MicroRNAs in Cardiac Diseases

IMAiA-Institute for Molecular Biology and RNA Technology, Faculty of Science and Engineering, Faculty of Health, Medicine and Life Sciences, Maastricht University, 6229 ER Maastricht, The Netherlands
*
Author to whom correspondence should be addressed.
Cells 2019, 8(7), 737; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070737
Submission received: 5 June 2019
/
Revised: 8 July 2019
/
Accepted: 16 July 2019
/
Published: 18 July 2019
(This article belongs to the Special Issue The Molecular and Cellular Basis of Cardiovascular Disease)
Abstract
:Since their discovery 20 years ago, microRNAs have been related to posttranscriptional regulation of gene expression in major cardiac physiological and pathological processes. We know now that cardiac muscle phenotypes are tightly regulated by multiple noncoding RNA species to maintain cardiac homeostasis. Upon stress or various pathological conditions, this class of non-coding RNAs has been found to modulate different cardiac pathological conditions, such as contractility, arrhythmia, myocardial infarction, hypertrophy, and inherited cardiomyopathies. This review summarizes and updates microRNAs playing a role in the different processes underlying the pathogenic phenotypes of cardiac muscle and highlights their potential role as disease biomarkers and therapeutic targets.
1. Introduction
Heart diseases are among the leading causes of morbidity and mortality worldwide [1]. Correct embryonic development, homeostasis, contraction-excitement coupling, and stress response of the cardiac muscle is controlled by precise spatiotemporal gene regulation. Alterations in these genetic expression patterns have been related to pathological cardiac conditions, which can lead to heart failure [2,3].
In addition to the regulation by transcription factors, microRNAs (miRNAs) are also involved in differential gene expression found in the pathophysiologic cardiac condition [4,5]. MiRNAs are evolutionarily conserved noncoding RNA molecules (~22 nucleotides long, single-stranded) that regulate gene expression through imperfect base-pairing with complementary sequences in their target mRNA leading to translational repression or transcript degradation [6]. Most miRNA genes are transcribed by RNA polymerase II from intergenic, intronic or polycistronic loci as a long primary miRNA transcript (pri-miRNA), which is then cleaved by the Drosha endoribonuclease to a 70-nt-long hairpin structure with 2-nt-3′ overhangs (pre-miRNA) [7]. Pre-miRNA is subsequently exported to the cytoplasm and processed by a second endoribonuclease, Dicer, to form a 22-nucleotide-long miRNA:miRNA* duplex with imperfect complementarity. One strand of this duplex, the guide strand, then combines with the Argonaute (AGO) protein into the RNA-induced silencing complex (RISC), while the passenger strand gets degraded [8]. Whether the 5p or the 3p (i.e., originating from the 5′ or 3′ end of the pre-miRNA hairpin) RNA strand of the duplex becomes the guide strand depends partially on the thermodynamic stability at the 5′ ends of the duplex [9]. In general, the strand with lower stability preferentially combines with the AGO (Figure 1).
This leads to certain miRNAs having both their strands loaded into the RISC with equal proportion, while for others one strand will dominate [9]. The targeting of a mRNA occurs through imperfect base-pairing between the transcript and the so-called seed sequence in the miRNA, usually covering the nucleotides in positions 2–7 of the latter [10]. As a consequence, a single miRNA can regulate multiple mRNA targets involved in diverse biological processes and, vice versa, a single mRNA can be regulated by several miRNAs.
Mature miRNAs are referred to with the prefix “miR-” followed by an identifying number reflecting their order of discovery. In case of miRNAs with sequences differing in only one or two nucleotides, an additional letter or number is added to the name, for example ‘miR-208a’ versus ‘miR-208b’ or ‘miR-133a-1′ versus ‘miR133a-2′ [4,7].
MiRNAs can be grouped into families, based on the mature miRNA or on the sequence and/or structure of the pre-miRNAs [11]. Many miRNAs are also grouped together into polycistronic clusters, in which several miRNAs are produced from one primary transcript [12]. Up to 60% of human protein coding genes are estimated to be post-transcriptionally regulated by miRNAs [13] and, currently, ~2500 miRNAs have been annotated in the human genome [14]. In recent years, the role of miRNAs in the control of cardiovascular events related both to key biological functions and disorders has been increasingly investigated.
This review summarizes the most recent studies highlighting the role of miRNAs in several cardiac pathogenic conditions (Table 1, Figure 2). We also provide an overview of the potential role of circulating miRNAs as disease biomarkers (Figure 3), as well as a description of the currently available approaches to use miRNAs as potential therapeutic tools for different cardiac conditions. This review limits its scope to studies of the myocardium itself; the roles of miRNAs in other types of cardiovascular diseases, like those involving the vasculature, diabetes, or aging, are not included.
2. Contractility Defects
Cardiac contractility consists of the fast and unidirectional development of mechanical force and motion, determined by a highly ordered organization of sarcomeric proteins. The progressive impairment of passive extension and active contraction after adverse structural remodeling (e.g., concentric hypertrophy) is an indicator of heart failure [87,88]. Cardiac contractility primarily relies on the expression of two cardiac myosin heavy chain (MHC) genes, α and β, also known as Myh6 and Myh7, respectively, which are regulated in an antithetical manner [89]. Together with cardiac stress, which induces the parallel downregulation of Myh6 and the upregulation of Myh7, endogenous factors regulate the expression of these genes. Thyroid hormone T3 signaling controls the expression of these two MHC genes by stimulating Myh6 expression and inhibiting Myh7 expression after birth [90]. In addition to the thyroid hormone signaling, some miRNAs have been related to the regulation of Myh6 and Myh7 genes. In vitro assays coupled to bioinformatic analysis showed that miR-27a targeted Myh7, but not Myh6, by inducing a strong upregulation of the gene upon thyroid hormone receptor β1 (Trβ1) signaling in neonatal rat ventricular cardiomyocytes and mouse embryonic stem cells [15]. In vivo studies in hypertrophic hearts in mice that underwent transverse aortic constriction (TAC) further highlighted this relationship, with the parallel upregulation of miR-27a as well as of Myh7 and the downregulation of Trβ1 [15].
Several studies also highlighted an important role of miR-208 in the regulation of MHC genes. MiR-208a is encoded by an intronic portion of Myh6 gene and its deletion in mice was related to the decreased expression of Myh7 gene in response to stress or hypothyroidism, after thoracic aortic banding [16]. A second study in mice further highlighted the regulatory role of miR-208a, which modulated the expression of Myh7 and Myh7b, as well as of miR-208b and miR-499, their respective intronic miRNAs. In turn, miR-208b and miR-499 demonstrated a dominant role in the specification of muscle fiber identity by activating slow and repressing fast myofiber gene programs [91]. Another study showed an increase in the expression of pro-hypertrophic β-MHC during early stages of diabetes in type-2 diabetic mouse hearts following the upregulation of miR-208a [17]. The upregulation of miR-208a appeared to precede the switch from α- to β-MHC isoforms and the development of systolic and diastolic dysfunction. Inhibition of this miRNA prevented the activation of β-MHC and the subsequent hypertrophic response. A similar upregulation of miR-208a was found in tissue of type-2 human diabetic hearts [17]. Taken together, these studies highlight the double role of MHC genes, which, in addition to producing a major cardiac contractile protein, regulate cardiac growth and gene expression in response to stress and hormonal signaling through miRNAs encoded in their introns.
Other studies reported other miRNAs involved in the regulation of cardiac contractility, with mechanisms not related to MHC. Mice under pressure overload conditions display a reduction of miR-22, which in turn has been related to the alteration of the intracellular calcium homeostasis due to a reduced sarcoplasmic reticulum (SR) Ca2+ load [18]. In these conditions there was a decrease in the expression of sarcoplasmic reticulum Ca2+ ATPase activity (Serca2a), a Ca2+ reuptake pump critical for cardiac contractility and whose reduced expression is a marker for heart failure, as well as of several genes encoding for proteins in the vicinity of the cardiac Z disk/titin cytoskeleton: Calsarcin-1, Casq2, Ldb3, Melusin and Titin. Furthermore, in miR-22-/- mice the transcription factor Purb is upregulated; this in turn negatively regulates Srf, itself a transcription factor that modulates Serca2a expression. This suggests that miR-22 is involved in the impaired contractility through Serca2a regulation [18]. Additionally, a transgenic mouse model for cardiac-specific overexpression of miR-1 showed age-dependent decrease of heart function associated with myofibril fragmentation and shorter sarcomeres [19]. Downregulation of genes involved in sarcomeric assembly, such as myosin light chain kinase (Mylk3) and cardiac calmodulin (Calm1/Calm2), was observed. This led to decreased phosphorylation of myosin light chain 2v (MLC2v), cardiac myosin binding protein-C (cMyBP-C) and calmodulin-activated protein kinase II (CaMKII). In the suggested mechanism, miR-1 overexpression led to the repression of Mylk3 and Calm1/Calm2 genes, which in turn attenuated the phosphorylation of MLC2v, CaMKII, and cMyBP-C, with subsequent sarcomeric disassembly, adverse structural remodeling, and impaired heart function [19].
3. Arrhythmias
Arrhythmias are abnormal deviations from the normal heart rate and/or rhythm and are generally caused by abnormal conduction or repolarization, or by a combination of both [92]. In diseased hearts, regional changes in electrophysiology can lead to non-uniform anisotropy of impulse propagation, and anisotropic re-entry lies at the basis of arrhythmic development [93]. Cardiac arrhythmias may arise from alterations in intracellular Ca2+ cycling in the SR. Normally, due to depolarization, Ca2+ enters the cardiomyocytes through voltage-dependent Ca2+ channels during the plateau phase of the action potential, which then triggers a release of more Ca2+ from the SR. This process is controlled through the phosphorylation of phospholamban (SR Ca2+-ATPase inhibitor), the ryanodyne receptor (RyR2) (the SR Ca2+ release channel) and the L-type Ca2+ channel, a process that has been shown to be regulated by miRNAs [94,95]. Overexpression of miR-1 in rat ventricular cardiomyocytes resulted in abnormal Ca2+ cycling and showed increased phosphorylation of RyR2, while decreasing the expression of a subunit of protein phosphatase 2A (PP2A), a phosphatase which can influence cardiac contractility through phosphorylation of proteins necessary for Ca2+ release [20]. Similarly, in cardiomyocytes of hearts isolated from canines with chronic heart failure that showed increased left ventricular (LV) dimensions and reduced LV contractility, the expression of both miR-1 and miR-133 was found to be increased [21]. Again, decreased PP2A, increased RyR2 phosphorylation and abnormal Ca2+ cycling were responsible for the phenotypic changes [21].
Arrhythmias can also be caused by abnormalities in the function of gap junctions, intercellular structures in the intercalated discs which provide a low-resistance pathway for direct cell-to-cell passage of the electrical stimulus [96]. Connexin-43 (Cx-43), encoded by the gap junction alpha-1 (GJA1) gene, is the most important component of the gap junction in the heart. The miRNA cluster miR-17-92 targets this gene, as well as the phosphatase and tensin homolog (Pten) gene, a lipid phosphatase that plays a role in cardiomyocyte size and cardiac contractility [22]. Conditional overexpression of the miRNA cluster in cardiac and smooth muscle tissues in a homozygous transgenic mouse model led to downregulation of these two genes resulting in spontaneous arrhythmias and a dramatic decrease in survival rate [22]. The authors hypothesized that the direct suppression of Gja1 by miR-17-92 in the heart may aberrantly regulate the electric impulse propagation with subsequent lethal arrhythmias. Cx-43 was also found to be a direct target of miR-206 [23]. The overexpression of this miRNA in HL-1 cells and in adult mouse heart caused suppression of Cx-43, abnormal heart rate and PR interval, and reduced life span in the animals.
4. Myocardial Infarction
Myocardial infarction (MI) refers to a decrease in blood flow to the heart, resulting in damage to cardiomyocytes due to lack of oxygen and it is often caused by the rupture of atherosclerotic plaques [97,98]. Acute MI is characterized by ischemic injury and cardiomyocyte apoptosis, followed by structural alterations, such as increase of extracellular matrix protein, fibrosis, and hypertrophy of cardiac myocytes, which leads to cardiac dysfunction and eventually causes heart failure [99]. Although the heart was long considered to be a non-mitotic organ, studies performed on cardiac tissue samples from patients who died 4 to 12 days after MI showed that 4% of myocyte nuclei express the proliferation marker Ki-67 in the regions adjacent to the infarct [100]. Interestingly, cardiomyocyte proliferation was shown to be involved in cardiac regeneration and both processes have been related to miRNA regulation. Several studies detected miRNAs as key regulators of cardiomyocyte proliferation and of improvement of cardiac function after MI. While miR-15, miR-195, and miR-497 were reported to negatively regulate these processes [24,25,26], miR-590-3p, miR-199a-3p and miR-294 are considered promoters of cell cycle re-entry in both neonatal and adult rat cardiomyocytes as well as in in vivo studies on MI adult mouse models [27,28].
A miRNA frequently associated with MI is miR-133. In a study on bone marrow-derived mesenchymal stem cells (MSCs), Chen et al. revealed the protective role on miR-133 against apoptosis under hypoxia [29]. Transplantation of miR-133-overexpressing MSCs in rat infarcted hearts resulted in lower inflammatory level and smaller infarct size, as well as in improved cardiac function, due to the repression of the expression of the fibrinogenesis-promoting gene 1 (Snail1) in cardiomyocytes [29]. A previous study, focused on the role of the β-blocker carvedilol in MI, showed that the upregulation of miR-133 by this drug ameliorates the impaired cardiac function and reduced apoptosis in the infarcted heart in rats and in the presence of oxidative stress [30]. The cytoprotective role of carvedilol was abolished by knocking down miR-133 by its antisense inhibitor, while it was mimicked by the overexpression of the miRNA, which reduced apoptosis by targeting Caspase-9, one of the first molecules activated in the apoptosis [101]. Very recently, the role of miR-133 in MI was further studied in cardiomyocytes or in hearts of mice treated with the anthraquinone aloe-emodin (AE) [31]. In this context, H2O2-mediated downregulation of miR-133 was inhibited by AE, which also prevented the increase of Caspase-3 activity. Accordingly, transfection with miR-133 inhibitor abolished the anti-apoptotic effects of AE. These results suggest the protective role of miR-133 in MI and corroborate its function in the prevention of apoptosis.
A group of miRNAs implicated in the cardiac response to MI maps in the so-called Dlk1-Dio3 noncoding RNA (ncRNA) locus [102,103]. This evolutionary conserved genomic region spans between the Delta-like homolog 1 (Dlk1) and type III iodothyronine deiodinase (Dio3) protein coding genes and hosts a large cluster of ncRNAs, including more than 50 miRNAs both in mice and in humans, as well as several long noncoding RNAs (lncRNAs) and small nucleolar RNAs [103]. Interestingly, immediately following the Dlk1 gene there are two intergenic differentially methylated DNA regions [104]. This epigenetically imprinted region regulates gene expression of the locus, with the methylated paternal allele expressing the protein-coding genes and the unmethylated maternal allele expressing the ncRNAs. One early study characterized the miRNA expression profile of a MI mouse model and found that more than a quarter of all differentially expressed miRNAs belonged to the Dlk1-Dio3 region [102]. All of these miRNAs were upregulated. Several reports corroborated these findings. Two independent studies showed that miR-539 is upregulated in response to anoxic conditions in vitro [32] and MI in vivo [33]. In the former, Wang et al. showed that, in MI mice, the cardiac apoptosis-related lncRNA (Carl) can suppress apoptosis and mitochondrial fission in anoxic cardiomyocytes by acting as a ‘sponge’ for miR-539, which in turn targets prohibitin-2 (Phb2), a gene important for mitochondrial function [32]. In the latter study, miR-539 was shown to target O-GlcNAcase (Oga), a protein-targeting deglycosylase that was downregulated in a MI mouse model [33]. Increased global levels of protein O-GlcNAcylation has been shown to improve cardiac response after acute stress such as ischemia-reperfusion, MI and oxidative stress. [105]. Two other miRNAs belonging to the Dlk1-Dio3 region are miR-410 and miR-495. These were shown to be able to promote proliferation of cardiomyocytes in vitro through targeting Cbp/P300 Interacting Transactivator With Glu/Asp Rich Carboxy-Terminal Domain 2 (Cited2), a transcriptional co-activator that plays a role in cardiac development and morphology [34]. A subsequent study by the same group showed that these two miRNAs are upregulated in mouse models for MI, cardiac hypertrophy and muscular dystrophy [35]. Finally, another study found increased expression of miR-433 in three models of cardiac injury featuring fibrosis [36]. Treatment of miR-433 antagomir before inducing MI in the animals led to better preservation of cardiac function and reduced fibrosis. MiR-433 downregulated antizyme inhibitor 1 (Azin1) and c-Jun N-terminal kinases 1 (Jnk1), leading to activation of the transforming growth factor-β (TGF-β) pathway and the mitogen-activated protein kinase 1 (Mapk1).
5. Hypertrophy
Cardiomyocyte hypertrophy (CH) indicates an increase in the size of cardiomyocytes without an increase in the amount of cells [106]. While heart muscle hypertrophy initially represents an adaptive response necessary for the maintenance of the cardiac output, prolonged hypertrophic growth is associated with adverse molecular and histological consequences. This often results in heart failure through cardiomyocyte degeneration and death [107,108]. Hypertrophy can either be physiologic, such as when it develops during exercise, or pathologic, when it leads to heart failure and other cardiovascular diseases. CH, hence, is a highly complex remodeling process, also regulated by miRNAs. Several reports support the pro-hypertrophic role of miR-22. Its cardiac-specific deletion in mouse was sufficient to blunt hypertrophy and cardiac remodeling in the presence of stressors, such as isoproterenol and activated calcineurin transgene [37]. Additionally, miR-22 overexpression in neonatal rat cardiomyocytes increased cell size and induced hypertrophic markers, such as the natriuretic peptide A (Nppa) gene, while knockdown of miR-22 attenuated the hypertrophy induced by phenylephrine, isoproterenol or angiotensin II (Ang-II) [45]. The Authors showed that miR-22 might influence CH by Pten inhibition, possibly involving phosphatidylinositol-3-kinase (PI3K)-protein kinase B (AKT) [45]. This data agrees with the findings observed both in neonatal rat cardiomyocytes as well as in a mouse model for CH obtained through agomir-22 treatment [39]. The upregulation of miR-22 not only induced CH, but also increased the production of the hypertrophic markers atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-MHC, and reduced PTEN protein levels.
The miR-212/132 family was found upregulated in cardiomyocytes following different hypertrophic stimuli such as Ang-II, phenylephrine or insulin-like growth factor and in the cardiac tissue of TAC mice [40]. While miR-212/312 overexpression turned out to be sufficient to induce CH in transgenic animals, miR-212/132 null mice presented a smaller heart-to-body-weight ratio than wild type (WT) animals and were protected against CH induced by TAC operation [40]. Both miRNAs appeared to target and negatively regulate the expression of the anti-hypertrophic forkhead boxO (FoxO3) transcription factor, with the subsequent hyperactivation of the pro-hypertrophic calcineurin/nuclear factor of activated T cell (NFAT) signaling pathway [40]. The same pathway correlated also with miR-199b, which was found upregulated not only in hearts from calcineurin transgenic mice, an animal model for heart failure, but also in TAC mice, and in biopsies from human patients who suffered from heart failure [41]. MiR-199b resulted to be overexpressed by the calcineurin/NFAT pathway in vivo and to target the dual specificity tyrosine phosphorylation regulated kinase 1A (Dyrk1a) gene, involved in the phosphorylation of NFAT factors in pathogenic feed forward mechanism. Accordingly, in vivo treatment with a chemically modified antisense oligonucleotide specific for miR-199b led to regression of hypertrophy, restoration of Dyrk1a expression levels, and normalized NFAT activity [41].
MiR-199a is also a pro-hypertropic miRNA and its cardiomyocyte-specific overexpression in mice induced CH and inhibition of autophagy [42]. In particular, miR-199a targeted the pro-autophagic and anti-hypertrophic factor glycogen synthase kinase 3β, known to suppress the mammalian target of rapamycin (mTOR), one of the major negative autophagic regulators. Moreover, the hypertrophic induction by miR-199a was attenuated by overexpressing the autophagy related gene 5 in cardiomyocytes, while treatment with rapamycin restored cardiac autophagy and decreased hypertrophy in miR-199a transgenic mice [42].
Another well-known cardiac miRNA is miR-206, which, if overexpressed, resulted in CH in both in vitro and in vivo [43]. Indeed, its suppression exacerbated ischemia/reperfusion injury and hindered CH. Mechanistically, miR-206 is positively regulated by Yes-associated protein (YAP), a key molecule of the Hippo pathway, which induces cardiomyocytes apoptosis and hypertrophy, and targets the forkhead box P1 (FoxP1), known to negatively regulate cardiac hypertrophy through inhibition of Nfat3 [109]. As with MI, several miRNAs from the Dlk1-Dio3 locus play a role also in CH. An example is given by miR-154, which was upregulated in a TAC mouse model [44]. Injection of an antimir against miR-154 reduced adverse cardiac remodeling and fibrosis after TAC. It also led to the reduced expression of cyclin-dependent kinase inhibitor 2B (Cdkn2b), a cell cycle inhibitor. The Authors speculated that regulation of Cdkn2b by miR-154 might contribute to the anti-fibrotic phenotype, although they did not show it was a direct target of the miRNA [44]. Also, miR-410 and miR-495 were upregulated in an Ang-II-induced mouse model for CH as well as in neonatal rat cardiomyocytes treated with phenylephrine, a pro-hypertrophic compound [35]. Knockdown of these two miRNAs in neonatal rat cardiomyocytes reduced the hypertrophic response after phenylephrine treatment. A later study using a rat model of pulmonary arterial hypertension found that the upregulation of miR-495 also increased hypertrophic markers, while the reduction of this miRNA attenuated the pathogenic phenotype [45]. While Dlk1-Dio3 miRNAs are usually upregulated in cardiac diseases, some are downregulated in the pathological conditions. Among them, miR-541 was downregulated in cardiomyocytes treated with Ang-II, as a model of CH [46]. Transgenic mice overexpressing miR-541 had reduced hypertrophy upon Ang-II treatment. While the target of miR-541 was not identified, the Authors found that the miRNA itself was negatively regulated by the pro-hypertrophic microphthalmia-associated transcription factor (MITF) [46].
A significant number of other miRNAs have been related to the negative regulation of CH. Among them the most known are miR-1 and miR-133. MiR-1 is known to reverse CH during the compensatory phase of heart failure. Restoration of miR-1 expression via adeno-associated virus (AAV) serotype 9 delivery in Sprague-Dawley rats subjected to ascending aortic stenosis resulted in the regression of the hypertrophy, reduction of myocardial fibrosis and apoptosis as well as inactivation of the MAPK signaling pathway [47]. In a mouse model treated with isoproterenol (used to induce heart failure), miR-1a-3p agomir (agomir-1) injection reduced CH, fibrosis and apopotosis. Also, the mitochondrial DNA-encoded proteins NADH dehydrogenase 1 and cytochrome c oxydase 1 increased after the treatment with agomir-1, suggesting novel possible therapeutic targets for the disease [48]. The potential role of miR-1 in CH was investigated also in relation with the cyclin-dependent kinases-Retinoblastoma (CDKs-Rb) pathway [49]. While miR-1 expression was decreased in hypertrophic myocardium of rats that underwent abdominal aortic constriction and in phenypephrine-treated neonatal rat cardiomyocytes, the cyclin-dependent kinase 6 (CDK6) level was increased in the same samples and was shown to be a target of miR-1. Treatment with miR-1 mimic or CDK6 siRNA reduced the hypertrophic phenotype in terms of reduction of cell size, expression of ANF and β-MHC and the phosphorylated pRb. These results suggest a role of miR-1 in the axis CDK6-Rb pathway in CH. [47,49]. MiR-133 was one of the first miRNAs described in CH: in 2007, its expression inversely correlated to CH in transverse aortic arch-constricted mice, transgenic mice with cardiac-restricted overexpression of a constitutively active mutant Akt kinase, and rats induced to exercise [50]. More recently, miR-133, which is an antagonist of inositol 1,4,5′-triphosphate receptor II calcium channel, was found downregulated in hypertrophy, leading to increased calcium signaling and thus inducing pathological remodeling [51]. In a separate study, miR-133 expression was investigated in rats subjected to hyperthyroidism [52]. Treatment with type 1 Angiotensin II receptor (AT1R) caused elevated levels of thyroid hormone, which in turn led to CH. In this context, miR-133 was downregulated and, accordingly, its targets SERCA2a and calcineurin were upregulated. These results suggest that miR-133 plays a key role in CH through different mechanisms.
6. MiRNAs and Inherited Cardiomyopathies
Genetically inherited cardiomyopathies represent a significant percentage of cardiovascular diseases [110]. These include disorders such as arrhythmogenic cardiomyopathy (ACM), hypertrophic cardiomyopathy (HCM), and dilated cardiomyopathy (DCM). Several miRNAs have been found altered also in these genetic diseases.
ACM is characterized by ventricular cardiomyocyte loss and subsequent replacement by fibro-fatty tissue, often leading to severe ventricular tachyarrhythmias and sudden cardiac death [111]. Most mutations causing ACM have been identified in genes coding for desmosomal proteins [111]. In a recent study, transgenic mice overexpressing the Q558* mutation in DSG2 gene, encoding for desmoglein-2, recapitulated several ACM features, such as fibro-fatty replacement, reduction in desmosomal size and number as well as the suppression of the Wnt/β-catenin signaling [53]. RNA sequencing of heart tissue revealed that miR-217-5p and miR-708-5p were the most upregulated miRNAs, while miR-499-5p was the most downregulated miRNA in the ACM model [53]. Another study reported that overexpression of miR-130a in murine myocardium induced ventricular arrhythmias and that this miRNA targets the gap junction protein connexin-43 [54]. Interestingly, in a separate study using the same model, the Authors identified the desmosomal protein desmocollin-2 as a target of miR-130a [55]. The work of Zhang et al. is the first one investigating the expression of miRNAs in human ACM samples [56]. The evaluation by S-Poly (T) Plus of the expression of 1078 miRNAs in 24 human cardiac ACM samples identified miR-21-5p and miR-135b as significantly up- and downregulated, respectively. Interestingly, in silico analyses suggested the correlation of these two miRNAs with Wnt and Hippo pathways, which have been associated with ACM pathogenesis [112,113]. Translational functional studies focused on these miRNAs are required to confirm this data and to support the role of these ncRNAs as a potential therapeutic target for this disease.
In genetic diseases, the definition of the underlying pathogenesis is often challenging, as mutations in a given gene can be associated with different phenotypic expressions. MiRNAs, and other non-coding RNAs, represent an additional layer that should be investigated in a homogenous group of patients to clarify the mechanisms associated with perturbations in a specific gene. Within this context, a study has been performed to investigate HCM, characterized by the so-called “myocardial disarray” involving the hypertrophic nondilated left ventricle, is frequently caused by mutations in genes coding for proteins that are part of the contractile components of the cardiac sarcomere or Z disk [114]. Kuster et al. found miR-204, embedded in the transient receptor potential cation channel subfamily M member 3 (TRPM3) gene, to be upregulated in heart samples from 6 HCM patients carrying a mutation in the myosin binding protein C (MYBPC3) gene, one of the most frequently mutated genes in the disease [57]. TRPM3 encodes for a cation-selective channel involved in calcium entry and, interestingly, was also upregulated in HCM samples. Accordingly, calcium homeostasis is dysregulated in HCM [115] and this finding suggests that TRPM3 might be involved in the disease pathogenesis caused by MYBPC3 mutations.
MiR-139-5p was found to be one of the most downregulated miRNAs in HCM patient hearts [58]. This result was confirmed in a recent study where the expression of miR-139-5p was reduced in left ventricular tissues of HCM patients [59].
Another study searched for miRNA-transcription factor feed-forward loops that were differentially regulated in HCM patients by integrating miRNA and gene expression profiles with experimentally verified transcription factor-target gene and miRNA-target gene interactions [60]. The Authors found that the most dysregulated feed-forward loop was between miR-17-5p and the fatty acid synthase (FASN) and the signal transducer and activator of transcription (STAT3) genes. FASN, a palmitate-synthesizing gene, has been linked to heart failure [116], while STAT3 is a cell-survival factor which protects cells from apoptosis during oxidative stress [117]. Interestingly, increased levels of phosphorylated STAT3 were found in a double-mutation murine model of familial HCM [118], while miR-17-5p has been shown to target STAT3 [119].
DCM is defined by an abnormally large left ventricle with poor contractility and is genetically very heterogeneous [120]. Most of the pathogenic mutations have been found in genes encoding for proteins related to the cytoskeleton, sarcomere and nuclear envelope [120,121]. Highlighting the complex regulatory roles that miRNAs can play, miR-148a was recently found to be downregulated in DCM while being upregulated in concentric hypertrophy in human cardiac biopsies [61]. These findings were corroborated in transgenic mouse models for DCM and concentric hypertrophy. Antagomir-mediated miR-148a knockdown in WT mice led to thinning of the cardiac wall, chamber dilation, increased left ventricle volume and reduced ejection fraction. On the other hand, upregulation of miR-148a through AAV-mediated delivery protected against systolic dysfunction caused by pressure overload [61].
MiR-208b was found upregulated in the myocardium of a heterozygous knock-in mouse model expressing a truncated titin, after induction of DCM by chronic exposure to angiotensin II or isoproterenol [62]. Inhibition of miR-208b with an antimir prevented DCM development in this mouse model and led to cardiac hypertrophy and only slight fibrosis, which was similar to WT mice treated with angiotensin II. Interestingly, the study also found that miR-208b was significantly upregulated in human DCM patients [62].
Another report on CD4+ T cells, which are known to play a role in the chronic inflammation of DCM, found that miR-451a was downregulated in the CD4+ T cells of DCM patients [63]. The overexpression of miR-451a in T cells inhibited their activation and proliferation, while the inhibition of this miRNA led to increased expression of activation markers. Myc was determined to be a target of miR-451a and was found to be upregulated in CD4+ T cells from patients with DCM, while knocking down Myc expression also suppressed the activation and proliferation of T cells [63]. Together with cardiac fibrosis, inflammation was investigated also in another study on DCM rats [64]. The downregulation of miR-132 was associated not only to elevated apoptosis and cardiac fibrosis, but also to higher levels of PTEN, Bcl-2 associated X protein, Ang II and aldosterone inflammatory markers. On the contrary, the upregulation of miR-132 caused reduction of PTEN and activated the PI3K/Akt pathway, which resulted in the repression of apoptosis, cardiac fibrosis and inflammatory response.
While they are distinct diseases, one study using left ventricular heart samples from both DCM and HCM patients revealed that miR-155, miR-10b and miR-23a were all overexpressed in both diseases compared to control samples [65]. On the other hand, miR-214 and miR-21 were respectively down- and upregulated in DCM patients specifically, while miR-1-3p and miR-27a were found to be downregulated only in HCM patients [65]. This result suggests that miRNAs are involved in very complex regulatory networks orchestrating the development of phenotypic features typical of specific different diseases.
Interestingly, also one major component of the miRNA biogenesis machinery, Dicer, has been linked to DCM. Targeted cardiac deletion of Dicer in a mouse model led to progressive DCM, heart failure and early postnatal lethality [122]. A miRNA microarray confirmed the reduction of mature miRNA levels, while levels of miRNA precursors were increased. Consistent with these results, the expression of DICER was found to be decreased in human patients with DCM or heart failure. However, after implantation of a left ventricular assist device, the expression of DICER was restored [122].
Long QT syndrome (LQTS) is a genetic channelopathy characterized by the prolongation of ventricular repolarization, susceptibility to Torsades de pointes, and risk for sudden death [123]. Several major cardiac components were found altered in LQTS. The miR-1/miR-133a cluster includes the most abundant miRNAs in the heart and is involved in the regulation of cardiac ion channels. The decrease of the miR-1/miR-133a cluster dosage in mice was sufficient to induce the LQTS phenotype, related to the abnormal impact of the β-adrenergic signaling on the depolarizing L-type calcium channel [66]. The authors were not able to detect a single miR-1 or miR-133a target gene involved in the alteration of the adrenergic signaling, but they hypothesized that multiple changes determined by the perturbed cluster can orchestrate the L-type calcium channel activity in their LQTS model [66]. The ether-a-go-go-related gene (hERG) is the major molecular component of the rapidly activating delayed rectifier K+ current (Ikr) and it is also modulated by miRNAs in LQTS. By coupling in silico and in vitro studies, Lian et al. correlated miR-134, miR-103a-1, miR-143, and miR-3619 overexpression with the downregulation of hERG mRNA and protein [67]. Moreover, the current activation and tail amplitude of hERG channel was reduced upon the increase of miR-103a-1 levels. Despite these promising results, further studies will have to be performed to demonstrate the mechanistic events linking these miRNAs and the alterations in hERG activity.
7. Circulating MiRNAs as Biomarkers for Cardiac Diseases
Due to their presence in various biological fluids, including plasma and serum, and the reliability in their measurements, miRNAs elicited particular attention as biomarkers for many diseases [124]. Several reports have investigated the diagnostic potential of circulating miRNAs as markers for the prediction of cardiovascular morbidity and mortality (Table 1, Figure 3).
Circulating miRNAs were abundantly studied in MI. In a prospective study, 70 patients underwent cardiac magnetic resonance at week 1 and 6 months after ST-segment elevation MI (STEMI) [68]. The analysis of miRNA expression in the plasma at admission revealed the prognostic value of miR-1254, which correlated with left ventricular remodeling and systolic function, such as changes in left ventricular volumes and ejection fraction at 6 months after STEMI. Moreover, circulating miR-22-5p and miR-150-3p were overexpressed in the early stage of acute MI, while miR-132-5p was reduced in plasma of 35 patients with the same condition [69]. Also miR-499 was associated with MI [70] and myocardial damage in cardiac diseases [71], while miR-34a was correlated with left ventricular remodeling after acute MI [72,73]. Finally, circulating miR-30a-5p expression was related with left ventricular dysfunction and heart failure after MI in 99 patients [74].
As far as arrhythmia is concerned, circulating miR-21 was associated with left atrial low-voltages areas in 102 patients with persistent atrial fibrillation undergoing catheter ablation [75], while circulating miR-483, miR-23a and miR-26a may be implicated in post-operative atrial fibrillation [76,77].
Circulating miRNAs have also been linked with inherited cardiomyopathies. MiR-142-5p, miR-143-3p, miR-27b and miR-126-3p were found significantly altered in 30 patients affected with childhood DCM [78]. In a separate study on 55 children affected with DCM, circulating miR-155 and miR-636 were found overexpressed, while miR-646 and miR-639 were downregulated in transplanted patients compared with affected patients who recovered their ventricular function [79]. Also, DCM was associated with altered plasma levels of miR-21, miR-26, miR-29, miR-30, miR-133a [80,81], and with the exosomal miR-92b-5p, found overexpressed in 43 affected patients [82]. Few studies have been performed on ACM, identifying miR-320a and miR-185 significantly down- and upregulated, respectively, in the plasma of affected patients [83,84]. In inherited cardiomyopathies, the circulating miRNA profiling coupled with the harbored mutation can potentially improve the risk stratification of the patients. In the study of Derda et al., increased mir-29a levels were detected in the blood of patients affected with hypertrophic non-obstructive cardiomyopathy carrying mutations in MYH7 gene, while miR-155 was downregulated in HCM patients with MYBPC3 mutations [85]. Also Roncarati et al. associated miR-29a with both hypertrophy and fibrosis in 41 HCM patients, but no correlation with the carried mutation was performed [86].
8. MiRNAs as Therapeutic Targets for Cardiac Diseases
The strong impact of miRNAs on the cardiac phenotype elicits particular interest in the possibility of targeting these molecules as therapeutic substrates. Depending on how miRNAs are dysregulated when the heart is upon stress, their individual manipulation is of great potential for novel treatment development to restore the normal phenotype. The technical details of the different therapeutic approaches have been reviewed previously [125]. Here we will summarize the latest data from preclinical studies that have employed viral or polymeric systems.
Recombinant viral systems such as lentiviruses, adenoviruses, and adeno-associated viruses, are commonly used to target genetic material into a given cell. Lentiviral overexpression of miR-99a in a mouse model for MI improved cardiac function via the mTOR/P70/S6K pathway [126]. Also, AAVs have been used to both knockdown or overexpress miRNAs. Tao et al. used AAV9 to transfer miR-433 sponge into the heart of mice after MI, resulting in an improvement of the phenotype [36]. Similarly, miR-378, a regulator of cardiac hypertrophy, was delivered via AAV9 in vivo, improving cardiac function [127]. AAV9 delivery has also been exploited to treat DCM into sarcoglycan-beta (Sgcb)-null mouse disease models. The resulting overexpression of miR-669a, downregulated in DCM, reduced cardiac fibrosis, hypertrophy and cardiomyocyte apoptosis for up to 18 months [128].
Different studies focused on the identification of delivery systems based on polymers to modulate miRNAs in specific cardiac conditions. Bejerano et al. delivered intravenously miR-21 mimic to treat MI in mice by using anionic nanoparticles spontaneously assembled upon complexation of the nucleic acid with hyaluronan-sulfate through calcium ion bridges [129]. This strategy succeeded in targeting macrophages in the infarcted area in mouse hearts, leading to pro-inflammatory-to-reparative switch and subsequent angiogenesis, reduced apoptosis, fibrosis and hypertrophy. Another attempt to improve cardiac function after MI includes injectable hyaluronic acid hydrogel miR-302 mimic delivery both in vitro and in a Confetti mouse model [130]. This system promoted cardiomyocytes proliferation and improved cardiac function, such as higher ejection fraction and functional shortening, together with a reduction in cardiac end-diastolic and end-systolic volumes 4 weeks after MI. Hydrogel was used also to deliver polymeric nanoparticles carrying miRNAs (miNPs), such as miR-199a-3p, in human embryonic stem cell-derived cardiomyocytes and endothelial cells or for intramyocardial injection in rats post-MI [131]. As a result, miNPs treatment promoted angiogenesis in hypoxia, with low cytotoxicity in vitro, and improved cardiac function in vivo after 4 weeks. Furthermore, cardiac hypertrophy was improved in mice with aortic coarctation using cholesterol-terminated ethanolamine-aminated poly(glycidyl methacrylate) (CHO-PGEA) to deliver miR-182 inhibitor to cardiomyocytes with a high efficiency [132]. In this context, the delivery of miR-182 inhibitor resulted in the restoration the level of FOXO3, an anti-hypertrophic transcription factor, in the heart of affected animals, which in turn led to alleviation of cardiac hypertrophy, without side effects in other organs.
9. Conclusions
Many different biological events interplay to determine the cardiovascular phenotype and its response to injury or stress, with a multitude of miRNAs exerting their influence over these processes. The studies on miRNAs in cardiovascular diseases provide a renewable benefit for the scientific and clinical communities. First, they can help elucidating the pathologic mechanisms by detecting altered target transcripts. The identification of perturbed pathways associated with cardiac phenotypes may allow developing novel drugs to antagonize specific pathogenic mechanisms. Also, in the context of inherited cardiomyopathies, miRNAs could provide insights to study the factors responsible for the wide variable expressivity as well as to bridge the genotype-phenotype gap, thus improving the risk stratification of patients.
Second, several studies identified a number of circulating miRNAs that are altered in the plasma or serum of different subtypes of patients. The potential of these molecules is undisputable to confirm the diagnosis in borderline patients but also to be used as non-invasive early biomarkers and to detect at risk subjects. However, several limitations still hinder their use in the daily clinical practice. Studies on bigger cohorts of patients should be performed to assess the reproducibility of the obtained results. Moreover, standardized protocols should be defined for the detection and the quantification of miRNAs. Finally, cross-comparisons among different patient cohorts should be considered to define the specificity of the miRNA profile for a given disease and so to help in the differential diagnosis.
Third, miRNAs can potentially be used as a new substrate for novel tailored therapies. Many reports highlight the efficacy of miRNA treatments in vivo; however, further studies are required to translate these results into clinical applications. For example, major effort should be address to test the safety of the delivery systems, the administration, the dosage and the duration of the treatment, as well as the occurrence and the prevention of side effects.
Author Contributions
Conceptualization, M.C. and R.C.; writing—original draft preparation, R.C. and M.C.; writing—review and editing, M.C. and R.C.; investigation, R.C. and M.C.; visualization, R.C.; resources, M.C.; supervision, M.C.; project administration, M.C.
Funding
We acknowledge the support from the DCVA, an initiative with support from the Dutch Heart Foundation, DCVA2017-2018 ARENA-PRIME.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics - 2018 update: A report from the American Heart Association. Circulation 2018, 137, e467–e492. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.L.; Horwich, T.B.; Fonarow, G.C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Clerk, A.; Cullingford, T.E.; Fuller, S.J.; Giraldo, A.; Markou, T.; Pikkarainen, S.; Sugden, P.H. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J. Cell. Physiol. 2007, 212, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, A.; Osiak, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today 2015, 20, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor Ambros The functions of animal microRNAs. Nature 2004, 431, 350–355. [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne). 2018. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of miRNA strand selection: follow the leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Kamanu, T.K.K.; Radovanovic, A.; Archer, J.A.C.; Bajic, V.B. Exploration of miRNA families for hypotheses generation. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. 2012, 18, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Leptidis, S.; el Azzouzi, H.; Lok, S.I.; de Weger, R.; Olieslagers, S.; Kisters, N.; Silva, G.J.; Heymans, S.; Cuppen, E.; Berezikov, E.; et al. A Deep Sequencing Approach to Uncover the miRNOME in the Human Heart. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Nishi, H.; Ono, K.; Horie, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Watanabe, S.; Takaya, T.; Tamaki, Y.; Takanabe-Mori, R.; et al. MicroRNA-27a Regulates Beta Cardiac Myosin Heavy Chain Gene Expression by Targeting Thyroid Hormone Receptor 1 in Neonatal Rat Ventricular Myocytes. Mol. Cell. Biol. 2011, 31, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science (80-.). 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Rawal, S.; Nagesh, P.T.; Coffey, S.; Van Hout, I.; Galvin, I.F.; Bunton, R.W.; Davis, P.; Williams, M.J.A.; Katare, R. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc. Diabetol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; Van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of MicroRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef]
- Ai, J.; Zhang, R.; Gao, X.; Niu, H.F.; Wang, N.; Xu, Y.; Li, Y.; Ma, N.; Sun, L.H.; Pan, Z.W.; et al. Overexpression of microRNA-1 impairs cardiac contractile function by damaging sarcomere assembly. Cardiovasc. Res. 2012, 95, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Györke, S. MiR-1 overexpression enhances ca2+release and promotes cardiac arrhythmogenesis by targeting pp2a regulatory subunit b56α and causing camkii-dependent hyperphosphorylation of RyR2. Circ. Res. 2009, 104, 514–521. [Google Scholar] [CrossRef]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunitomo, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.S.; Park, D.S.; Rotllan, N.; Chamorro-Jorganes, A.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.K.L.; Hernando, E. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, T.Y.; Cao, J.N.; Feng, Q.T.; Fu, Y.J.; Xu, X.; Yang, C.J. MicroRNA-206 Downregulates Connexin43 in Cardiomyocytes to Induce Cardiac Arrhythmias in a Transgenic Mouse Model. Hear. Lung Circ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Badalian-Very, G. Clinical applications of microRNAs. F1000Research 2013. [Google Scholar] [CrossRef]
- Li, X.; Zeng, Z.; Li, Q.; Xu, Q.; Xie, J.; Hao, H.; Luo, G.; Liao, W.; Bin, J.; Huang, X.; et al. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget 2015, 6, 18829–18844. [Google Scholar] [CrossRef] [Green Version]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef]
- Borden, A.; Kurian, J.; Nickoloff, E.; Yang, Y.; Troupes, C.D.; Ibetti, J.; Lucchese, A.M.; Gao, E.; Mohsin, S.; Koch, W.J.; et al. Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry After Injury. Circ. Res. 2019, 125, 14–25. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Chen, W.; Xie, L.; Zhao, Z.-A.; Yang, J.; Chen, Y.; Lei, W.; Shen, Z. MicroRNA-133 overexpression promotes the therapeutic efficacy of mesenchymal stem cells on acute myocardial infarction. Stem Cell Res. Ther. 2017. [Google Scholar] [CrossRef]
- Xu, C.; Hu, Y.; Hou, L.; Ju, J.; Li, X.; Du, N.; Guan, X.; Liu, Z.; Zhang, T.; Qin, W.; et al. β-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J. Mol. Cell. Cardiol. 2014, 75, 111–121. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, H.; Yang, D.; He, F.; Yuan, Y.; Guo, J.; Hu, J.; Yu, J.; Yan, X.; Wang, S.; et al. Aloe-Emodin Attenuates Myocardial Infarction and Apoptosis Via Up-Regulating MiR-133 Expression. Pharmacol. Res. 2019. [Google Scholar] [CrossRef]
- Wang, K.; Long, B.; Zhou, L.-Y.; Liu, F.; Zhou, Q.-Y.; Liu, C.-Y.; Fan, Y.-Y.; Li, P.-F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; DeMartino, A.M.; Watson, L.J.; Brittian, K.R.; Zafir, A.; Dassanayaka, S.; Hong, K.U.; Jones, S.P. MicroRNA-539 Is Up-regulated in Failing Heart, and Suppresses O-GlcNAcase Expression. J. Biol. Chem. 2014, 289, 29665–29676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.L.; Naya, F.J. MicroRNAs in the Myocyte Enhancer Factor 2 (MEF2)-regulated Gtl2-Dio3 Noncoding RNA Locus Promote Cardiomyocyte Proliferation by Targeting the Transcriptional Coactivator Cited2. J. Biol. Chem. 2015, 290, 23162–23172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.L.; Maruyama, S.; Sano, S.; Accorsi, A.; Girgenrath, M.; Walsh, K.; Naya, F.J. miR-410 and miR-495 Are Dynamically Regulated in Diverse Cardiomyopathies and Their Inhibition Attenuates Pathological Hypertrophy. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.P.; et al. Crucial Role of miR-433 in Regulating Cardiac Fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.P.; Chen, J.; Seok, H.Y.; Zhang, Z.; Kataoka, M.; Hu, X.; Wang, D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013, 112, 1234–1243. [Google Scholar] [CrossRef]
- Xu, X.-D.; Song, X.-W.; Li, Q.; Wang, G.-K.; Jing, Q.; Qin, Y.-W. Attenuation of MicroRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J. Cell. Physiol. 2012, 227, 1391–1398. [Google Scholar] [CrossRef]
- Tu, Y.; Wan, L.; Bu, L.; Zhao, D.; Dong, D.; Huang, T.; Cheng, Z.; Shen, B. MicroRNA-22 downregulation by atorvastatin in a mouse model of cardiac hypertrophy: A new mechanism for antihypertrophic intervention. Cell. Physiol. Biochem. 2013, 31, 997–1008. [Google Scholar] [CrossRef]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012. [Google Scholar] [CrossRef]
- Da Costa Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; El Azzouzi, H.; Hansen, A.; Coenen-De Roo, C.J.; Bierhuizen, M.F.; Van Der Nagel, R.; et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010, 12, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. MiR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef]
- Yang, Y.; Del Re, D.P.; Nakano, N.; Sciarretta, S.; Zhai, P.; Park, J.; Sayed, D.; Shirakabe, A.; Matsushima, S.; Park, Y.; et al. miR-206 Mediates YAP-Induced Cardiac Hypertrophy and Survival. Circ. Res. 2015, 117, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.-M.; Tham, Y.K.; Ooi, J.Y.Y.; Patterson, N.L.; Kiriazis, H.; Su, Y.; Thomas, C.J.; Lin, R.C.Y.; et al. Inhibition of miR-154 Protects Against Cardiac Dysfunction and Fibrosis in a Mouse Model of Pressure Overload. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Fu, J.; Chen, Y.; Li, F. Attenuation of MicroRNA-495 Derepressed PTEN to Effectively Protect Rat Cardiomyocytes from Hypertrophy. Cardiol. 2018, 139, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, N.; Long, B.; Fan, Y.-Y.; Liu, C.-Y.; Zhou, Q.-Y.; Murtaza, I.; Wang, K.; Li, P.-F. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Ding, C.; Yin, P.; He, L.; Xu, Q.; Wu, Z.; Shi, Y.; Su, L. MiR-1a-3p mitigates isoproterenol-induced heart failure by enhancing the expression of mitochondrial ND1 and COX1. Exp. Cell Res. 2019, 378, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Tang, C.; Zhu, W.; Zhu, J.; Lin, Q.; Fu, Y.; Deng, C.; Xue, Y.; Yang, M.; Wu, S.; et al. CDK6 mediates the effect of attenuation of miR-1 on provoking cardiomyocyte hypertrophy. Mol. Cell. Biochem. 2016, 412, 289–296. [Google Scholar] [CrossRef]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Drawnel, F.M.; Wachten, D.; Molkentin, J.D.; Maillet, M.; Aronsen, J.M.; Swift, F.; Sjaastad, I.; Liu, N.; Catalucci, D.; Mikoshiba, K.; et al. Mutual antagonism between IP3RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Biol. 2012, 199, 783–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, G.P.; Lino, C.A.; Guedes, E.C.; do Nascimento Moreira, L.; Barreto-Chaves, M.L.M. Cardiac microRNA-133 is down-regulated in thyroid hormone-mediated cardiac hypertrophy partially via Type 1 Angiotensin II receptor. Basic Res. Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signaling and miRNA dysregulation. Cardiovasc. Res. 2018, 115, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J. Mol. Cell. Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, S.R.; Calway, T.; Harmon, C.; Farrell, P.; Kim, G.H. MicroRNA-130a Regulation of Desmocollin 2 in a Novel Model of Arrhythmogenic Cardiomyopathy. MicroRNA 2017, 6, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, S.; Dong, T.; Yang, J.; Xie, Y.; Wu, Y.; Kang, K.; Hu, S.; Gou, D.; Wei, Y. Profiling of differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kuster, D.W.D.; Mulders, J.; ten Cate, F.J.; Michels, M.; dos Remedios, C.G.; da Costa Martins, P.A.; van der Velden, J.; Oudejans, C.B.M. MicroRNA transcriptome profiling in cardiac tissue of hypertrophic cardiomyopathy patients with MYBPC3 mutations. J. Mol. Cell. Cardiol. 2013, 65, 59–66. [Google Scholar] [CrossRef]
- Song, L.; Su, M.; Wang, S.; Zou, Y.; Wang, X.; Wang, Y.; Cui, H.; Zhao, P.; Hui, R.; Wang, J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 2014, 18, 2266–2274. [Google Scholar] [CrossRef]
- Ming, S.; Shui-Yun, W.; Wei, Q.; Jian-Hui, L.; Ru-Tai, H.; Lei, S.; Mei, J.; Hui, W.; Ji-Zheng, W. miR-139-5p inhibits isoproterenol-induced cardiac hypertrophy by targetting c-Jun. Biosci. Rep. 2018. [Google Scholar] [CrossRef]
- Shi, H.; Li, J.; Song, Q.; Cheng, L.; Sun, H.; Fan, W.; Li, J.; Wang, Z.; Zhang, G. Systematic identification and analysis of dysregulated miRNA and transcription factor feed-forward loops in hypertrophic cardiomyopathy. J. Cell. Mol. Med. 2019, 23, 306–316. [Google Scholar] [CrossRef]
- Raso, A.; Dirkx, E.; Philippen, L.E.; Fernandez-Celis, A.; De Majo, F.; Sampaio-Pinto, V.; Sansonetti, M.; Juni, R.; el Azzouzi, H.; Calore, M.; et al. Therapeutic Delivery of miR-148a Suppresses Ventricular Dilation in Heart Failure. Mol. Ther. 2018, 27, 584–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Schötterl, S.; Backes, D.; Brunner, E.; Hahn, J.K.; Ionesi, E.; Aidery, P.; Sticht, C.; Labeit, S.; Kandolf, R.; et al. Inhibition of miR-208b improves cardiac function in titin-based dilated cardiomyopathy. Int. J. Cardiol. 2017, 230, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wang, K.; Li, Y.; Xia, N.; Nie, S.; Lv, B.; Zhang, M.; Tu, X.; Li, Q.; Tang, T.; et al. Down-regulation of microRNA-451a facilitates the activation and proliferation of CD4+ T cells by targeting Myc in patients with dilated cardiomyopathy. J. Biol. Chem. 2017, 292, 6004–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Huang, Y.; Lu, J.; Lin, J.; Ge, Z.; Huang, H. Upregulated microRNA-132 rescues cardiac fibrosis and restores cardiocyte proliferation in dilated cardiomyopathy through the phosphatase and tensin homolog–mediated PI3K/Akt signal transduction pathway. J. Cell. Biochem. 2019, 120, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.; Chen, L.; Chen, K.; Zhou, J.; Song, J. MiR-1-3p that correlates with left ventricular function of HCM can serve as a potential target and differentiate HCM from DCM. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Besser, J.; Malan, D.; Wystub, K.; Bachmann, A.; Wietelmann, A.; Sasse, P.; Fleischmann, B.K.; Braun, T.; Boettger, T. MiRNA-1/133a Clusters Regulate Adrenergic Control of Cardiac Repolarization. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Guo, J.; Huang, X.; Yang, X.; Huang, G.; Mao, H.; Sun, H.H.; Ba, Y.; Zhou, J. miRNAs Regulate hERG. J. Cardiovasc. Electrophysiol. 2016, 27, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Cediel, G.; Bär, C.; Núñez, J.; Revuelta-Lopez, E.; Gavara, J.; Ríos-Navarro, C.; Llorente-Cortes, V.; Bodí, V.; Thum, T.; et al. Circulating miR-1254 predicts ventricular remodeling in patients with ST-Segment-Elevation Myocardial Infarction: A cardiovascular magnetic resonance study. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, P.; Li, F.; Yuan, G.; Wang, X.; Zhang, A.; Li, F. Plasma miR-22-5p, miR-132-5p, and miR-150-3p Are Associated with Acute Myocardial Infarction. Biomed Res. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Yang, C.; Han, Z. Circulating miR-499 as a potential biomarker for acute myocardial infarction. Ann. Transl. Med. 2016. [Google Scholar] [CrossRef]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 Reflect Myocardial Damage in Cardiovascular Disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.; Zhou, M.; He, J.; Meng, W.; Ma, X.; Dong, S.; Meng, X.; Zhao, X.; Wang, X.; He, F. Circulating miR-208b and miR-34a are associated with left ventricular remodeling after acute myocardial infarction. Int. J. Mol. Sci. 2014, 15, 5774–5788. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, H.-W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.-D.; Fisch, S.; Unno, K.; Sereti, K.-I.; Liao, R. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Maciejak, A.; Kostarska-Srokosz, E.; Gierlak, W.; Dluzniewski, M.; Kuch, M.; Marchel, M.; Opolski, G.; Kiliszek, M.; Matlak, K.; Dobrzycki, S.; et al. Circulating miR-30a-5p as a prognostic biomarker of left ventricular dysfunction after acute myocardial infarction. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Maleck, C.; von Ungern-Sternberg, S.N.I.; Neupane, B.; Heinzmann, D.; Marquardt, J.; Duckheim, M.; Scheckenbach, C.; Stimpfle, F.; Gawaz, M.; et al. Circulating MicroRNA-21 Correlates With Left Atrial Low-Voltage Areas and Is Associated With Procedure Outcome in Patients Undergoing Atrial Fibrillation Ablation. Circ. Arrhythm. Electrophysiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Harling, L.; Lambert, J.; Ashrafian, H.; Darzi, A.; Gooderham, N.J.; Athanasiou, T. Elevated serum microRNA 483-5p levels may predict patients at risk of post-operative atrial fibrillation. Eur. J. Cardio-Thoracic Surg. 2017, 51, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Moreira, D.A.R.; Gun, C.; Wang, H.-T.L.; Hirata, M.H.; de Freitas Germano, J.; Leite, G.G.S.; Farsky, P. Analysis of Circulating miR-1, miR-23a, and miR-26a in Atrial Fibrillation Patients Undergoing Coronary Bypass Artery Grafting Surgery. Ann. Hum. Genet. 2017, 81, 99–105. [Google Scholar] [CrossRef]
- Jiao, M.; You, H.-Z.; Yang, X.-Y.; Yuan, H.; Li, Y.-L.; Liu, W.-X.; Jin, M.; Du, J. Circulating microRNA signature for the diagnosis of childhood dilated cardiomyopathy. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.D.; Karimpour-Fard, A.; Peterson, V.; Auerbach, S.R.; Stenmark, K.R.; Stauffer, B.L.; Sucharov, C.C. Circulating microRNA as a biomarker for recovery in pediatric dilated cardiomyopathy. J. Hear. Lung Transplant. 2015, 34, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Rubiś, P.; Totoń-Żurańska, J.; Wiśniowska-Śmiałek, S.; Holcman, K.; Kołton-Wróż, M.; Wołkow, P.; Wypasek, E.; Natorska, J.; Rudnicka-Sosin, L.; Pawlak, A.; et al. Relations between circulating microRNAs (miR-21, miR-26, miR-29, miR-30 and miR-133a), extracellular matrix fibrosis and serum markers of fibrosis in dilated cardiomyopathy. Int. J. Cardiol. 2017, 231, 201–206. [Google Scholar] [CrossRef]
- Rubiś, P.; Totoń-Żurańska, J.; Wiśniowska-Śmiałek, S.; Dziewięcka, E.; Kołton-Wróż, M.; Wołkow, P.; Pitera, E.; Rudnicka-Sosin, L.; Garlitski, A.C.; Gackowski, A.; et al. The relationship between myocardial fibrosis and myocardial microRNAs in dilated cardiomyopathy: A link between mir-133a and cardiovascular events. J. Cell. Mol. Med. 2018, 22, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, Y.; Du, Y.; Tao, J.; Zhou, Z.; Yang, Z. Serum Exosomal MiR-92b-5p as a Potential Biomarker for Acute Heart Failure Caused by Dilated Cardiomyopathy. Cell. Physiol. Biochem. 2018, 46, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; D’Alessandra, Y.; Farina, F.M.; Casella, M.; Cattaneo, F.; Catto, V.; Chiesa, M.; Stadiotti, I.; Brambilla, S.; Dello Russo, A.; et al. MiR-320a as a Potential Novel Circulating Biomarker of Arrhythmogenic CardioMyopathy. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hsiao, Y.W.; Chang, S.L.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chiang, S.J.; Hu, Y.F.; Tuan, T.C.; Chao, T.F.; et al. Circulating microRNAs in arrhythmogenic right ventricular cardiomyopathy with ventricular arrhythmia. EP Eur. 2018, 20, f37–f45. [Google Scholar] [CrossRef] [PubMed]
- Derda, A.A.; Thum, S.; Lorenzen, J.M.; Bavendiek, U.; Heineke, J.; Keyser, B.; Stuhrmann, M.; Givens, R.C.; Kennel, P.J.; Christian Schulze, P.; et al. Blood-based microRNA signatures differentiate various forms of cardiac hypertrophy. Int. J. Cardiol. 2015, 196, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, Among Other Up-Regulated MicroRNAs, Is the Only Biomarker for Both Hypertrophy and Fibrosis in Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M. The sarcomeric cytoskeleton: Who picks up the strain? Curr. Opin. Cell Biol. 2011, 23, 39–46. [Google Scholar] [CrossRef]
- Agarkova, I.; Perriard, J.C. The M-band: An elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 2005, 15, 477–485. [Google Scholar] [CrossRef]
- Weiss, A.; Leinwand, L.A. The mammalian myosin heavy chain gene family. Annu. Rev. Cell Dev. Biol. 1996, 12, 417–439. [Google Scholar] [CrossRef]
- Morkin, E. Control of cardiac myosin heavy chain gene expression. Microsc. Res. Tech. 2000, 50, 522–531. [Google Scholar] [CrossRef]
- van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antzelevitch, C.; Burashnikov, A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card. Electrophysiol. Clin. 2011, 3, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Janse, M.J. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc. Res. 2004, 61, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Despa, S. Cardiac Excitation-Contraction Coupling. In Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier Inc.: London, UK, 2013. [Google Scholar]
- Fabiato, A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 247–289. [Google Scholar] [CrossRef]
- Delmar, M.; Liang, F.X. Connexin43 and the regulation of intercalated disc function. Hear. Rhythm 2012, 9, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Thygesen, K.; Alpert, J.S.; White, H.D.; Jaffe, A.S.; et al. Third universal definition of myocardial infarction. Eur. Heart J. 2012, 33, 2551–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Liu, M.; Sun, R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biochem. Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; Van Der Made, I.; Herias, V.; Van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. Molecular Medicine miR-133 and miR-30 Regulate Connective Tissue Growth Factor Implications for a Role of MicroRNAs in Myocardial Matrix Remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Urbanek, K.; Kajstura, J.; Yan, S.M.; Finato, N.; Bussani, R.; Nadal-Ginard, B.; Silvestri, F.; Leri, A.; Beltrami, C.A.; et al. Evidence That Human Cardiac Myocytes Divide after Myocardial Infarction. N. Engl. J. Med. 2001, 344, 1750–1757. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Janssen, R.; Zuidwijk, M.; Muller, A.; Mulders, J.; Oudejans, C.B.M.; Simonides, W.S. Cardiac Expression of Deiodinase type 3 (Dio3) Following Myocardial Infarction Is Associated With the Induction of a Pluripotency microRNA Signature from the Dlk1-Dio3 Genomic Region. Endocrinology 2013, 154, 1973–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, T.; Naya, F.; Dill, T.L.; Naya, F.J. A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.S.; Yevtodiyenko, A.; Schmidt, C.L.; Schmidt, J.V. Allele-specific histone modifications regulate expression of the Dlk1-Gtl2 imprinted domain. Genomics 2007, 89, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Denis, M.; Persello, A.; Rathagirishnan, R.; Lauzier, B. Protein O-GlcNAcylation in Cardiac Pathologies: Past, Present, Future. Front. Endocrinol. (Lausanne). 2019. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Seidman, J.G.; Seidman, C.E. The genetic basis for cardiac remodeling. Annu. Rev. Genomics Hum. Genet. 2005, 6, 185–216. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Invest. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac Plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Kerppola, T.K. Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy. Mol. Cell. Biol. 2011, 31, 3068–3080. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Calore, M.; Lorenzon, A.; De Bortoli, M.; Poloni, G.; Rampazzo, A. Arrhythmogenic cardiomyopathy: a disease of intercalated discs. Cell Tissue Res. 2015, 360, 491–500. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; McKenna, W.J.; Watkins, H. Disease Pathways and Novel Therapeutic Targets in Hypertrophic Cardiomyopathy. Circ. Res. 2011, 109, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd Alla, J.; Graemer, M.; Fu, X.; Quitterer, U. Inhibition of G-protein-coupled Receptor Kinase 2 Prevents the Dysfunctional Cardiac Substrate Metabolism in Fatty Acid Synthase Transgenic Mice. J. Biol. Chem. 2016, 291, 2583–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Duan, W.; Jin, Z.; Yi, W.; Yan, J.; Zhang, S.; Wang, N.; Liang, Z.; Li, Y.; Chen, W.; et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J. Pineal Res. 2013, 55, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsman, T.; Kelly, M.; Ng, D.C.H.; Tan, J.-E.; Tu, E.; Lam, L.; Bogoyevitch, M.A.; Seidman, C.E.; Seidman, J.G.; Semsarian, C. Severe Heart Failure and Early Mortality in a Double-Mutation Mouse Model of Familial Hypertrophic Cardiomyopathy. Circulation 2008, 117, 1820–1831. [Google Scholar] [PubMed] [Green Version]
- Du, W.; Pan, Z.; Chen, X.; Wang, L.; Zhang, Y.; Li, S.; Liang, H.; Xu, C.; Zhang, Y.; Wu, Y.; et al. By Targeting Stat3 microRNA-17-5p Promotes Cardiomyocyte Apoptosis in Response to Ischemia Followed by Reperfusion. Cell. Physiol. Biochem. 2014, 34, 955–965. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy. Circ. Res. 2017, 121, 731–748. [Google Scholar]
- Dellefave, L.; McNally, E.M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, A.J.; Schwartz, P.J.; Crampton, R.S.; Tzivoni, D.; Locati, E.H.; MacCluer, J.; Hall, W.J.; Weitkamp, L.; Vincent, G.M.; Garson, A. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991, 84, 1136–1144. [Google Scholar] [CrossRef]
- Ghai, V.; Wang, K. Recent progress toward the use of circulating microRNAs as clinical biomarkers. Arch. Toxicol. 2016, 90, 2959–2978. [Google Scholar] [CrossRef]
- De Majo, F.; De Windt, L.J. RNA therapeutics for heart disease. Biochem. Pharmacol. 2018, 155, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xie, J.; Li, R.; Shi, J.; Sun, J.; Gu, R.; Ding, L.; Wang, L.; Xu, B. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell. Mol. Med. 2014, 18, 919–928. [Google Scholar] [CrossRef]
- Ganesan, J.; Ramanujam, D.; Sassi, Y.; Ahles, A.; Jentzsch, C.; Werfel, S.; Leierseder, S.; Loyer, X.; Giacca, M.; Zentilin, L.; et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation 2013, 127, 2097–2106. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Crippa, S.; Montecchiani, C.; Camps, J.; Cornaglia, A.I.; Boldrin, L.; Morgan, J.; Calligaro, A.; Casasco, A.; Orlacchio, A.; et al. Long-term miR-669a therapy alleviates chronic dilated cardiomyopathy in dystrophic mice. J. Am. Heart Assoc. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohen, S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Liu, Y.; Chung, J.J.; Wang, T.; Gaffey, A.C.; Lu, M.; Cavanaugh, C.A.; Zhou, S.; Kanade, R.; Atluri, P.; et al. Sustained miRNA delivery from an injectable hydrogel promotes cardiomyocyte proliferation and functional regeneration after ischaemic injury. Nat. Biomed. Eng. 2017, 1, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Qin, X.; Wang, H.; Zhao, X.; Liu, Y.; Wo, H.T.; Liu, C.; Nishiga, M.; Chen, H.; Ge, J.; et al. An in Vivo miRNA Delivery System for Restoring Infarcted Myocardium. ACS Nano 2019. [Google Scholar] [CrossRef]
- Zhi, Y.; Xu, C.; Sui, D.; Du, J.; Xu, F.; Li, Y. Effective Delivery of Hypertrophic miRNA Inhibitor by Cholesterol-Containing Nanocarriers for Preventing Pressure Overload Induced Cardiac Hypertrophy. Adv. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MicroRNA biogenesis. First the microRNA (miRNA) gene is transcribed to create the pri-miRNA, a single-stranded RNA hairpin with imperfect base pairing. Then, Drosha will cleave the pri-miRNA into a 70-nucleotide hairpin with a 2-nucleotide-3′ overhang, the pre-miRNA. After export to the cytoplasm, Dicer will further process the molecule and form a double-stranded miRNA:miRNA* duplex 22 nucleotides long. One strand of this duplex, called the guide strand, combines with the Argonaute (AGO) protein and the target messenger RNA into the RNA-induced silencing complex (RISC). The other strand, the passenger strand, is degraded.
Figure 1.
MicroRNA biogenesis. First the microRNA (miRNA) gene is transcribed to create the pri-miRNA, a single-stranded RNA hairpin with imperfect base pairing. Then, Drosha will cleave the pri-miRNA into a 70-nucleotide hairpin with a 2-nucleotide-3′ overhang, the pre-miRNA. After export to the cytoplasm, Dicer will further process the molecule and form a double-stranded miRNA:miRNA* duplex 22 nucleotides long. One strand of this duplex, called the guide strand, combines with the Argonaute (AGO) protein and the target messenger RNA into the RNA-induced silencing complex (RISC). The other strand, the passenger strand, is degraded.

Figure 2.
MiRNAs and the relative targets associated with cardiac diseases. Green miRNAs are upregulated, while red miRNAs are downregulated in the different diseases. NA, not available.
Figure 2.
MiRNAs and the relative targets associated with cardiac diseases. Green miRNAs are upregulated, while red miRNAs are downregulated in the different diseases. NA, not available.

Figure 3.
Circulating miRNAs are associated with different cardiac diseases. Green miRNAs are upregulated, while red miRNAs are downregulated in the different diseases.
Figure 3.
Circulating miRNAs are associated with different cardiac diseases. Green miRNAs are upregulated, while red miRNAs are downregulated in the different diseases.


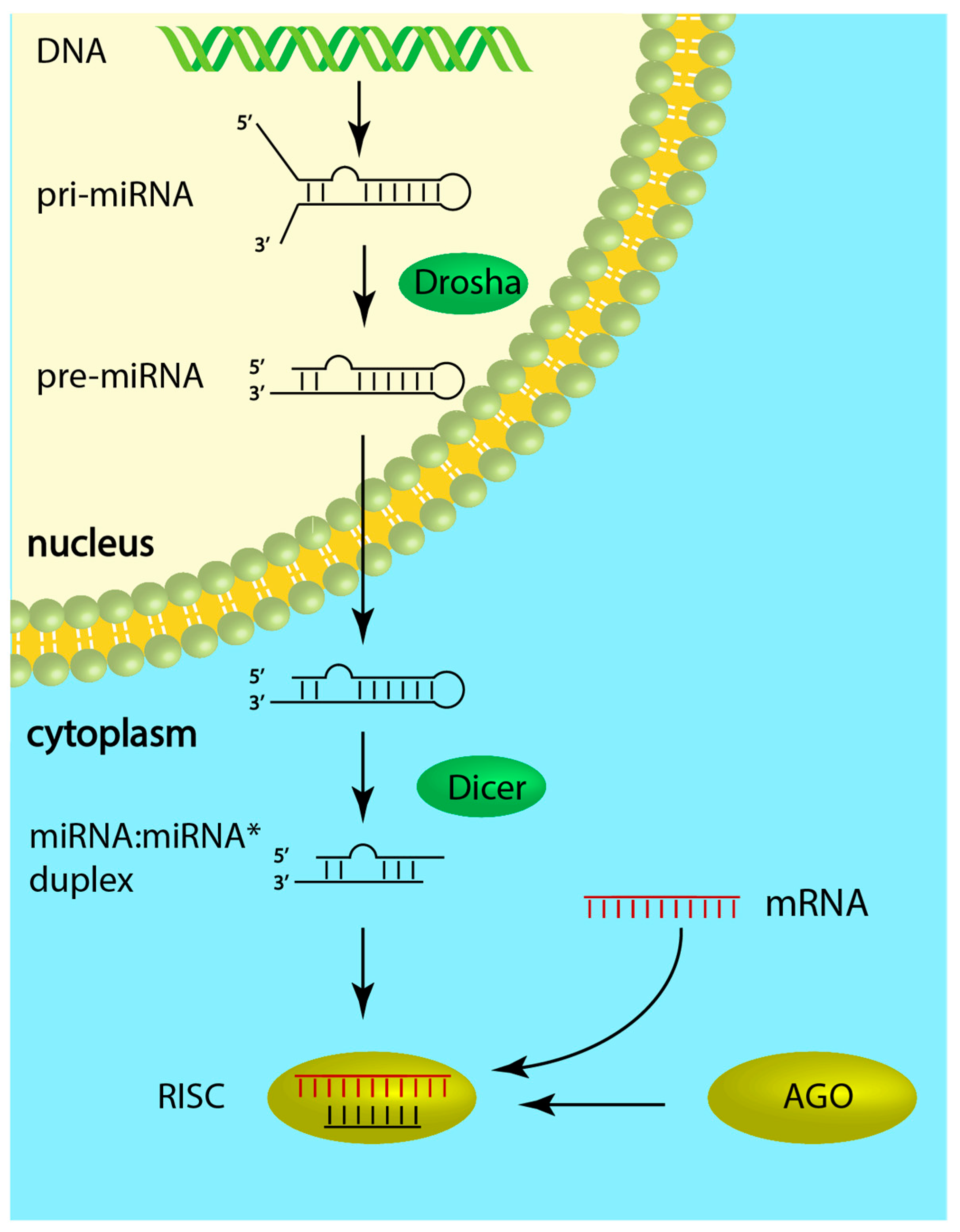{kind=link}
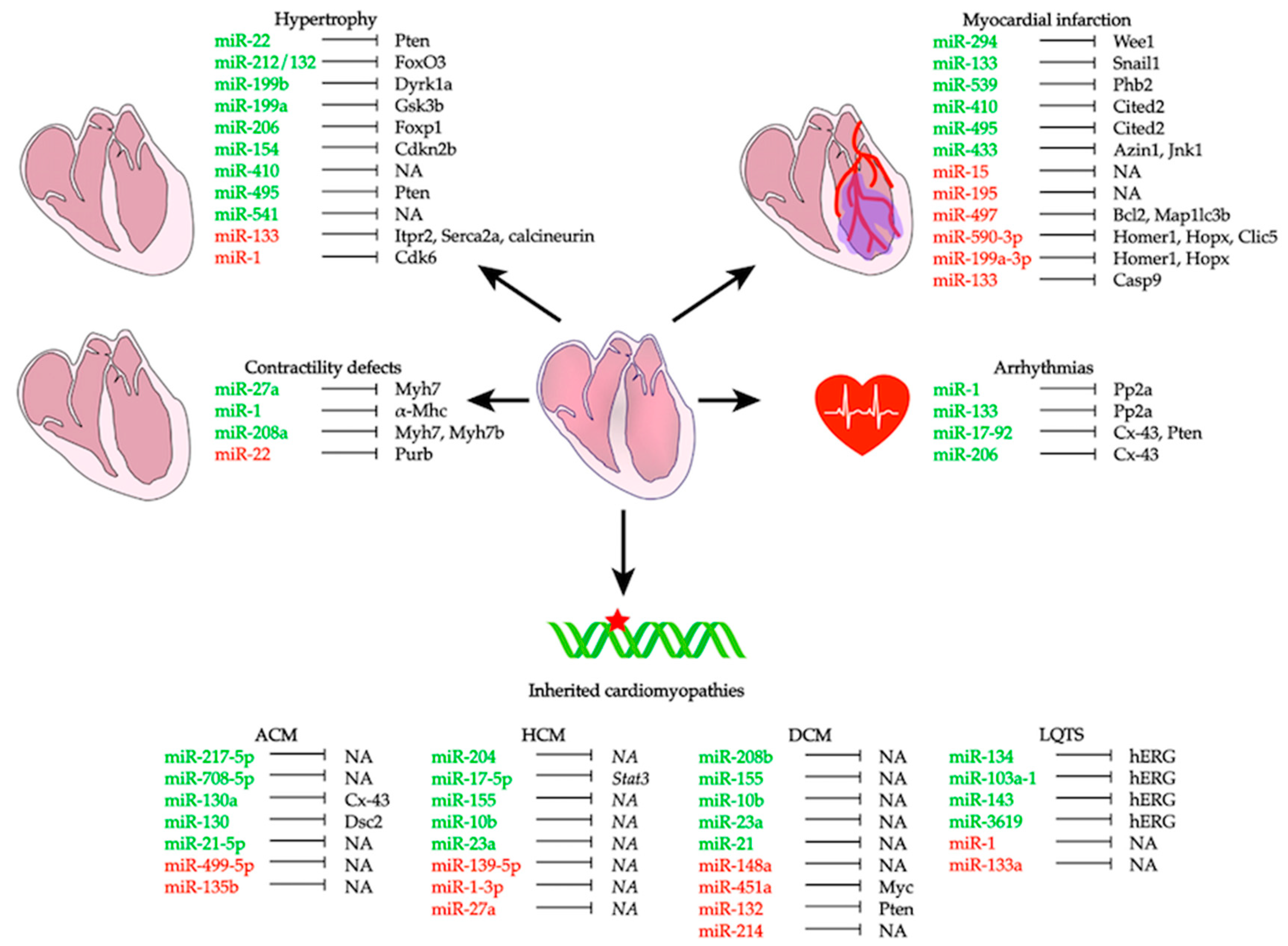{kind=link}
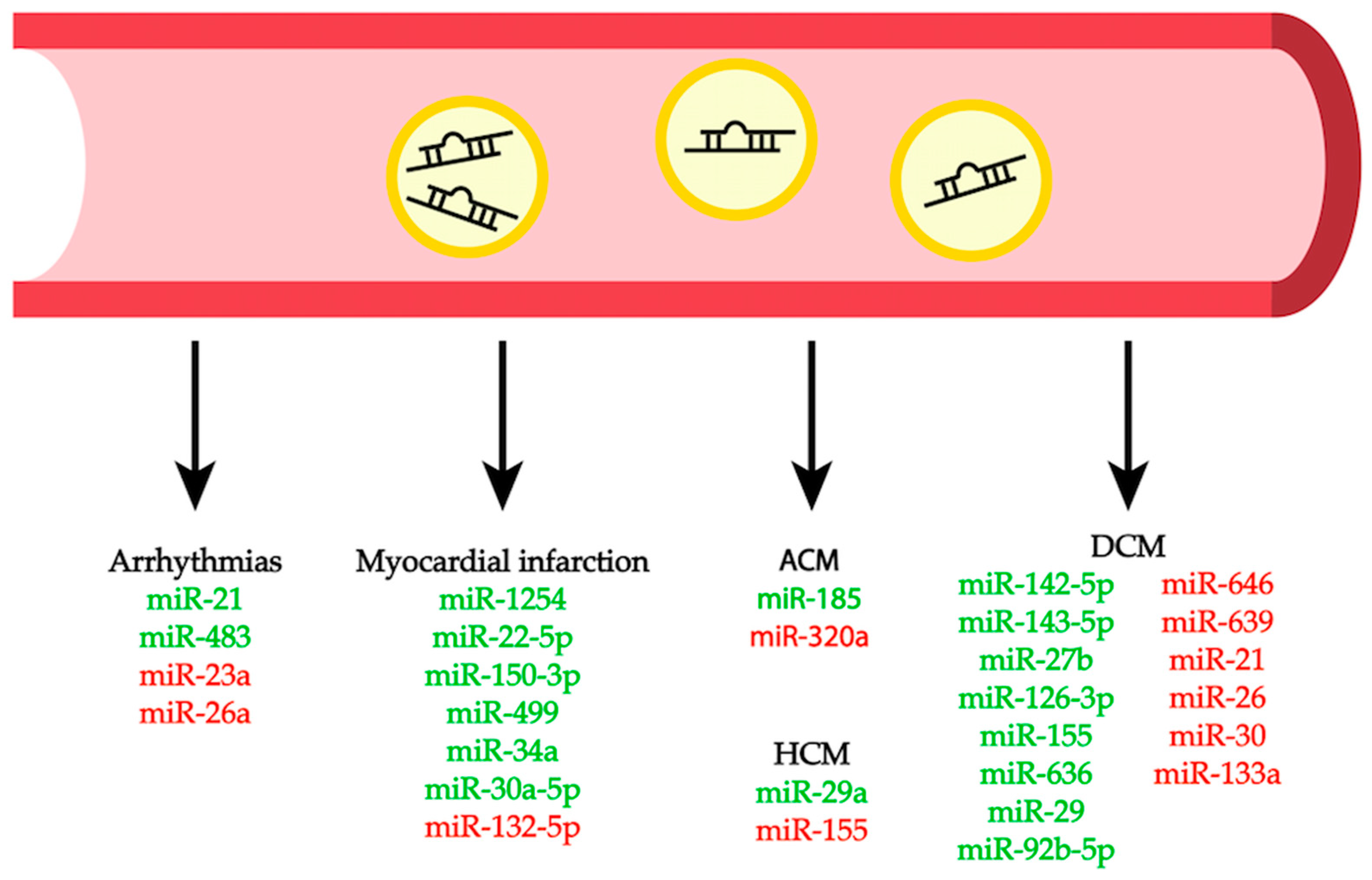{kind=link}
Table 1.
MicroRNAs involved in cardiac health and disease. Overview of several microRNAs that play a role in different cardiac phenotypes. MI, myocardial infarction; ACM, arrhythmogenic cardiomyopathy; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy, LQTS, long QT syndrome.
Table 1.
MicroRNAs involved in cardiac health and disease. Overview of several microRNAs that play a role in different cardiac phenotypes. MI, myocardial infarction; ACM, arrhythmogenic cardiomyopathy; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy, LQTS, long QT syndrome.
| Cardiac miRNA | |||
| miRNA | Up- or Downregulated | Disease | Reference |
| miR-27a | Upregulated | Contractility | [15] |
| miR-208a | Upregulated | Contractility | [16,17] |
| miR-22 | Downregulated | Abnormal Ca2+ cycling | [18] |
| miR-1 | Upregulated Upregulated Upregulated | Contractility Abnormal Ca2+ cycling Chronic heart failure | [19] [20] [21] |
| miR-133 | Upregulated | Chronic heart failure | [21] |
| miR-17-92 cluster | Upregulated | Arrhythmia | [22] |
| miR-206 | Upregulated | Arrhythmia | [23] |
| miR-15 family | Downregulated | MI | [24,25] |
| miR-195 | Downregulated | MI | [25] |
| miR-497 | Downregulated | MI | [26] |
| miR-590-3p miR-199a-3p | Downregulated | MI | [27] |
| miR-294 | Upregulation | MI | [28] |
| miR-133 | Upregulated Downregulated | MI | [29,30] [31] |
| miR-539 | Upregulated | MI | [32,33] |
| miR-410 miR-495 | Upregulated | MI Hypertrophy | [34,35] |
| miR-433 | Upregulated | MI/fibrosis | [36] |
| miR-22 | Downregulated Upregulated | Hypertrophy | [37] [38,39] |
| miR-212/132 family | Upregulated | Hypertrophy | [40] |
| miR-199b | Upregulated | Hypertrophy | [41] |
| miR-199a | Upregulated | Hypertrophy | [42] |
| miR-206 | Upregulated | Hypertrophy | [43] |
| miR-154 | Upregulated | Hypertrophy | [44] |
| miR-410 | Upregulated | Hypertrophy | [35] |
| miR-495 | Upregulated | Hypertrophy | [35,45] |
| miR-541 | Downregulated | Hypertrophy | [46] |
| miR-1 | Downregulated | Hypertrophy | [47,48,49] |
| miR-133 | Downregulated | Hypertrophy | [48,50,51,52] |
| miR-217-5p miR-708-5p | Upregulated | ACM | [53] |
| miR-499-5p | Downregulated | ACM | [53] |
| miR-130a | Upregulated | ACM | [54,55] |
| miR-21-5p | Upregulated | ACM | [56] |
| miR-135b | Downregulated | ACM | [56] |
| miR-204 | Upregulated | HCM | [57] |
| miR-139-5p | Downregulated | HCM | [58,59] |
| miR-17-5p | Upregulated | HCM | [60] |
| miR-148a | Downregulated | DCM | [61] |
| miR-208b | Upregulated | DCM | [62] |
| miR-451a | Downregulated | DCM | [63] |
| miR-132 | Downregulation | DCM | [64] |
| miR-155 miR-10b miR-23a | Upregulated | HCM/DCM | [65] |
| miR-214 miR-21 | Downregulated Upregulated | DCM DCM | [65] |
| miR-1-3p miR-27a | Downregulated | HCM | [65] |
| miR-1/miR-133a | Downregulated | LQTS | [66] |
| miR-134 miR-103a-1 miR-143 miR-3619 | Upregulated | LQTS | [67] |
| Circulating miRNA | |||
| miRNA | Up- or Downregulated | Disease | Reference |
| miR-1254 | Upregulated | MI | [68] |
| miR-22-5p miR-150-3p | Upregulated | MI | [69] |
| miR-132-5p | Downregulated | MI | [69] |
| miR-499 | Upregulated | MI | [70,71] |
| miR-34a | Upregulated | MI | [72,73] |
| miR-30a-5p | Upregulated | MI | [74] |
| miR-21 | Upregulated | Arrhythmia | [75] |
| miR-483 | Upregulated | Arrhythmia | [76] |
| miR-23a miR-26a | Downregulated | Arrhythmia | [77] |
| miR-142-5p miR-143-3p miR-27b miR-126-3p | Upregulated | DCM | [78] |
| miR-155 miR-636 | Upregulated | DCM | [79] |
| miR-646 miR-639 | Downregulated | DCM | [79] |
| miR-29 | Upregulated | DCM | [80,81] |
| miR-21 miR-26 miR-30 miR-133a | Downregulated | DCM | [80,81] |
| miR-92b-5p | Upregulated | DCM | [82] |
| miR-320a | Downregulated | ACM | [83] |
| miR-185 | Upregulated | ACM | [84] |
| miR-29a | Upregulated | HCM | [85,86] |
| miR-155 | Downregulated | HCM | [85] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Colpaert, R.M.W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8, 737. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070737
AMA Style
Colpaert RMW, Calore M. MicroRNAs in Cardiac Diseases. Cells. 2019; 8(7):737. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070737
Chicago/Turabian StyleColpaert, Robin M.W., and Martina Calore. 2019. "MicroRNAs in Cardiac Diseases" Cells 8, no. 7: 737. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070737
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.