Stromal Cell Signature Associated with Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer


Abstract
:1. Introduction
2. Patients and Methods
Stromal Cell Selection and Microarray Analysis
3. Results
3.1. Patients
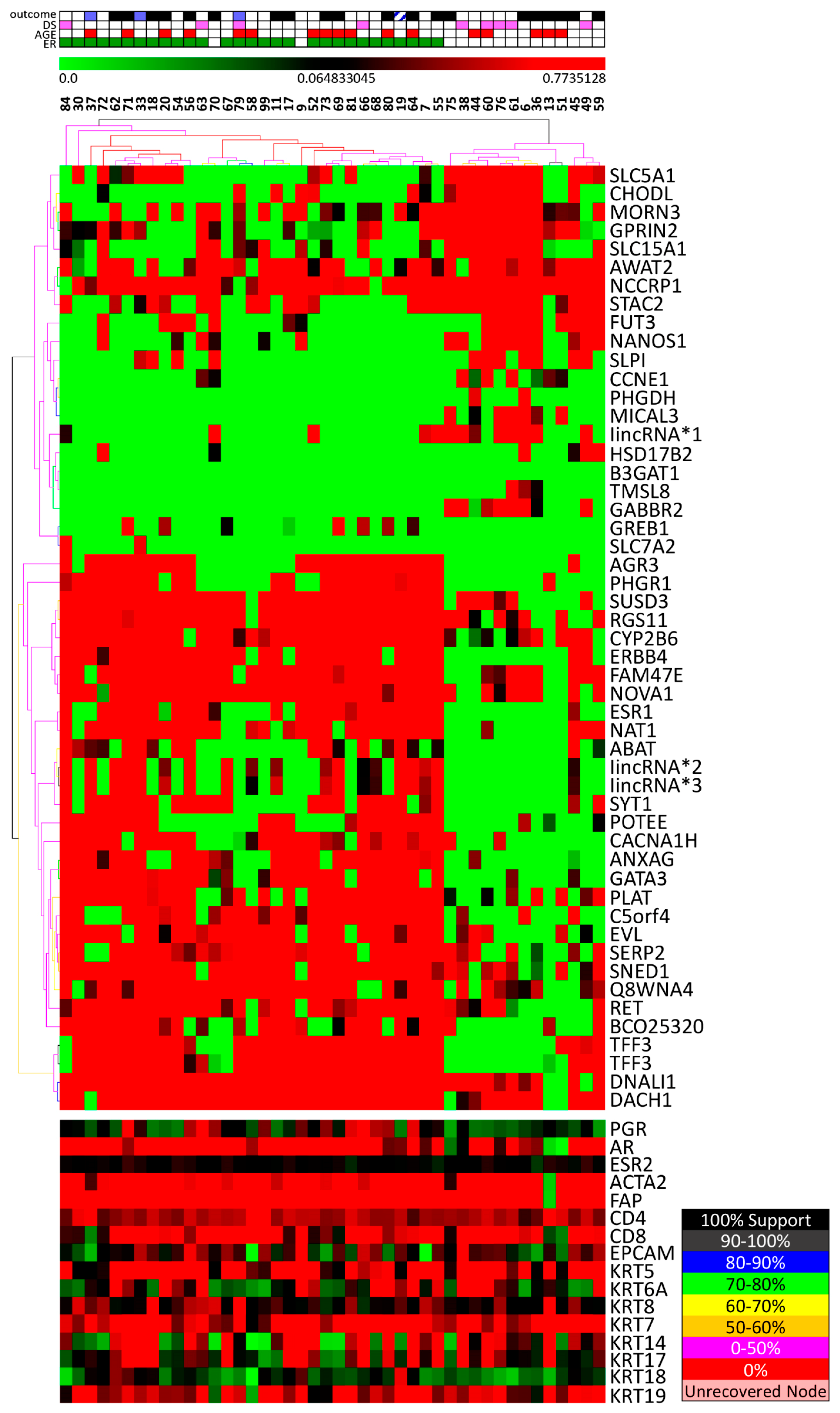
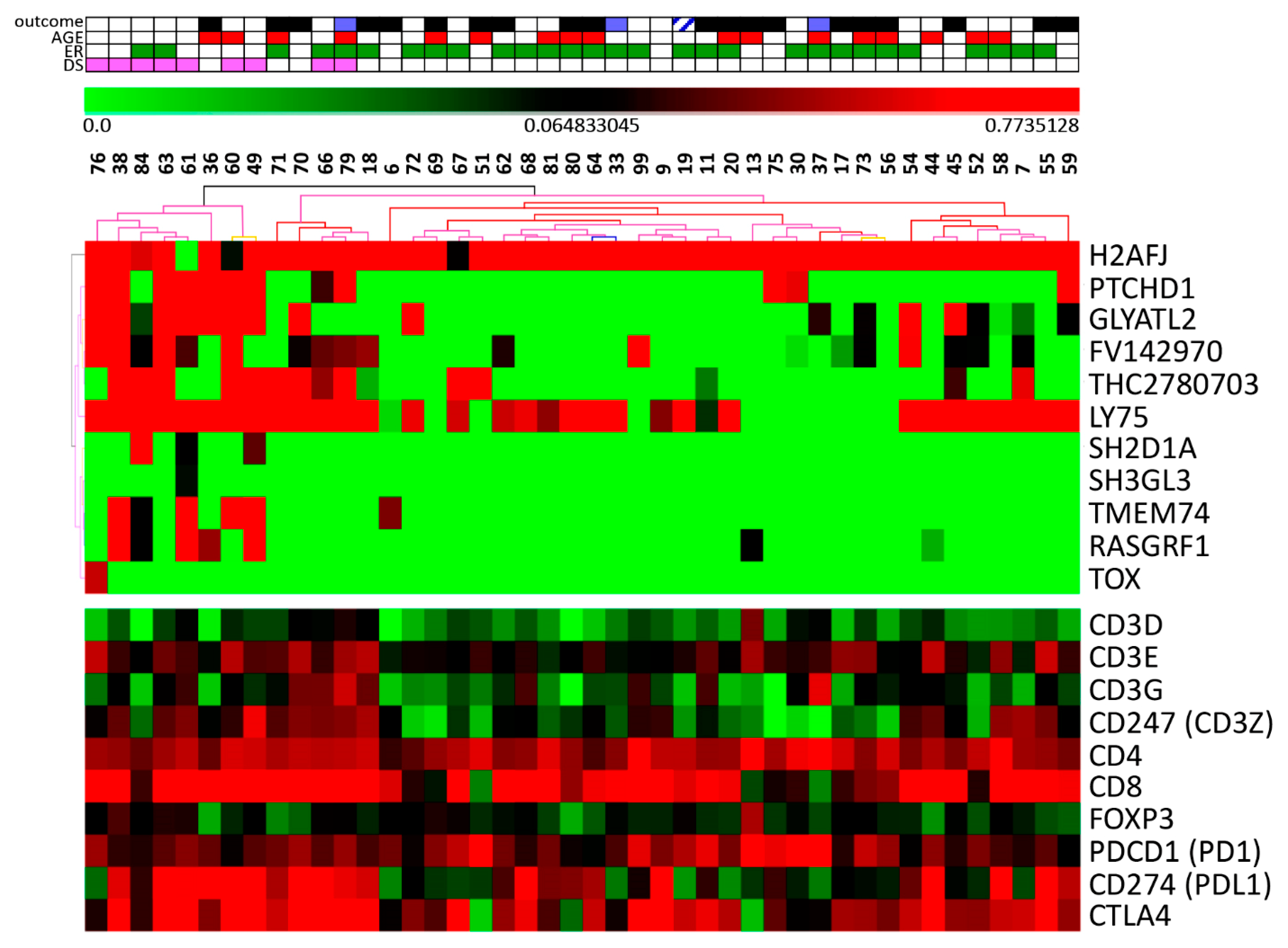
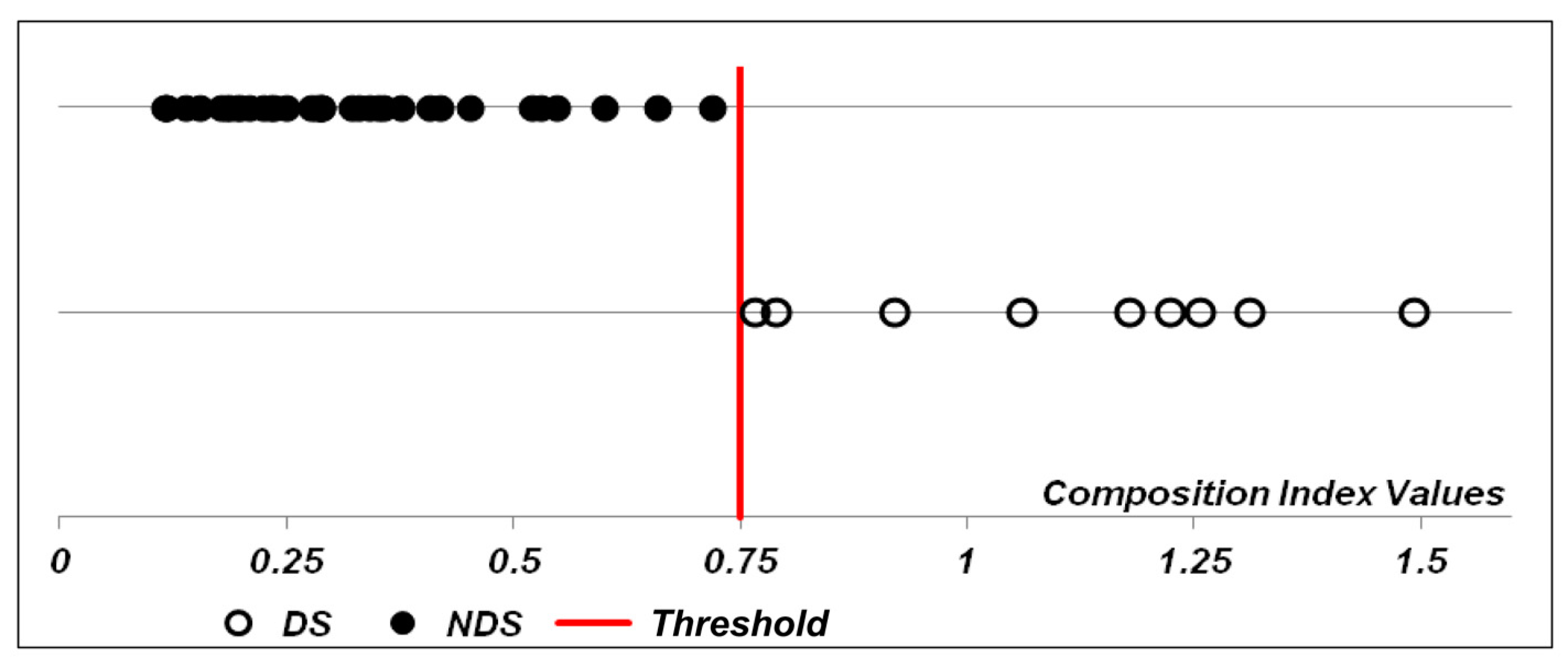
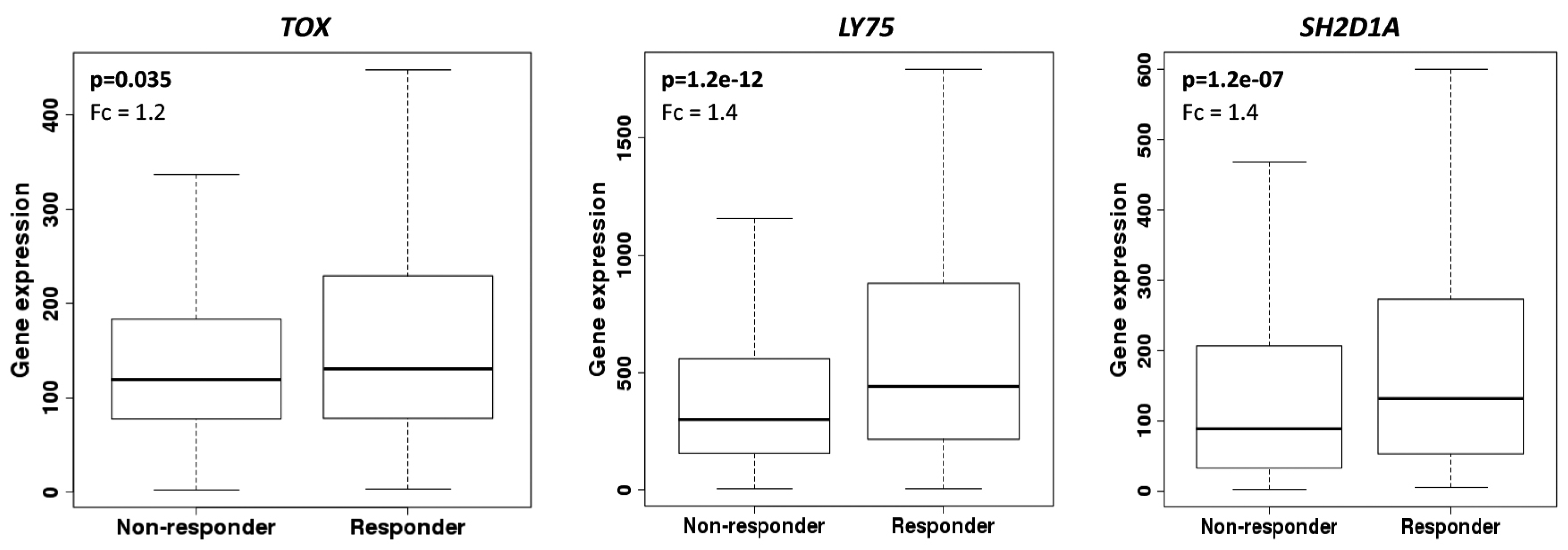
3.2. Stromal Cells Transcriptional Profile
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Elledge, R.; Mohsin, S.; Osborne, C.K.; Chamness, G.C.; Allred, D.C.; et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 2003, 362, 362–369. [Google Scholar] [CrossRef]
- Folgueira, M.A.; Carraro, D.M.; Brentani, H.; Patrão, D.F.; Barbosa, E.M.; Netto, M.M.; Caldeira, J.R.; Katayama, M.L.; Soares, F.A.; Oliveira, C.T.; et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin. Cancer Res. 2005, 11, 7434–7443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, J.; Leyh, B. The impact of tumor stroma on drug response in breast cancer. Semin. Cancer Boil. 2015, 31, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Rozenchan, P.B.; Carraro, D.M.; Brentani, H.; Mota, L.D.D.C.; Bastos, E.P.; Ferreira, E.N.E.; Torres, C.H.; Katayama, M.L.H.; Roela, R.A.; Lyra, E.C.; et al. Reciprocal changes in gene expression profiles of cocultured breast epithelial cells and primary fibroblasts. Int. J. Cancer 2009, 125, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.P.; Benvenuti, T.T.; Honda, S.T.; Del Valle, P.R.; Katayama, M.L.; Brentani, H.P.; Carraro, D.M.; Rozenchan, P.B.; Brentani, M.M.; de Lyra, E.C.; et al. Influence of the interaction between nodal fibroblast and breast cancer cells on gene expression. Tumour Biol. 2011, 32, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.-H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.-X.; Qiu, J.; et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Dekker, T.; Charehbili, A.; Smit, V.; Dijke, P.T.; Kranenbarg, E.M.-K.; Van De Velde, C.; Nortier, J.; Tollenaar, R.; Mesker, W.; Kroep, J. Disorganised stroma determined on pre-treatment breast cancer biopsies is associated with poor response to neoadjuvant chemotherapy: Results from the NEOZOTAC trial. Mol. Oncol. 2015, 9, 1120–1128. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy With or Without Carboplatin in Human Epidermal Growth Factor Receptor 2–Positive and Triple-Negative Primary Breast Cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef]
- Eryılmaz, M.; Mutlu, H.; Ünal, B.; Salim, D.; Müsri, F.; Coşkun, H. The importance of stromal and intratumoral tumor lymphocyte infiltration for pathologic complete response in patients with locally advanced breast cancer. J. Cancer Res. Ther. 2018, 14, 619. [Google Scholar] [CrossRef] [PubMed]
- De Campos, C.P.; Pereira, C.A.D.B.; Rancoita, P.M.V.; Polpo, A.; Pereira, C.D.B. Ordering Quantiles through Confidence Statements. Entropy 2016, 18, 357. [Google Scholar] [CrossRef] [Green Version]
- Marques, F.P.C.; Pereira, C.A.d.B. Predictive Analysis of Microarray Data. Open J. Genet. 2014, 4, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef]
- FAP Fibroblast Activation Protein Alpha. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/gene/2191 (accessed on 23 October 2019).
- McNamara, K.M.; Moore, N.L.; Hickey, T.E.; Sasano, H.; Tilley, W.D. Complexities of androgen receptor signalling in breast cancer. Endocr.-Relat. Cancer 2014, 21, T161–T181. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [Green Version]
- Tozlu, S.; Girault, I.; Vacher, S.; Vendrell, J.; Andrieu, C.; Spyratos, F.; Cohen, P.; Lidereau, R.; Bieche, I. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr.-Relat. Cancer 2006, 13, 1109–1120. [Google Scholar] [CrossRef]
- Yang, F.; Foekens, J.A.; Yu, J.; Sieuwerts, A.M.; Timmermans, M.; Klijn, J.G.; Atkins, D.; Wang, Y.; Jiang, Y. Laser microdissection and microarray analysis of breast tumors reveal ER-alpha related genes and pathways. Oncogene 2006, 25, 1413–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielson, M.; Chiesa, F.; Paulsson, J.; Strell, C.; Behmer, C.; Rönnow, K.; Czene, K.; Östman, A.; Hall, P. Amount of stroma is associated with mammographic density and stromal expression of oestrogen receptor in normal breast tissues. Breast Cancer Res. Treat. 2016, 158, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, M.; Giovannelli, P.; Cernera, G.; Di Santi, A.; Marino, I.; Bilancio, A.; Galasso, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front Endocrinol. 2015, 5, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, P.; Gunn, D.; McBryan, T.; Dirks, R.W.; Van Heemst, D.; Lim, F.-L.; Jochemsen, A.G.; Vries, M.V.-D.; Nagel, J.; Adams, P.D.; et al. Microarray-based identification of age-dependent differences in gene expression of human dermal fibroblasts. Mech. Ageing Dev. 2012, 133, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Neville, M.C. Stages in the development of mammary function. In Lactation. Physiology, Nutrition and Breast Feeding; Margaret, C.N., Marianne, R.N., Eds.; Plenun Press: New York, NY, USA, 1983; pp. 104–116. [Google Scholar]
- Yao, J.; Weremowicz, S.; Feng, B.; Gentleman, R.C.; Marks, J.R.; Gelman, R.; Brennan, C.; Polyak, K. Combined cDNA Array Comparative Genomic Hybridization and Serial Analysis of Gene Expression Analysis of Breast Tumor Progression. Cancer Res. 2006, 66, 4065–4078. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ghareeb, W.M.; Lu, X.; Huang, Y.; Huang, S.; Chi, P. Coexpression network analysis linked H2AFJ to chemoradiation resistance in colorectal cancer. J. Cell Biochem. 2019, 120, 10351–10362. [Google Scholar] [CrossRef]
- Noor, A.; Whibley, A.; Marshall, C.R.; Gianakopoulos, P.J.; Piton, A.; Carson, A.R.; Orlic-Milacic, M.; Lionel, A.C.; Sato, D.; Pinto, D.; et al. Disruption at the PTCHD1 locus on Xp22.11 in autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010, 2, 49ra68. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Lim, H.W.; Kim, J.R.; Rott, L.; Hillsamer, P.; Butcher, E.C. Unique gene expression program of human germinal center T helper cells. Blood 2004, 104, 1952–1960. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Kato, M.; McDonald, K.J.; Ross, I.L.; Vuckovic, S.; Munster, D.; Macdonald, K.P.; Hart, D.N.; Khan, S.; Chen, K. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int. Immunol. 2006, 18, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Swiggard, W.J.; Heufler, C.; Peng, M.; Mirza, A.; Steinman, R.M.; Nussenzweig, M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 1995, 375, 151–155. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, E.; Gil, G.L.; Benito, A.C.; González-Billalabeitia, E.; Conesa, M.A.V.; García, T.G.; García-Garre, E.; Vicente, V.; De La Peña, F.A. Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Res. 2014, 16, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Id | Age (y) | HT | ER | PR | HER2 | Ki67 (%) | T dim preCT (cm) | T dim postCT (cm) | ypTN (postCT) |
|---|---|---|---|---|---|---|---|---|---|
| 52 | ≤40 | D | + | + | − | 5 | 6.0 | 3.5 | ypT2N0 |
| 84 | >40 | L | + | − | − | 5 | 8.0 | 0.0 | ypT0N0 |
| 67 | >40 | D/L | + | + | − | 10 | 8.3 | 18.0 | ypT4N3 |
| 20 | ≤40 | D | + | + | − | 10 | 6.0 | 6.0 | ypT4N2 |
| 71 | ≤40 | D/IS | + | + | − | 10 | 7.6 | 4.5 | ypT2N3 |
| 66 | >40 | D | + | + | − | 60 | 11.0 | 0.8 | ypT1bN0 |
| 68 | >40 | D | + | + | − | 80 | 8.0 | 5.4 | ypT3N2 |
| 33 | >40 | D | + | + | − | 20 | 6.0 | 0.6 | ypT1bN2 |
| 64 | ≤40 | D | + | + | − | 20 | 8.0 | 1.8 | ypT1cN1 |
| 54 | >40 | D | + | + | − | 20 | 9.0 | 8.3 | ypT3N0 |
| 19 | >40 | D | + | + | − | 20 | 7.0 | 8.0 | ypT3N2 |
| 37 | ≤40 | D | + | + | − | 20 | 6.5 | 6.5 | ypT3N1 |
| 58 | ≤40 | D | + | − | − | 30 | 10.0 | 11.0 | ypT4N3 |
| 7 | >40 | D | + | + | − | 30 | 7.8 | 1.5 | ypT1cN3 |
| 79 | ≤40 | D | + | + | − | 70 | 5.2 | 0.0 | ypT0N0 |
| 9 | >40 | D | − | + | − | 70 | 4.0 | 4.4 | ypT2N0 |
| 6 | >40 | D | − | + | − | 80 | 6.0 | 7.8 | ypT4N2 |
| 73 | ≤40 | D | + | + | − | 80 | 8.0 | 5.8 | ypT3N2 |
| 63 | >40 | D | + | + | − | 90 | 6.5 | 0.6 | ypT1bN0 |
| 80 | ≤40 | D | + | + | − | 90 | 6.1 | 2.8 | ypT4Nx |
| 81 | ≤40 | D | + | − | ND | 100 | 6.4 | 5.0 | ypT2N1 |
| 30 | >40 | L | + | + | − | ND | 6.0 | 5.5 | ypT3N1 |
| 56 | ≤40 | D | + | + | + | ND | 14.0 | 6.8 | ypT3N1 |
| 62 | >40 | D | + | + | − | 10 | 6.0 | 2.0 | ypT1cN0 |
| 55 | >40 | D | + | + | − | 10 | 5.5 | 6.3 | ypT4N2 |
| 99 | >40 | D | + | + | + | 10 | 12.0 | 0.8 | ypT4N0 |
| 17 | >40 | D | + | − | + | 20 | 8.0 | 9.0 | ypT4N2 |
| 69 | ≤40 | D | + | − | − | 90 | 5.5 | 9.0 | ypT3N3 |
| 18 | >40 | D | + | − | − | 90 | 4.0 | 1.7 | ypT1N2 |
| 11 | >40 | D | + | + | + | ND | 7.0 | 2.2 | ypT2N2 |
| 72 | >40 | D | + | + | + | ND | 5.5 | 0.0 | ypT0N1 |
| 59 | >40 | D | − | − | + | 60 | 6.0 | 5.8 | ypT3N1 |
| 49 | >40 | D | − | − | + | 90 | 8.0 | 0.0 | ypT1miN0 |
| 60 | ≤40 | D | − | − | - | ND | 3.5 | 0.0 | ypT0N0 |
| 44 | ≤40 | D | − | − | + | 100 | 5.4 | 2.0 | ypT1cN0 |
| 70 | >40 | D/O | − | − | − | 40 | 5.1 | 4.5 | ypT2N2 |
| 75 | >40 | D | − | − | − | 60 | 7.0 | 7.5 | ypT4N3 |
| 51 | ≤40 | D | − | − | + | 80 | 8.9 | 4.5 | ypT4N1 |
| 38 | >40 | D | − | − | − | 90 | 5.2 | 1.0 | ypT1bN0 |
| 36 | ≤40 | D | − | − | − | 90 | 6.5 | 4.0 | ypT4N3 |
| 76 | >40 | D | − | − | − | 90 | 8.5 | 0.0 | ypTisN0 |
| 45 | >40 | L | − | − | − | ND | 7.5 | 4.0 | ypT4N3 |
| 13 | ≤40 | D | − | − | − | ND | 6.5 | 4.2 | ypT2N1 |
| 61 | >40 | D | − | − | − | 100 | 5.5 | 0.3 | ypT1aN0 |
| GO Gene Sets (Biological Process) | Genes (CORE ENRICHMENT) |
|---|---|
| ADAPTIVE_IMMUNE_RESPONSE_GO_0002460 | FOXP3 CD74 CRTAM C2 TRAF2 MAP3K7 TNFSF13 SOCS5 TRAF6 IL18 TLR8 EBI3 PTPRC CD40LG |
| ADAPTIVE_IMMUNE_RESPONSE | FOXP3 CD74 CRTAM C2 TRAF2 MAP3K7 TNFSF13 SOCS5 TRAF6 IL18 BCL10 TLR8 EBI3 PTPRC CD40LG |
| POSITIVE_REGULATION_OF_LYMPHOCYTE_ACTIVATION | CD47 LCK NCK1 CD24 CD3E ICOSLG TNFSF13 ZAP70 SOCS5 IL18 EBI3 PTPRC SIRPG |
| REGULATION_OF_IMMUNE_SYSTEM_PROCESS | FOXP3 APOBEC3G CD47 LCK TGFB2 NCK1 LAX1 CD24 CD3E ICOSLG CRTAM C2 TRAF2 MAP3K7 TNFSF13 ZAP70 SOCS5 TRAT1 TRAF6 IL18 EREG UBE2N TLR8 EBI3 PTPRC SIRPG NCR1 FYN NFAM1 LAT2 INHBA CD28 |
| POSITIVE_REGULATION_OF_IMMUNE_SYSTEM_PROCESS | CD47 LCK TGFB2 NCK1 CD24 CD3E ICOSLG CRTAM C2 TRAF2 MAP3K7 TNFSF13 ZAP70 SOCS5 TRAT1 TRAF6 IL18 EREG UBE2N TLR8 EBI3 PTPRC SIRPG FYN NFAM1 LAT2 CD28 |
| POSITIVE_REGULATION_OF_T_CELL_ACTIVATION | CD47 LCK NCK1 CD24 CD3E ICOSLG ZAP70 SOCS5 IL18 EBI3 PTPRC SIRPG |
| IMMUNE_RESPONSE | LY75 FOXP3 APOBEC3G IL15 CTSS TRIM22 TLR7 PTGER4 POU2AF1 PRKRA CD74 TGFB2 IL10RB DEFB1 TAPBP LAX1 CXCL13 HLA-DRB3 FYB BLNK NFIL3 CD96 SKAP1 CRTAM C2 TRAF2 IRF8 CD83 CTSC TCF7 MAP3K7 TNFSF13 CHUK ZAP70 IL2 YTHDF2 SOCS5 TRAT1 CCL5 TRAF6 IL6 AIM2 IL18 CCL25 BCL10 IKBKAP EREG LCP2 CXCR4 OPRK1 UBE2N CCL20 TNFAIP1 CCL2 LTB4R TLR8 CEBPB WAS CD164 SECTM1 GTPBP1 EBI3 CD7 TCF12 CD79B IL2RG GEM PTPRC GZMA CCR5 NCR1 CCL23 GPR65 FYN CD40LG XBP1 DPP4 CCR2 MAP4K2 APOA1 NFAM1 NCF4 LAT2 |
| _S_TRANSITION_OF_MITOTIC_CELL_CYCLE | CUL2 CUL1 GFI1 CDKN2A LATS2 CDKN2C PPP6C ACVR1 CDKN1B INHBA CDCA5 CDKN1A GSPT1 ACVR1B CDKN2D RCC1 |
| DNA_DEPENDENT_DNA_REPLICATION | GTPBP4 MSH5 RFC4 RAD17 CCDC88A RFC3 MSH6 MSH2 PRIM1 POLA1 TSPYL2 RFC1 PRIM2 GMNN POLB EREG HMGB2 CDK2AP1 REV3L S100A11 EXO1 NBN CDC6 MLH1 RPAIN |
| T_CELL_ACTIVATION | FOXP3 CD47 LCK NLRC3 NCK1 LAX1 CD24 CD3E ICOSLG CRTAM NHEJ1 ZAP70 IL2 SOCS5 IL18 EBI3 CD7 PTPRC SIRPG |
| APOPTOSIS (APOPTOSIS_GO) | CASP1 BAX NFKB1 IRF1 TNFRSF21 IRF4 GZMB FAS BID NFKBIA CASP3 TRAF2 CASP7 CHUK BIRC2 MDM2 TP53 TRAF3 TNF NFKBIE FASLG CASP4 APAF1 BIRC3 CASP6 TRAF1 CYCS |
| REGULATION_OF_IMMUNE_EFFECTOR_PROCESS | FOXP3 APOBEC3G CRTAM TRAF2 MAP3K7 TRAF6 PTPRC NCR1 |
| LYMPHOCYTE_ACTIVATION | FOXP3 CD47 LCK NLRC3 NCK1 LAX1 CD24 CD3E ICOSLG CRTAM TPD52 NHEJ1 TNFSF13 ZAP70 IL2 SOCS5 IL18 EBI3 CD7 PTPRC SIRPG CD40LG NFAM1 LAT2 INHBA CD28 CD3D |
| POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION | TRAF2 NOD2 MAP3K7 TRAF6 EREG IFNG ATP6AP2 CD40LG |
| POSITIVE_REGULATION_OF_MULTICELLULAR_ORGANISMAL_PROCESS | CD47 LCK TGFB2 NCK1 CD24 CD3E ICOSLG CRTAM C2 TRAF2 NOD2 MAP3K7 TNFSF13 ZAP70 SOCS5 TRAT1 TRAF6 IL18 EREG IFNG UBE2N TLR8 EBI3 PTPRC ATP6AP2 SIRPG BMP4 FYN CD40LG NFAM1 LAT2 CD28 MC4R |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katayama, M.L.H.; Vieira, R.A.d.C.; Andrade, V.P.; Roela, R.A.; Lima, L.G.C.A.; Kerr, L.M.; Campos, A.P.d.; Pereira, C.A.d.B.; Serio, P.A.d.M.P.; Encinas, G.; et al. Stromal Cell Signature Associated with Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer. Cells 2019, 8, 1566. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121566
Katayama MLH, Vieira RAdC, Andrade VP, Roela RA, Lima LGCA, Kerr LM, Campos APd, Pereira CAdB, Serio PAdMP, Encinas G, et al. Stromal Cell Signature Associated with Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer. Cells. 2019; 8(12):1566. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121566
Chicago/Turabian StyleKatayama, Maria Lucia Hirata, René Aloísio da Costa Vieira, Victor Piana Andrade, Rosimeire Aparecida Roela, Luiz Guilherme Cernaglia Aureliano Lima, Ligia Maria Kerr, Adriano Polpo de Campos, Carlos Alberto de Bragança Pereira, Pedro Adolpho de Menezes Pacheco Serio, Giselly Encinas, and et al. 2019. "Stromal Cell Signature Associated with Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer" Cells 8, no. 12: 1566. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121566