
Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding

 ,
,
Abstract
:1. Introduction
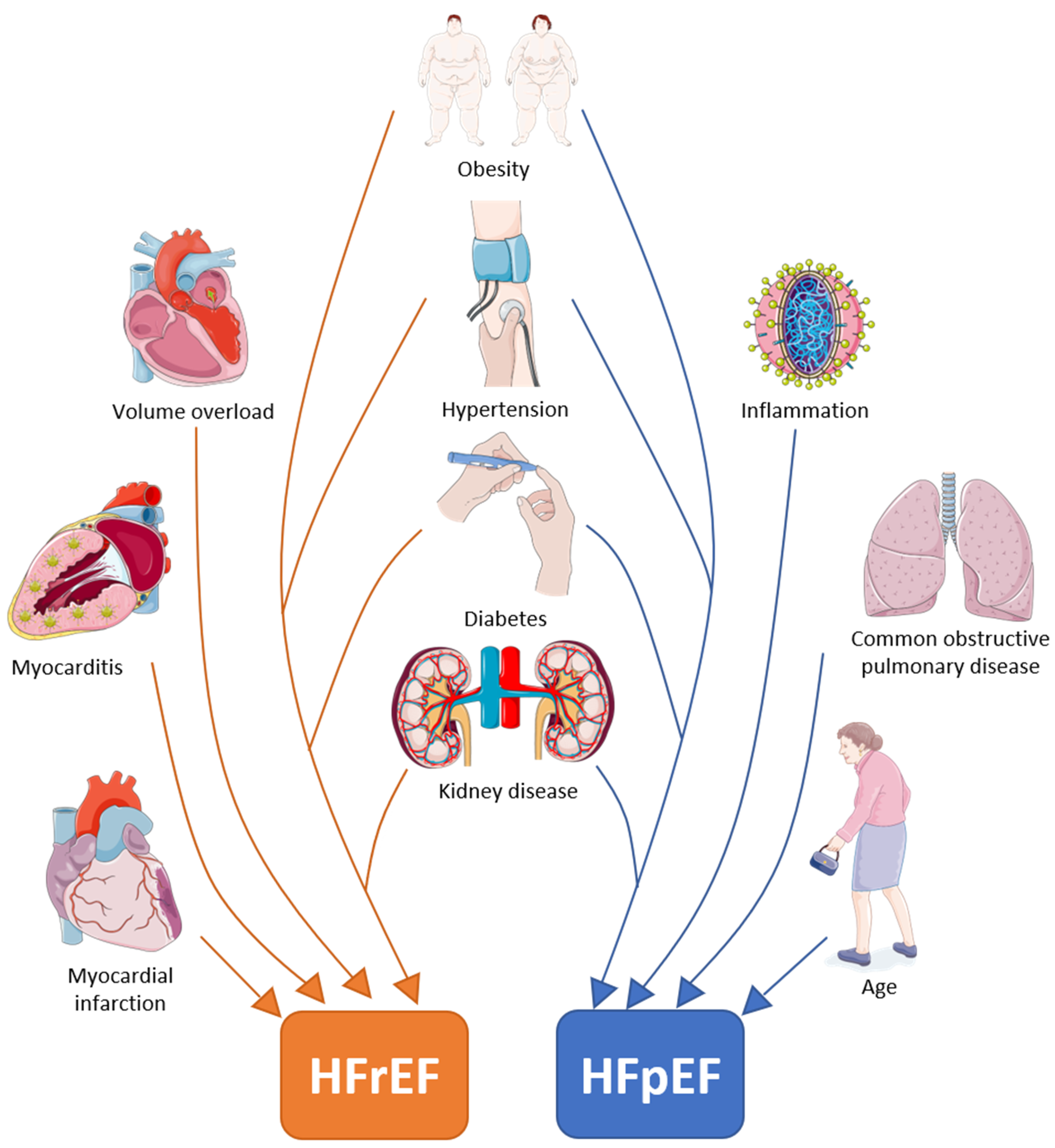
2. Differences in Comorbidities/Risk Factors in HFrEF and HFpEF
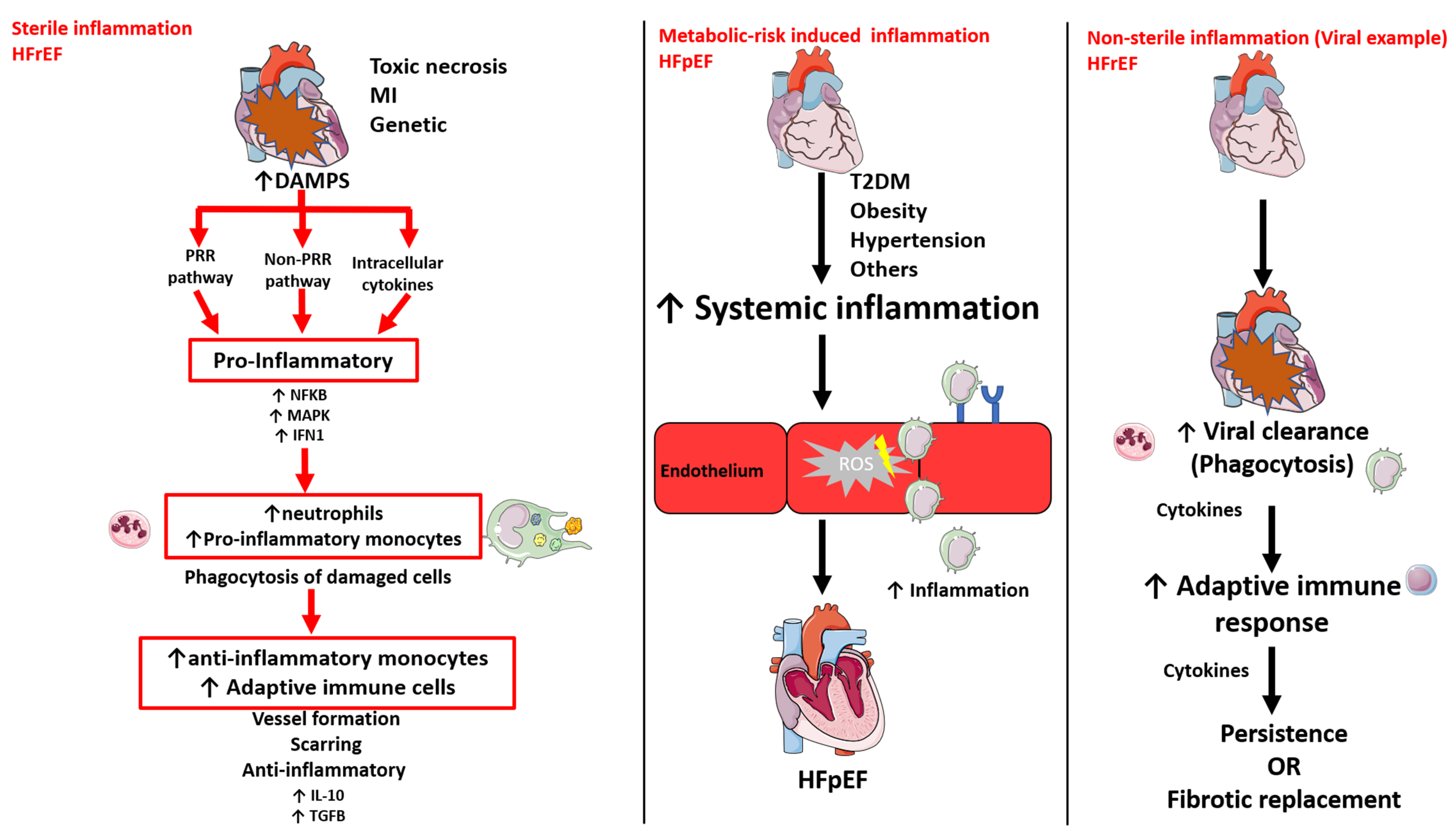
3. Systemic and Cardiac Inflammation has a Differential Role in HFrEF and HFpEF
3.1. Sterile Inflammation
3.2. Metabolic Risk-Induced Inflammation
3.3. Non-sterile inflammation
4. Endothelial Dysfunction: a Main Player in Both HFrEF and HFpEF
5. Cardiomyocyte Alterations in HFrEF and HFpEF: Eccentric Versus Concentric Hypertrophy
6. Cardiomyocyte Cell Death: a Typical Characteristic of HFrEF
7. Different Types of Cardiac Fibrosis in HFrEF and HFpEF
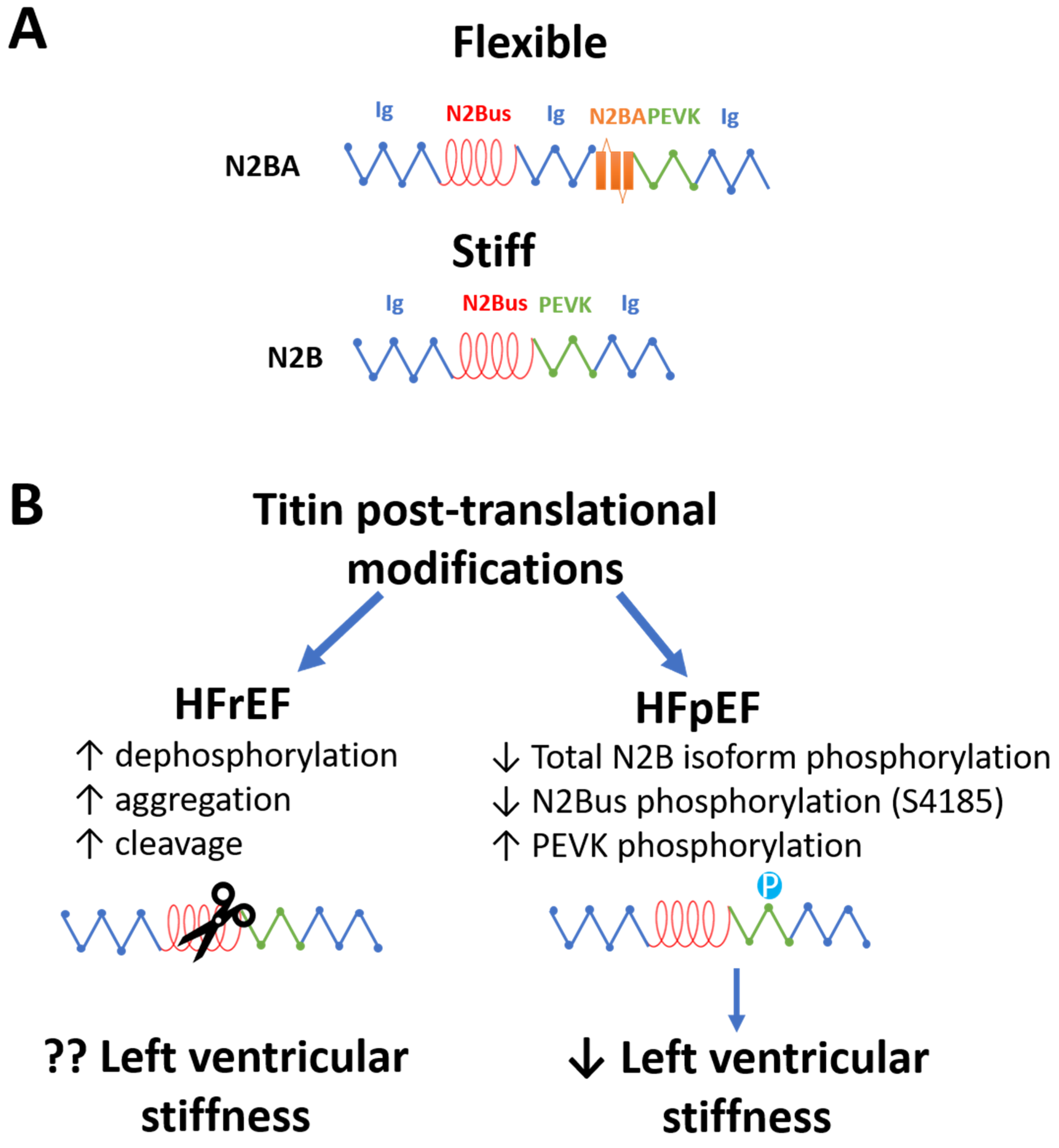
8. Left Ventricular Stiffness in HFrEF and HFpEF: Cardiac Titin and Ca2+ Levels are Differently Affected in HFpEF and HFrEF
8.1. Titin Modifications in HFrEF and HFpEF
8.2. Cardiac Ca2+ Levels in HFpEF and HFrEF
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation end products |
| CAD | coronary artery disease |
| CO | cardiac output |
| CD36 | cluster of differentiation 36 |
| cGMP | cyclic guanosine monophosphate |
| CRP | C-reactive protein |
| DAMPs | damage-associated molecular patterns |
| ECM | extracellular matrix |
| EF | ejection fraction |
| eNOS | endothelial nitric oxide synthase |
| ET-1 | endothelin-1 |
| Fpassive | cardiomyocyte passive stiffness |
| HSCs | hemopoietic stem cells |
| HF | heart failure |
| HFmrEF | heart failure with mid-range EF |
| HFpEF | heart failure with preserved EF |
| HFrEF | heart failure with reduced EF |
| HSPs | heat shock proteins |
| ICAM | intercellular adhesion molecule |
| IL-6 | interleukin-6 |
| IGF-1 | insulin-like growth factor 1 |
| LOX | lysyl-oxidase |
| MAPK | mitogen-activated kinase |
| MMPs | matrix metalloproteinases |
| NFκB | nuclear factor kappa-B |
| N2Bus | titin isoform N2B unique sequence |
| NCX | Na+/Ca2+ exchanger |
| NO | nitric oxide |
| PEVK | titin region rich in proline, glutamate, valine, and lysine |
| PKA | protein kinase A |
| PKG | protein kinase G activity |
| PP1 | protein phosphatase 1 |
| PP2a | protein phosphatases 2a |
| PRR | pathogen recognition receptor |
| IFN | type I interferon |
| RAGE | receptor for advanced glycation end products |
| SERCA2a | Ca2+ ATPase 2a |
| SR | sarcoplasmic reticulum |
| STEMI | ST-segment elevation MI |
| TAC | transverse aortic constriction |
| TIMP-1 | tissue inhibitors of matrix proteinase 1 |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| TGFβ | transforming growth factor β |
| TSP-1 | thrombospondin-1 |
| TNF-α | tumour necrosis factor alpha |
| VM | viral myocarditis |
References
- Ambrosy, A.P.; Fonarow, G.C.; Butler, J.; Chioncel, O.; Greene, S.J.; Vaduganathan, M.; Nodari, S.; Lam, C.S.; Sato, N.; Shah, A.N.; et al. The global health and economic burden of hospitalizations for heart failure: Lessons learned from hospitalized heart failure registries. J. Am. Coll. Cardiol. 2014, 63, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Vedin, O.; Lam, C.S.P.; Koh, A.S.; Benson, L.; Teng, T.H.K.; Tay, W.T.; Braun, O.O.; Savarese, G.; Dahlstrom, U.; Lund, L.H. Significance of Ischemic Heart Disease in Patients With Heart Failure and Preserved, Midrange, and Reduced Ejection Fraction: A Nationwide Cohort Study. Circ. Heart Fail 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.S.; Tay, W.T.; Teng, T.H.K.; Vedin, O.; Benson, L.; Dahlstrom, U.; Savarese, G.; Lam, C.S.P.; Lund, L.H. A comprehensive population-based characterization of heart failure with mid-range ejection fraction. Eur. J. Heart Fail 2017, 19, 1624–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, J.R.; Kapoor, R.; Ju, C.; Heidenreich, P.A.; Eapen, Z.J.; Hernandez, A.F.; Butler, J.; Yancy, C.W.; Fonarow, G.C. Precipitating Clinical Factors, Heart Failure Characterization, and Outcomes in Patients Hospitalized With Heart Failure With Reduced, Borderline, and Preserved Ejection Fraction. JACC Heart Fail 2016, 4, 464–472. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch. Intern. Med. 2001, 161, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Borlaug, B.A.; Melenovsky, V.; Russell, S.D.; Kessler, K.; Pacak, K.; Becker, L.C.; Kass, D.A. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation 2006, 114, 2138–2147. [Google Scholar] [CrossRef] [Green Version]
- Kass, D.A.; Bronzwaer, J.G.; Paulus, W.J. What mechanisms underlie diastolic dysfunction in heart failure? Circ. Res. 2004, 94, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal Trends in the Incidence of and Mortality Associated With Heart Failure With Preserved and Reduced Ejection Fraction. JACC Heart Fail 2018, 6, 678–685. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.M.; Dudley, S.C., Jr. Diastolic dysfunction. Circ. J. 2015, 79, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.E.; Lyass, A.; Lee, D.S.; Vasan, R.S.; Kannel, W.B.; Larson, M.G.; Levy, D. Predictors of new-onset heart failure: Differences in preserved versus reduced ejection fraction. Circ. Heart Fail 2013, 6, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.S.; Gona, P.; Vasan, R.S.; Larson, M.G.; Benjamin, E.J.; Wang, T.J.; Tu, J.V.; Levy, D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: Insights from the framingham heart study of the national heart, lung, and blood institute. Circulation 2009, 119, 3070–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumholz, H.M.; Larson, M.; Levy, D. Sex differences in cardiac adaptation to isolated systolic hypertension. Am. J. Cardiol. 1993, 72, 310–313. [Google Scholar] [CrossRef]
- Ergatoudes, C.; Schaufelberger, M.; Andersson, B.; Pivodic, A.; Dahlstrom, U.; Fu, M. Non-cardiac comorbidities and mortality in patients with heart failure with reduced vs. preserved ejection fraction: A study using the Swedish Heart Failure Registry. Clin. Res. Cardiol. 2019, 108, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ather, S.; Chan, W.; Bozkurt, B.; Aguilar, D.; Ramasubbu, K.; Zachariah, A.A.; Wehrens, X.H.; Deswal, A. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J. Am. Coll. Cardiol. 2012, 59, 998–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felker, G.M.; Shaw, L.K.; Stough, W.G.; O’Connor, C.M. Anemia in patients with heart failure and preserved systolic function. Am. Heart J. 2006, 151, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Thorp, M.L.; Gurwitz, J.H.; McManus, D.D.; Goldberg, R.J.; Allen, L.A.; Hsu, G.; Sung, S.H.; Magid, D.J.; Go, A.S. Chronic kidney disease and outcomes in heart failure with preserved versus reduced ejection fraction: The Cardiovascular Research Network PRESERVE Study. Circ. Cardiovasc. Qual. Outcomes. 2013, 6, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Streng, K.W.; Nauta, J.F.; Hillege, H.L.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; Lang, C.C.; Metra, M.; Ng, L.L.; et al. Non-cardiac comorbidities in heart failure with reduced, mid-range and preserved ejection fraction. Int. J. Cardiol. 2018, 271, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Yajima, T. Viral myocarditis: Potential defense mechanisms within the cardiomyocyte against virus infection. Future Microbiol 2011, 6, 551–566. [Google Scholar] [CrossRef] [Green Version]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis--diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Lim, S.L.; Tay, W.T.; Teng, T.K.; Chandramouli, C.; Ouwerkerk, W.; Wander, G.S.; Sawhney, J.P.S.; Yap, J.; MacDonald, M.R.; et al. Microvascular Disease in Patients With Diabetes With Heart Failure and Reduced Ejection Versus Preserved Ejection Fraction. Diabetes Care 2019, 42, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seferovic, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Janicki, J.S.; Brower, G.L.; Gardner, J.D.; Chancey, A.L.; Stewart, J.A., Jr. The dynamic interaction between matrix metalloproteinase activity and adverse myocardial remodeling. Heart Fail Rev. 2004, 9, 33–42. [Google Scholar] [CrossRef]
- Gonzalez, A.; Ravassa, S.; Beaumont, J.; Lopez, B.; Diez, J. New targets to treat the structural remodeling of the myocardium. J. Am. Coll. Cardiol. 2011, 58, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Gurusamy, N.; Das, D.K. Autophagy, redox signaling, and ventricular remodeling. Antioxid Redox Signal 2009, 11, 1975–1988. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Penn, M.S. The role of leukocyte-generated oxidants in left ventricular remodeling. Am. J. Cardiol. 2008, 101, 30D–33D. [Google Scholar] [CrossRef]
- Zile, M.R.; Gaasch, W.H.; Carroll, J.D.; Feldman, M.D.; Aurigemma, G.P.; Schaer, G.L.; Ghali, J.K.; Liebson, P.R. Heart failure with a normal ejection fraction: Is measurement of diastolic function necessary to make the diagnosis of diastolic heart failure? Circulation 2001, 104, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Gottdiener, J.S.; Hetzel, S.J.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Baicu, C.F.; Massie, B.M.; Carson, P.E.; Investigators, I.P. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation 2011, 124, 2491–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessner, A.; Hohensinner, P.J.; Rychli, K.; Neuhold, S.; Zorn, G.; Richter, B.; Hulsmann, M.; Berger, R.; Mortl, D.; Huber, K.; et al. Prognostic value of apoptosis markers in advanced heart failure patients. Eur. Heart J. 2009, 30, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Huang, Y.; Hunyor, S.; dos Remedios, C.G. Cardiomyocyte apoptosis is associated with increased wall stress in chronic failing left ventricle. Eur. Heart J 2003, 24, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.Y.; Zhu, X.F.; Yang, Y.; Ye, P. High-sensitive cardiac troponin T. J. Geriatr. Cardiol. 2013, 10, 102–109. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc. Res. 2009, 81, 465–473. [Google Scholar] [CrossRef]
- Brouwers, F.P.; de Boer, R.A.; van der Harst, P.; Voors, A.A.; Gansevoort, R.T.; Bakker, S.J.; Hillege, H.L.; van Veldhuisen, D.J.; van Gilst, W.H. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur. Heart J. 2013, 34, 1424–1431. [Google Scholar] [CrossRef]
- Dai, Z.; Aoki, T.; Fukumoto, Y.; Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 2012, 60, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Roy, C.; Slimani, A.; de Meester, C.; Amzulescu, M.; Pasquet, A.; Vancraeynest, D.; Beauloye, C.; Vanoverschelde, J.L.; Gerber, B.L.; Pouleur, A.C. Associations and prognostic significance of diffuse myocardial fibrosis by cardiovascular magnetic resonance in heart failure with preserved ejection fraction. J. Cardiovasc. Magn. Reson. 2018, 20, 55. [Google Scholar] [CrossRef] [Green Version]
- Kasner, M.; Westermann, D.; Lopez, B.; Gaub, R.; Escher, F.; Kuhl, U.; Schultheiss, H.P.; Tschope, C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 2011, 57, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Fukumoto, Y.; Sugimura, K.; Oikawa, M.; Satoh, K.; Nakano, M.; Nakayama, M.; Shimokawa, H. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. -Comparison between preserved and reduced ejection fraction heart failure. Circ J 2011, 75, 2605–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune-stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Bielecka-Dabrowa, A.; Sakowicz, A.; Misztal, M.; von Haehling, S.; Ahmed, A.; Pietrucha, T.; Rysz, J.; Banach, M. Differences in biochemical and genetic biomarkers in patients with heart failure of various etiologies. Int. J. Cardiol. 2016, 221, 1073–1080. [Google Scholar] [CrossRef]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G.; Martos, R.; Baugh, J.A.; Ledwidge, M.T.; McDonald, K.M. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail 2011, 13, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.J.; Yeghiazarians, Y.; Shih, H.; Hwang, J.; Ye, J.; Sievers, R.; Zheng, D.; Palasubramaniam, J.; Palasubramaniam, D.; Karschimkus, C.; et al. Myocardial production and release of MCP-1 and SDF-1 following myocardial infarction: Differences between mice and man. J. Transl. Med. 2011, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Nakamura, K.; Kusano, K.F.; Nakamura, Y.; Ohta-Ogo, K.; Nagase, S.; Sakuragi, S.; Ohe, T. Expression of monocyte chemoattractant protein-1 in idiopathic dilated cardiomyopathy. Int. J. Cardiol. 2008, 126, 427–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broder, M.I.; Cohn, J.N. Evolution of abnormalities in left ventricular function after acute myocardial infarction. Circulation 1972, 46, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Diamond, G.; Forrester, J.S. Effect of coronary artery disease and acute myocardial infarction on left ventricular compliance in man. Circulation 1972, 45, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.O., Jr.; Hunt, D.; Rackley, C.E. Left ventricular hemodynamics in anterior and inferior myocardial infarction. Am. J. Cardiol. 1973, 32, 8–16. [Google Scholar] [CrossRef]
- Aoyagi, T.; Pouleur, H.; Van Eyll, C.; Rousseau, M.F.; Mirsky, I. Wall motion asynchrony is a major determinant of impaired left ventricular filling in patients with healed myocardial infarction. Am. J. Cardiol. 1993, 72, 268–272. [Google Scholar] [CrossRef]
- Smith, M.; Ratshin, R.A.; Harrell, F.E., Jr.; Russell, R.O., Jr.; Rackley, C.E. Early sequential changes in left ventricular dimensions and filling pressure in patients after myocardial infarction. Am. J. Cardiol. 1974, 33, 363–369. [Google Scholar] [CrossRef]
- Bertrand, M.E.; Rousseau, M.F.; Lefebvre, J.M.; Lablanche, J.M.; Asseman, P.H.; Carre, A.G.; Lekieffre, J.P. Left ventricular compliance in acute transmural myocardial infarction in man. Eur. J. Cardiol. 1978, 7, 179–193. [Google Scholar]
- Paulus, W.J.; Dal Canto, E. Distinct Myocardial Targets for Diabetes Therapy in Heart Failure With Preserved or Reduced Ejection Fraction. JACC Heart Fail 2018, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Borbely, A.; Niessen, H.W.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Linke, W.A.; Laarman, G.J.; Paulus, W.J. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 2006, 113, 1966–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, A.T.; Aronsen, J.M.; Skardal, K.; Hamdani, N.; Linke, W.A.; Danielsen, H.E.; Sejersted, O.M.; Sjaastad, I.; Louch, W.E. Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc. Res. 2017, 113, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, C.; Kole, J.; Musters, R.; Hamdani, N.; Paulus, W.J. alpha-B Crystallin Reverses High Diastolic Stiffness of Failing Human Cardiomyocytes. Circ Heart Fail 2017, 10, e003626. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Derks, K.W.J.; Barandiaran Aizpurua, A.; Merken, J.J.; Wang, P.; Bierau, J.; van den Wijngaard, A.; Schalla, S.M.; Abdul Hamid, M.A.; et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur. Heart J. 2018, 39, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Cho, W.J.; Hudson, B.; Kassiri, Z.; Granzier, H.; Schulz, R. Titin is a target of matrix metalloproteinase-2: Implications in myocardial ischemia/reperfusion injury. Circulation 2010, 122, 2039–2047. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Maier, L.S.; Piacentino, V., 3rd; Weisser, J.; Hasenfuss, G.; Houser, S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002, 106, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Limas, C.J.; Olivari, M.T.; Goldenberg, I.F.; Levine, T.B.; Benditt, D.G.; Simon, A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc. Res. 1987, 21, 601–605. [Google Scholar] [CrossRef]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercadier, J.J.; Lompre, A.M.; Duc, P.; Boheler, K.R.; Fraysse, J.B.; Wisnewsky, C.; Allen, P.D.; Komajda, M.; Schwartz, K. Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J. Clin. Investig. 1990, 85, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Schillinger, W.; Pieske, B.; Holubarsch, C.; Heilmann, C.; Posival, H.; Kuwajima, G.; Mikoshiba, K.; Just, H.; Hasenfuss, G.; et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 1995, 92, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Munch, G.; Bolck, B.; Karczewski, P.; Krause, E.G.; Erdmann, E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 1999, 31, 479–491. [Google Scholar] [CrossRef]
- Westermann, D.; Kasner, M.; Steendijk, P.; Spillmann, F.; Riad, A.; Weitmann, K.; Hoffmann, W.; Poller, W.; Pauschinger, M.; Schultheiss, H.P.; et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation 2008, 117, 2051–2060. [Google Scholar] [CrossRef]
- Kruger, M.; Babicz, K.; von Frieling-Salewsky, M.; Linke, W.A. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 910–916. [Google Scholar] [CrossRef]
- Warren, C.M.; Jordan, M.C.; Roos, K.P.; Krzesinski, P.R.; Greaser, M.L. Titin isoform expression in normal and hypertensive myocardium. Cardiovasc. Res. 2003, 59, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Odhiambo, J.F.; Ghnesis, A.; Ford, S.P.; Nathanielsz, P.W.; Ren, J.F.; Guo, W. Maternal Obesity (OB) Increases the Stiffer, Shorter Titin Isoform in the Maternal and Fetal Heart. Reprod. Sci. 2016, 23, 146–147. [Google Scholar]
- Hopf, A.E.; Andresen, C.; Kotter, S.; Isic, M.; Ulrich, K.; Sahin, S.; Bongardt, S.; Roll, W.; Drove, F.; Scheerer, N.; et al. Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1. Circ. Res. 2018, 123, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Borbely, A.; Falcao-Pires, I.; van Heerebeek, L.; Hamdani, N.; Edes, I.; Gavina, C.; Leite-Moreira, A.F.; Bronzwaer, J.G.; Papp, Z.; van der Velden, J.; et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ. Res. 2009, 104, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdani, N.; Franssen, C.; Lourenco, A.; Falcao-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; Lopez, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ. Heart Fail 2013, 6, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Runte, K.E.; Bell, S.P.; Selby, D.E.; Haussler, T.N.; Ashikaga, T.; LeWinter, M.M.; Palmer, B.M.; Meyer, M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium From Patients With Hypertensive Heart Disease and Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Tschope, C. Inflammation - Cause or Consequence of Heart Failure or Both? Curr. Heart Fail Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezbradica, J.S.; Coll, R.C.; Schroder, K. Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell Mol. Immunol. 2017, 14, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M.; Swirski, F.K. Innate immune cells in ischaemic heart disease: Does myocardial infarction beget myocardial infarction? Eur. Heart J. 2016, 37, 868–872. [Google Scholar] [CrossRef]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Nunez, J.; Nunez, E.; Bodi, V.; Sanchis, J.; Minana, G.; Mainar, L.; Santas, E.; Merlos, P.; Rumiz, E.; Darmofal, H.; et al. Usefulness of the neutrophil to lymphocyte ratio in predicting long-term mortality in ST segment elevation myocardial infarction. Am. J. Cardiol. 2008, 101, 747–752. [Google Scholar] [CrossRef]
- Dutta, P.; Nahrendorf, M. Monocytes in myocardial infarction. Arter. Thromb. Vasc. Biol. 2015, 35, 1066–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Laan, A.M.; Nahrendorf, M.; Piek, J.J. Republished: Healing and adverse remodelling after acute myocardial infarction: Role of the cellular immune response. Postgrad Med. J. 2013, 89, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Mann, D.L.; Entman, M.L.; Spinale, F.G. Extracellular matrix remodeling following myocardial injury. Ann. Med. 2003, 35, 316–326. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Tang, T.T.; Yuan, J.; Zhu, Z.F.; Zhang, W.C.; Xiao, H.; Xia, N.; Yan, X.X.; Nie, S.F.; Liu, J.; Zhou, S.F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 232. [Google Scholar] [CrossRef]
- Amin, M.N.; Mosa, A.A.; El-Shishtawy, M.M. Clinical study of advanced glycation end products in egyptian diabetic obese and non-obese patients. Int. J. Biomed Sci. 2011, 7, 191–200. [Google Scholar]
- Beyan, H.; Riese, H.; Hawa, M.I.; Beretta, G.; Davidson, H.W.; Hutton, J.C.; Burger, H.; Schlosser, M.; Snieder, H.; Boehm, B.O.; et al. Glycotoxin and autoantibodies are additive environmentally determined predictors of type 1 diabetes: A twin and population study. Diabetes 2012, 61, 1192–1198. [Google Scholar] [CrossRef] [Green Version]
- Chao, P.C.; Huang, C.N.; Hsu, C.C.; Yin, M.C.; Guo, Y.R. Association of dietary AGEs with circulating AGEs, glycated LDL, IL-1alpha and MCP-1 levels in type 2 diabetic patients. Eur. J. Nutr. 2010, 49, 429–434. [Google Scholar] [CrossRef]
- Miyata, T.; Inagi, R.; Iida, Y.; Sato, M.; Yamada, N.; Oda, O.; Maeda, K.; Seo, H. Involvement of beta 2-microglobulin modified with advanced glycation end products in the pathogenesis of hemodialysis-associated amyloidosis. Induction of human monocyte chemotaxis and macrophage secretion of tumor necrosis factor-alpha and interleukin-1. J. Clin. Investig. 1994, 93, 521–528. [Google Scholar] [CrossRef]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory markers and incident heart failure risk in older adults: The Health ABC (Health, Aging, and Body Composition) study. J. Am. Coll Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuBrock, H.M.; AbouEzzeddine, O.F.; Redfield, M.M. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS ONE 2018, 13, e0201836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card Fail 2015, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Haraldsen, G.; Kvale, D.; Lien, B.; Farstad, I.N.; Brandtzaeg, P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J. Immunol. 1996, 156, 2558–2565. [Google Scholar] [PubMed]
- Bonfanti, R.; Furie, B.C.; Furie, B.; Wagner, D.D. PADGEM (GMP140) is a component of Weibel-Palade bodies of human endothelial cells. Blood 1989, 73, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Obesity and Hypertension: Roles in the Cardiorenal Metabolic Syndrome. Med. Clin. North. Am. 2017, 101, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Pastore, L.; Tessitore, A.; Martinotti, S.; Toniato, E.; Alesse, E.; Bravi, M.C.; Ferri, C.; Desideri, G.; Gulino, A.; Santucci, A. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation 1999, 100, 1646–1652. [Google Scholar] [CrossRef] [Green Version]
- Grafe, M.; Auch-Schwelk, W.; Zakrzewicz, A.; Regitz-Zagrosek, V.; Bartsch, P.; Graf, K.; Loebe, M.; Gaehtgens, P.; Fleck, E. Angiotensin II-induced leukocyte adhesion on human coronary endothelial cells is mediated by E-selectin. Circ. Res. 1997, 81, 804–811. [Google Scholar] [CrossRef]
- Tummala, P.E.; Chen, X.L.; Sundell, C.L.; Laursen, J.B.; Hammes, C.P.; Alexander, R.W.; Harrison, D.G.; Medford, R.M. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation 1999, 100, 1223–1229. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. (Berl) 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Alem, M.M. Endothelial Dysfunction in Chronic Heart Failure: Assessment, Findings, Significance, and Potential Therapeutic Targets. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Rossa, S.; Landmesser, U.; Spiekermann, S.; Engberding, N.; Hornig, B.; Drexler, H. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur. Heart J. 2005, 26, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.D.; Biasucci, L.; Sabba, C.; Strom, J.A.; Jondeau, G.; Galvao, M.; Solomon, S.; Nikolic, S.D.; Forman, R.; LeJemtel, T.H. Impaired endothelium-mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J. Am. Coll. Cardiol. 1992, 19, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Kubo, S.H.; Rector, T.S.; Bank, A.J.; Williams, R.E.; Heifetz, S.M. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation 1991, 84, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Witman, M.A.; Fjeldstad, A.S.; McDaniel, J.; Ives, S.J.; Zhao, J.; Barrett-O’Keefe, Z.; Nativi, J.N.; Stehlik, J.; Wray, D.W.; Richardson, R.S. Vascular function and the role of oxidative stress in heart failure, heart transplant, and beyond. Hypertension 2012, 60, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Treasure, C.B.; Vita, J.A.; Cox, D.A.; Fish, R.D.; Gordon, J.B.; Mudge, G.H.; Colucci, W.S.; Sutton, M.G.; Selwyn, A.P.; Alexander, R.W.; et al. Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation 1990, 81, 772–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351, 2112–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantalis, V.; Schulman, I.H.; Hare, J.M. Nitroso-redox imbalance affects cardiac structure and function. J. Am. Coll. Cardiol. 2013, 61, 933–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Mattei, P.; Sudano, I.; Bernini, G.; Pinto, S.; Salvetti, A. Menopause is associated with endothelial dysfunction in women. Hypertension 1996, 28, 576–582. [Google Scholar] [CrossRef]
- Kawano, H.; Motoyama, T.; Kugiyama, K.; Hirashima, O.; Ohgushi, M.; Yoshimura, M.; Ogawa, H.; Okumura, K.; Yasue, H. Menstrual cyclic variation of endothelium-dependent vasodilation of the brachial artery: Possible role of estrogen and nitric oxide. Proc. Assoc. Am. Physicians 1996, 108, 473–480. [Google Scholar]
- David, F.L.; Carvalho, M.H.; Cobra, A.L.; Nigro, D.; Fortes, Z.B.; Reboucas, N.A.; Tostes, R.C. Ovarian hormones modulate endothelin-1 vascular reactivity and mRNA expression in DOCA-salt hypertensive rats. Hypertension 2001, 38, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Nevzati, E.; Shafighi, M.; Bakhtian, K.D.; Treiber, H.; Fandino, J.; Fathi, A.R. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir. Suppl. 2015, 120, 141–145. [Google Scholar] [CrossRef]
- Pedersen, S.H.; Nielsen, L.B.; Mortensen, A.; Nilas, L.; Ottesen, B. Progestins oppose the effects of estradiol on the endothelin-1 receptor type B in coronary arteries from ovariectomized hyperlipidemic rabbits. Menopause 2008, 15, 503–510. [Google Scholar] [CrossRef]
- Du, X.J.; Riemersma, R.A.; Dart, A.M. Cardiovascular protection by oestrogen is partly mediated through modulation of autonomic nervous function. Cardiovasc. Res. 1995, 30, 161–165. [Google Scholar] [CrossRef]
- Matthews, K.A.; Crawford, S.L.; Chae, C.U.; Everson-Rose, S.A.; Sowers, M.F.; Sternfeld, B.; Sutton-Tyrrell, K. Are changes in cardiovascular disease risk factors in midlife women due to chronological aging or to the menopausal transition? J. Am. Coll. Cardiol. 2009, 54, 2366–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivritas, D.; Becher, M.U.; Ebrahimian, T.; Arfa, O.; Rapp, S.; Bohner, A.; Mueller, C.F.; Umemura, T.; Wassmann, S.; Nickenig, G.; et al. Antiproliferative effect of estrogen in vascular smooth muscle cells is mediated by Kruppel-like factor-4 and manganese superoxide dismutase. Basic Res. Cardiol. 2011, 106, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Gros, R.; Hussain, Y.; Chorazyczewski, J.; Pickering, J.G.; Ding, Q.; Feldman, R.D. Extent of Vascular Remodeling Is Dependent on the Balance Between Estrogen Receptor alpha and G-Protein-Coupled Estrogen Receptor. Hypertension 2016, 68, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.Y.; Kim, M.J.; Jo, H.H.; Hwang, S.J.; Chae, B.; Chung, J.E.; Kwon, D.J.; Lew, Y.O.; Lim, Y.T.; Kim, J.H.; et al. Antioxidant effect of estrogen on bovine aortic endothelial cells. J. Steroid Biochem Mol. Biol. 2009, 117, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Rozentryt, P.; Ponikowska, B.; Hartmann, O.; Kustrzycka-Kratochwil, D.; Reczuch, K.; Nowak, J.; Borodulin-Nadzieja, L.; Polonski, L.; Banasiak, W.; et al. Circulating estradiol and mortality in men with systolic chronic heart failure. JAMA 2009, 301, 1892–1901. [Google Scholar] [CrossRef]
- Xu, Y.; Arenas, I.A.; Armstrong, S.J.; Plahta, W.C.; Xu, H.; Davidge, S.T. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc. Res. 2006, 69, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Alecrin, I.N.; Aldrighi, J.M.; Caldas, M.A.; Gebara, O.C.; Lopes, N.H.; Ramires, J.A. Acute and chronic effects of oestradiol on left ventricular diastolic function in hypertensive postmenopausal women with left ventricular diastolic dysfunction. Heart 2004, 90, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Dhot, J.; Prat, V.; Stevant, D.; Ferron, M.; Aillerie, V.; Erraud, E.; Erfanian, M.; De Waard, M.; Rozec, B.; Trochu, J.; et al. Phytoestrogen: Protective effect in HFpEF through ageing? Arch. Cardiovasc. Dis. Suppl. 2019, 11, 226. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcao-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Faxen, U.L.; Hage, C.; Benson, L.; Zabarovskaja, S.; Andreasson, A.; Donal, E.; Daubert, J.C.; Linde, C.; Brismar, K.; Lund, L.H. HFpEF and HFrEF Display Different Phenotypes as Assessed by IGF-1 and IGFBP-1. J. Card Fail 2017, 23, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M.; Santini, M.P.; Claycomb, W.C.; Ladurner, A.G.; Rosenthal, N. Local IGF-1 isoform protects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging 2009, 2, 43–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klovekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umansky, S.R.; Cuenco, G.M.; Khutzian, S.S.; Barr, P.J.; Tomei, L.D. Post-ischemic apoptotic death of rat neonatal cardiomyocytes. Cell Death Differ. 1995, 2, 235–241. [Google Scholar] [PubMed]
- Cheng, W.; Li, B.; Kajstura, J.; Li, P.; Wolin, M.S.; Sonnenblick, E.H.; Hintze, T.H.; Olivetti, G.; Anversa, P. Stretch-induced programmed myocyte cell death. J. Clin. Investig. 1995, 96, 2247–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.; Friedman, D.B.; Hill, S.; Caprioli, R.M.; Smith, H.M.; Hill, M.F. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. J. Mol. Cell Cardiol. 2007, 42, 884–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Antras, J.; Picatoste, B.; Ramirez, E.; Egido, J.; Tunon, J.; Lorenzo, O. Targeting metabolic disturbance in the diabetic heart. Cardiovasc Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Nelson, M.D.; Szczepaniak, E.W.; Smith, L.; Mehta, P.K.; Thomson, L.E.; Berman, D.S.; Li, D.; Bairey Merz, C.N.; Szczepaniak, L.S. Myocardial steatosis as a possible mechanistic link between diastolic dysfunction and coronary microvascular dysfunction in women. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H14–H19. [Google Scholar] [CrossRef] [Green Version]
- Sparagna, G.C.; Hickson-Bick, D.L.; Buja, L.M.; McMillin, J.B. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 2124–2132. [Google Scholar] [CrossRef]
- Yagyu, H.; Chen, G.; Yokoyama, M.; Hirata, K.; Augustus, A.; Kako, Y.; Seo, T.; Hu, Y.; Lutz, E.P.; Merkel, M.; et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Investig. 2003, 111, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.C.; Kovacs, A.; Blanton, R.M.; Han, X.; Courtois, M.; Weinheimer, C.J.; Yamada, K.A.; Brunet, S.; Xu, H.; Nerbonne, J.M.; et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 2005, 96, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic Res. 2019, 25, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef] [Green Version]
- Baicu, C.F.; Stroud, J.D.; Livesay, V.A.; Hapke, E.; Holder, J.; Spinale, F.G.; Zile, M.R. Changes in extracellular collagen matrix alter myocardial systolic performance. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H122–H132. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J., Jr.; Tryggvason, K.; et al. Cardiomyopathy associated with microcirculation dysfunction in laminin alpha4 chain-deficient mice. J. Biol. Chem. 2006, 281, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Echegaray, K.; Andreu, I.; Lazkano, A.; Villanueva, I.; Saenz, A.; Elizalde, M.R.; Echeverria, T.; Lopez, B.; Garro, A.; Gonzalez, A.; et al. Role of Myocardial Collagen in Severe Aortic Stenosis With Preserved Ejection Fraction and Symptoms of Heart Failure. Rev. Esp. Cardiol. (Engl. Ed.) 2017, 70, 832–840. [Google Scholar] [CrossRef]
- Fowlkes, V.; Clark, J.; Fix, C.; Law, B.A.; Morales, M.O.; Qiao, X.; Ako-Asare, K.; Goldsmith, J.G.; Carver, W.; Murray, D.B.; et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci. 2013, 92, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Willems, I.E.; Havenith, M.G.; De Mey, J.G.; Daemen, M.J. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 1994, 145, 868–875. [Google Scholar] [PubMed]
- Leslie, K.O.; Taatjes, D.J.; Schwarz, J.; vonTurkovich, M.; Low, R.B. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am. J. Pathol. 1991, 139, 207–216. [Google Scholar] [PubMed]
- Szardien, S.; Nef, H.M.; Troidl, C.; Willmer, M.; Voss, S.; Liebetrau, C.; Hoffmann, J.; Rolf, A.; Rixe, J.; Elsasser, A.; et al. Bone marrow-derived cells contribute to cell turnover in aging murine hearts. Int. J. Mol. Med. 2012, 30, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Rifkin, D.B. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: Activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J. Cell Biol. 1989, 109, 309–315. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Involvement of alphavbeta5 integrin-mediated activation of latent transforming growth factor beta1 in autocrine transforming growth factor beta signaling in systemic sclerosis fibroblasts. Arthritis Rheum 2005, 52, 2897–2905. [Google Scholar] [CrossRef]
- Morishita, T.; Uzui, H.; Mitsuke, Y.; Amaya, N.; Kaseno, K.; Ishida, K.; Fukuoka, Y.; Ikeda, H.; Tama, N.; Yamazaki, T.; et al. Association between matrix metalloproteinase-9 and worsening heart failure events in patients with chronic heart failure. ESC Heart Fail 2017, 4, 321–330. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Furuichi, K.; Shimizu, K.; Kokubo, S.; Hara, A.; Yamahana, J.; Okumura, T.; Matsushima, K.; Yokoyama, H.; et al. MCP-1/CCR2-dependent loop for fibrogenesis in human peripheral CD14-positive monocytes. J. Leukoc. Biol. 2006, 79, 555–563. [Google Scholar] [CrossRef]
- Kruglov, E.A.; Nathanson, R.A.; Nguyen, T.; Dranoff, J.A. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am. J. Physiol. Gastrointest Liver Physiol. 2006, 290, G765–G771. [Google Scholar] [CrossRef] [Green Version]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudek, S.B.; Cheng, J.; Du, J.; Wang, Y.; Hermosillo-Rodriguez, J.; Trial, J.; Taffet, G.E.; Entman, M.L. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell Cardiol. 2010, 49, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jugdutt, B.I. Aging and Heart Failure: Mechanisms and Management; Springer Science + Buisness Media: New York, NY, USA, 2014; pp. 360–362. [Google Scholar]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Weber, K.T. Infarct scar: A dynamic tissue. Cardiovasc. Res. 2000, 46, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Little, W.C.; Zile, M.R.; Kitzman, D.W.; Hundley, W.G.; O’Brien, T.X.; Degroof, R.C. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J. Card Fail 2005, 11, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Hartog, J.W.; Willemsen, S.; van Veldhuisen, D.J.; Posma, J.L.; van Wijk, L.M.; Hummel, Y.M.; Hillege, H.L.; Voors, A.A.; investigators, B. Effects of alagebrium, an advanced glycation endproduct breaker, on exercise tolerance and cardiac function in patients with chronic heart failure. Eur. J. Heart Fail 2011, 13, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Tromp, J.; van der Pol, A.; Klip, I.T.; de Boer, R.A.; Jaarsma, T.; van Gilst, W.H.; Voors, A.A.; van Veldhuisen, D.J.; van der Meer, P. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ. Heart Fail 2014, 7, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.D.; Baicu, C.F.; Rentz, T.J.; Van Laer, A.O.; Boggs, J.; Lacy, J.M.; Zile, M.R. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: Role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 2009, 119, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.D.; Baicu, C.F.; Rentz, T.J.; Van Laer, A.O.; Bonnema, D.D.; Zile, M.R. Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: Role of SPARC in post-synthetic procollagen processing. Am. J. Physiol. Heart Circ Physiol. 2010, 298, H614–H622. [Google Scholar] [CrossRef]
- Zuo, C.; Li, X.; Huang, J.; Chen, D.; Ji, K.; Yang, Y.; Xu, T.; Zhu, D.; Yan, C.; Gao, P. Osteoglycin attenuates cardiac fibrosis by suppressing cardiac myofibroblast proliferation and migration through antagonizing lysophosphatidic acid 3/matrix metalloproteinase 2/epidermal growth factor receptor signalling. Cardiovasc. Res. 2018, 114, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, M.F.; Pearson, A.C.; Kern, M.J.; Ratcliff, J.; Mrosek, D.G.; Labovitz, A.J. Left ventricular diastolic function: Comparison of pulsed Doppler echocardiographic and hemodynamic indexes in subjects with and without coronary artery disease. J. Am. Coll. Cardiol. 1989, 13, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Raya, T.E.; Gay, R.G.; Lancaster, L.; Aguirre, M.; Moffett, C.; Goldman, S. Serial changes in left ventricular relaxation and chamber stiffness after large myocardial infarction in rats. Circulation 1988, 77, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Baicu, C.F.; Gaasch, W.H. Diastolic heart failure--abnormalities in active relaxation and passive stiffness of the left ventricle. N. Engl. J. Med. 2004, 350, 1953–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Heerebeek, L.; Paulus, W.J. Understanding heart failure with preserved ejection fraction: Where are we today? Neth. Heart J. 2016, 24, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbely, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Giordano, F.J.; Hilal-Dandan, R.; Choi, D.J.; Rockman, H.A.; McDonough, P.M.; Bluhm, W.F.; Meyer, M.; Sayen, M.R.; Swanson, E.; et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J. Clin. Investig. 1997, 100, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Flesch, M.; Schwinger, R.H.; Schiffer, F.; Frank, K.; Sudkamp, M.; Kuhn-Regnier, F.; Arnold, G.; Bohm, M. Evidence for functional relevance of an enhanced expression of the Na(+)-Ca2+ exchanger in failing human myocardium. Circulation 1996, 94, 992–1002. [Google Scholar] [CrossRef]
- Makino, N.; Panagia, V.; Gupta, M.P.; Dhalla, N.S. Defects in sarcolemmal Ca2+ transport in hearts due to induction of calcium paradox. Circ. Res. 1988, 63, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [Green Version]
- Nuss, H.B.; Houser, S.R. Sodium-calcium exchange-mediated contractions in feline ventricular myocytes. Am. J. Physiol. 1992, 263, H1161–H1169. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HFrEF | HFpEF | |
|---|---|---|
| Comorbidities/risk factors | Obesity Hypertension Diabetes Kidney disease Male Volume overload Myocarditis Myocardial infarction [11] | Obesity Hypertension Diabetes Kidney disease Female COPD Age Anaemia Inflammation Liver disease Sleep apnoea Gout Cancer [10,11,12,14] |
| Systemic and cardiac inflammation | Sterile [19] Non-sterile [20,21] | Metabolic-risk induced [9] |
| Endothelial dysfunction | Late stage symptom [9] ↓ Prevalence [22] ↓ Microvascular density [23,24] ↓ NO bioavailability [25] | Early stage symptom [9] ↑ Prevalence [22] ↓ Microvascular density [23,24] ↓↓ NO bioavailability [25] |
| Cardiac hypertrophy | Eccentric [26,27,28,29,30] | Concentric [31,32] |
| Cardiomyocyte cell death | Present [9,33,34,35,36] | Absent [37] |
| Cardiac fibrosis | ↑ Perivascular fibrosis [38] Interstitial fibrosis [39,40,41] Replacement fibrosis [42] Collagen crosslinking? Fibrotic signalling: ↑ Serum TGFβ1 [43] ↓ MCP-1 [44,45,46] | ↑↑ Perivascular fibrosis [38] Interstitial fibrosis [39,40,41] ↑ Collagen crosslinking [40] Fibrotic signalling: ↑↑ Serum TGFβ1 [43] ↑ MCP-1 [44,45,46] |
| Left ventricular stiffness | Left ventricular stiffness? [47,48,49,50,51,52,53] Titin: Cardiomyocyte Fpassive? [54,55] ↑ N2BA [56] Titin dephosphorylation [57] Titin aggregation [57] Titin cleavage [58,59] Cardiac calcium signalling: ↑ Myocardial [Na+]I [60] ↓ SERCA2a activity [61,62,63,64,65] | ↑ Left ventricular stiffness [66] Titin: ↑ Cardiomyocyte Fpassive [54,55] N2BA → N2B [67,68,69,70,71] N2B hypophosphorylation [69,72,73] N2Bus hyperphosphorylation at S4185 [74] PEVK hyperphosphorylation at S11878 [74] Cardiac calcium signalling: ↑ Myocardial Ca2+ levels [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010242
Simmonds SJ, Cuijpers I, Heymans S, Jones EAV. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells. 2020; 9(1):242. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010242
Chicago/Turabian StyleSimmonds, Steven J., Ilona Cuijpers, Stephane Heymans, and Elizabeth A. V. Jones. 2020. "Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding" Cells 9, no. 1: 242. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9010242