeng. Biotechnol. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadekar, D.; Rangole, S.; Kale, V.; Limaye, L. Conditioned Medium from Placental Mesenchymal Stem Cells Reduces Oxidative Stress during the Cryopreservation of Ex Vivo Expanded Umbilical Cord Blood Cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Vaish, V.; Feng, M.; Field, K.; Chatzistamou, I.; Shim, M. Transgenic expression of cyclooxygenase-2 (COX2) causes premature aging phenotypes in mice. Aging 2016, 8, 2392–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, K.M.; Wade, F.M.; Wakade, C.; Mahesh, V.B.; Brann, D.W. Neuroprotection by stem cell factor in rat cortical neurons involves AKT and NFkappaB. J. Neurochem. 2005, 95, 9–19. [Google Scholar] [CrossRef]
- Saldana, L.; Valles, G.; Bensiamar, F.; Mancebo, F.J.; Garcia-Rey, E.; Vilaboa, N. Paracrine interactions between mesenchymal stem cells and macrophages are regulated by 1,25-dihydroxyvitamin D3. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S.; Muallem, S. ROS and Ca(2+)-Partners in sickness and in health. Cell Calcium 2016, 60, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Leon-Pinzon, C.; Cercos, M.G.; Noguez, P.; Trueta, C.; De-Miguel, F.F. Exocytosis of serotonin from the neuronal soma is sustained by a serotonin and calcium-dependent feedback loop. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Miguel, F.F.; Leon-Pinzon, C.; Noguez, P.; Mendez, B. Serotonin release from the neuronal cell body and its long-lasting effects on the nervous system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhazzani, A.; Rajagopalan, P.; Albarqi, Z.; Devaraj, A.; Mohamed, M.H.; Al-Hakami, A.; Chandramoorthy, H.C. Mesenchymal Stem Cells (MSCs) Coculture Protects [Ca2+]i Orchestrated Oxidant Mediated Damage in Differentiated Neurons In Vitro. Cells 2018, 7, 250. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7120250
Alhazzani A, Rajagopalan P, Albarqi Z, Devaraj A, Mohamed MH, Al-Hakami A, Chandramoorthy HC. Mesenchymal Stem Cells (MSCs) Coculture Protects [Ca2+]i Orchestrated Oxidant Mediated Damage in Differentiated Neurons In Vitro. Cells. 2018; 7(12):250. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7120250
Chicago/Turabian StyleAlhazzani, Adel, Prasanna Rajagopalan, Zaher Albarqi, Anantharam Devaraj, Mohamed Hessian Mohamed, Ahmed Al-Hakami, and Harish C. Chandramoorthy. 2018. "Mesenchymal Stem Cells (MSCs) Coculture Protects [Ca2+]i Orchestrated Oxidant Mediated Damage in Differentiated Neurons In Vitro" Cells 7, no. 12: 250. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7120250