MYC’s Fine Line Between B Cell Development and Malignancy

Lymphocyte Development and Disease Group, Josep Carreras Leukaemia Research Institute, IJC Building, Campus ICO-Germans Trias i Pujol, Ctra de Can Ruti, 08916 Barcelona, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(2), 523; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020523
Submission received: 31 January 2020
/
Revised: 20 February 2020
/
Accepted: 21 February 2020
/
Published: 24 February 2020
(This article belongs to the Special Issue Regulation and Function of the Myc Oncogene)
Abstract
:The transcription factor MYC is transiently expressed during B lymphocyte development, and its correct modulation is essential in defined developmental transitions. Although temporary downregulation of MYC is essential at specific points, basal levels of expression are maintained, and its protein levels are not completely silenced until the B cell becomes fully differentiated into a plasma cell or a memory B cell. MYC has been described as a proto-oncogene that is closely involved in many cancers, including leukemia and lymphoma. Aberrant expression of MYC protein in these hematological malignancies results in an uncontrolled rate of proliferation and, thereby, a blockade of the differentiation process. MYC is not activated by mutations in the coding sequence, and, as reviewed here, its overexpression in leukemia and lymphoma is mainly caused by gene amplification, chromosomal translocations, and aberrant regulation of its transcription. This review provides a thorough overview of the role of MYC in the developmental steps of B cells, and of how it performs its essential function in an oncogenic context, highlighting the importance of appropriate MYC regulation circuitry.
1. The Role of MYC in B Cell Differentiation
Hematopoietic stem cells (HSCs) give rise to mature B cells through the sequential differentiation of lymphoid progenitors. Long-term HSCs (LT-HSCs) have the ability to self-renew and reconstitute the entire immune system by differentiating into short-term HSCs (ST-HSCs). ST-HSCs differentiate into multipotent progenitors (MPPs) that branch later into common myeloid progenitors (CMPs) and lymphoid-primed multipotent progenitors (LMPPs) [1]. LMPPs become common lymphoid progenitors (CLPs) [2], which have the potential to differentiate into B and T lymphocytes, as well as natural killer (NK) cells [2]. Once committed to the lymphoid lineage, additional differentiation steps lead to the formation of pro-B and pre-B cells, which are the early B cell precursors for immature and germinal center (GC) B cells. Bone marrow-escaping mature naïve B cells receiving T cell-dependent signals become activated and localize to the GCs. At this point, they undergo massive proliferation and programmed Ig mutation coupled to antibody affinity-based selection, a process triggered by somatic hypermutation (SHM) and class switch recombination (CSR). Finally, they differentiate into memory B cells or plasma cells (PCs) [3,4] (Figure 1).
The inhibition of erythroid differentiation was the first evidence of MYC activity in vitro, leading to the suggestion that it could have a role in hematopoietic cell development [5,6]. Moreover, the findings that some type of retroviruses expressing MYC provoke the formation of hematopoietic tumors, such as myeloid leukemia [7], and that its expression is deregulated in Burkitt lymphoma [8], reinforced the idea of the potential involvement of MYC in hematopoiesis. In the specific case of B lymphocytes, the use of transgenic mice overexpressing MYC revealed a developmental blockade at the B cell stage, before the onset of lymphoma [9].
Given the importance of MYC deregulation in human leukemia and lymphoma, it is not surprising that its correct modulation is essential throughout the whole B lymphocyte development [10]. At the LT-HSC stage, there is a combined expression of c-MYC and N-MYC isoforms, but there is a complete absence of L-MYC family members [11]. Interestingly, MYC expression allows LT-HSCs and MPPs to be distinguished [10]. On the one hand, LT-HSCs display low levels of MYC to maintain a tight equilibrium between cell self-renewal capacity and differentiation. On the other hand, the activation of MYC expression promotes the differentiation of LT-HSCs into MPPs, which present increased proliferating activity [10,12].
Despite its role in maintaining the self-renewal capacity of LT-HSCs, MYC is also essential for controlling proper hematopoiesis. In fact, Myc-deficient murine embryos exhibit impaired hematopoiesis and die before mid-gestation [13]. At this developmental stage, the role of MYC proteins is hierarchical. N-MYC and L-MYC cannot be expressed alone and require the concomitant expression of c-MYC. For instance, the single deletion of N-MYC does not affect the quiescent state of HSCs or hematopoiesis, whereas the deletion of c-MYC in HSCs alters proliferation and survival [11]. In summary, c-MYC is essential for balancing self-renewal and differentiation at the HSC stage. Sustained expression of MYC encompasses the transition from HSCs to lymphoid-committed cells since its extensive ability to bind to promoter and enhancer regions endows it with an extensive gene transcription role in both developmental stages [10,14].
N-MYC and c-MYC are both expressed in lymphocyte progenitors, meanwhile only the expression of c-MYC is maintained during the rest of the differentiation process, despite being reduced in precursor and mature B cell stages [15]. MYC expression is induced in pre-B cells in response to B-cell receptor (BCR) stimulation [16,17]. MYC expression peaks coincide with the stages of higher proliferative rates in B lymphocyte generation [18]. In consequence, MYC has an essential role in the expansion of pro-B cells and differentiation to the pre-B stage [10]. Conditional knockout of c-MYC or N-MYC using the Cd19-Cre transgenic mouse model blocks the transition from pro-B to pre-B cells, confirming its role at this stage of lineage development [19].
In connection with these data, aberrant expression of MYC in transgenic mice results in a reduction of mature B lymphocyte numbers relative to those of pre-B cells [9]. In a similar way, the regulation of MYC expression may be altered by the presence of the antiapoptotic factor BCL2 [20], or the stimulation with cytokines, such as interleukin 7 (IL-7) [21], resulting in a tumorigenic outcome, given the ability of these two proteins to enhance cell survival. Conversely, Myc-null B lymphocytes have an impaired proliferation capacity when treated with stimulatory cytokines, such as the B-cell surface antigen CD40 and IL-4 [22].
During the complex program that naïve B cells undergo in the GC before they differentiate to memory B cells or PCs, the expression of MYC is maintained, though being depleted when the B cell exits de GC reaction [15,21]. In this context, MYC is basically restricted to specific phases of the GC reaction development and is mainly expressed during naïve B cell expansion and at stages preceding the light zone (LZ) to dark zone (DZ) transition [23,24].
B-cell lymphoma 6 (BCL6) is a direct repressor of MYC during the GC reaction [23]. BCL6 binds to the promoter region of MYC in pre-B and differentiated B cells [25,26,27]. Therefore, the expression pattern of both factors is mutually exclusive in most GC B cells, with 91% of those cells expressing either BCL6 or MYC, and only 8% showing co-expression of both proteins [23]. In GCs, when B cells interact with antigens and access T-helper (Th) cells, they transiently express MYC due to the transcriptional inhibition of BCL6 by the repressive machinery comprising BCR, IL-2, and interferon regulatory factor 4 (IRF4), the latter being induced upon CD40 activation [24,28,29]. In the LZ, the BCR also synergizes with CD40 to activate MYC and induce p-S6, allowing cell-cycle entry [30,31].
In these early stages of GC formation, MYC-expressing B cells express cyclin D2 (CCND2) [32,33] and D3 (CCND3) [34,35], which possibly contributes to their hyperproliferative phenotype during the initial rounds of cell division that give rise to the bulk of the GC B cells [36]. As described by Victora et al., B cell clonal expansion is restricted to the DZ, and cells move to the LZ in a bi-directional process controlled by T cells. Based on the amount of Ag captured, Th cells at the LZ determine whether MYC+ B cells re-enter the DZ for additional rounds of positive selection, or if they remain in the LZ [37].
MYC+ B cells at the LZ subsequently undergo transcription, whereby BCL6 binds the transcription factor (TF) MYC-interacting zing-finger protein 1 (MIZ1) [38], an MYC partner that acts to suppress CDK inhibitor p21 and thereby induce cell-cycle entry. At this stage, BCL6 and MYC are co-expressed in the LZ [23]. BCL6 also inhibits CCND2 expression [32,33], which is an MYC target. CCND3, which is not controlled by MYC [34,35], is expressed alone in these LZ GC B cells. The TF TCF3 (also called E2A) is intrinsically regulated by the induction of its own inhibitor ID3 (inhibitor of DNA binding 3), is expressed in the GC B cells, and activates CCND3 and E2F2, replacing CCND2-dependent proliferation in the LZ MYC+ B cells [27,39].
Logically, MYC expression must be tightly controlled in the DZ to limit cell divisions before each round of antigen affinity-based selection, as MYC controls the transcriptional pause release of RNA polymerase II, which is essential for activation-induced cytidine deaminase (AID)-induced somatic hypermutation (SHM) [40,41]. After several rounds of positive selection, the MYC- B cells finally exit the GC and become either B memory cells or plasmablasts. B-lymphocyte-induced maturation protein 1 (BLIMP1) suppresses MYC expression in plasmablasts and induces PC differentiation [42]. This dependency effect between MYC and B cell proliferation is known as “cyclic re-entry” [23]. A schematic summary of the role of MYC in B lymphocyte differentiation is shown in Figure 1.
2. MYC Role in Leukemogenesis
Unlike other proto-oncogenes, MYC is not activated by oncogenic mutations in the coding sequence. MYC transforms cells via aberrant overexpression of intact MYC protein by three main mechanisms: gene amplification, chromosomal translocation, and aberrant regulation of its expression. In the following sections, we describe the role of MYC in several types of leukemia.
2.1. B lymphoblastic Leukemia with t(9;22) BCR-ABL1 Rearrangement
The B-cell receptor – ABL proto-oncogene 1 (BCR-ABL1) fusion (a translocation widely known as the Philadelphia chromosome, Ph) protein product can activate Myc in bone marrow-derived murine pre-B cells [43]. The activation of MYC, combined with other oncoproteins, such as RAS, c-RAF, and c-JUN, promotes the activation of signaling pathways, leading to malignant cell transformation [44]. Remarkably, the repression of MYC impairs BCR-ABL1-mediated transformation, indicating that MYC not only has a complementary function but also is essential for ensuring leukemic transformation [43,45].
Whereas the activation of MYC in lymphomas is partially caused by an elevated mutation frequency in several cases, B-cell precursor leukemia has an almost negligible mutation rate [46]. However, BCR-ABL rearranged pre-B-acute lymphoblastic leukemia (ALL) is driven by an aberrant expression of AID [47], which is expressed at such an early stage of B lymphocyte development [48], as a consequence of the enhanced kinase activity of BCR-ABL1 fusion protein (i.e., tyrosine kinase P210) [47,49]. Nevertheless, the proportion of patients harboring mutations at the MYC gene itself among Ph+ ALL cases remains low and stable compared with that of Ph- patients [47].
In line with these data, MYC-IGH translocation, which is a common alteration in B-cell lymphomas [50], is not frequently present in the B-cell precursor ALL. However, when analyzing the genetic deletion of CDKN2, a common B-ALL feature, it was found that patients with the wild-type CDKN2 experienced a higher rate of MYC-IGH translocation [51], suggesting that the two genetic alterations may be mutually exclusive.
MYC is induced through different pathways triggered by the BCR-ABL1 fusion protein. For instance, the MYC gene is one of the pre-BCR downstream effectors whose signaling is transduced through spleen tyrosine kinase (SYK) [52,53]. The inhibition of SYK impairs cell viability via the repressed transcription of MYC oncogene [53]. In parallel, the pro-inflammatory marker sphingosine kinase 2 (SK2) promotes the activation of MYC in murine models of B-ALL by increasing its acetylation profile. The inhibition of SK2 provokes a drastic reduction in ALL cell proliferation through concomitant repression of MYC target genes [54]. Recently, the use of purinostat mesylate (a first-in-class histone deacetylase (HDAC) inhibitor with reported antitumor activity [55]) has also been shown to downregulate the BCR-ABL1 fusion protein targeting of MYC through the alteration of global histone 3 (H3) and histone 4 (H4) acetylation [56]. These studies reveal chromatin remodeling to be a promising therapeutic strategy in BCR-ABL1+ ALL. For instance, as described in greater detail below, the combination of HDAC and PI3K inhibition impairs MYC-dependent growth in hematological malignancies [57].
The Wnt signaling cascade is a well-characterized oncogenic pathway that can drive MYC oncogene activation. The BCR-ABL1 protein phosphorylates specific tyrosine residues of γ-catenin, thereby enhancing the direct binding of this effector to the MYC promoter [58]. The role of this kinase activity differs from that in HSCs, where BCR-ABL1 phosphorylates β-catenin, giving rise to initial forms of chronic myeloid leukemia (CML), without requiring MYC induction [58,59]. Instead, BCR-ABL1-driven activation of the JAK/STAT pathway through the phosphorylation of JAK2 has similar effects on both chronic myeloid leukemia (CML) and ALL, whereby pJAK2 and pSTAT5 cooperate to maintain elevated levels of MYC by protecting it from ubiquitin-dependent degradation [60,61].
MYC is also regulated at several post-transcriptional levels in Ph+ B-ALL. Since the highly structured 5’-UTR of MYC determines its translation rate, eIFs are key translation factors that enable MYC mRNA translation [62]. IgM signaling, which is active in chronic lymphoid leukemia (CLL) cells, promotes increased translation of MYC mRNA, together with the induction of eIF4 and eIF4GI [63,64]. eIF4 and MYC participate in a feedforward loop that enhances both activities [65]. This is not the only mechanism through which MYC reinforces its own expression. For instance, MYC, in cooperation with its TF partner MAX, binds to the promoter of BCR-ABL1, activating its transcription [66].
The aberrantly activated function of MYC in ALL also depends on protein stabilization. Some of the first evidence demonstrated that the induction of the Ras pathway prevents proteasomal-mediated degradation of MYC [67,68]. Moreover, most leukemia cell lines harbor an altered MYC form with a prolonged half-life, without possessing genetic mutations or chromosomal alterations [47,69]. The increased stability of MYC is explained by an excess of phosphorylation at Ser62, combined with low levels of pThr58, which promote glycogen synthase kinase 3 beta GSK3β-mediated ubiquitylation and proteasomal degradation [69,70].
Apart from its main function in driving tumor progression, MYC also induces apoptosis, since it targets some genes involved in the BCL2 network [71,72]. As part of this network, the apoptosis-inducer protein BIM acts as a major antagonist of BCL2. In fact, the Eµ-myc murine leukemia model has demonstrated that the deletion of Bim counteracts the potential induction of cell death by MYC, worsening the B-cell leukemia-associated prognosis of these mice [73]. The regulation of BIM is partially mediated by the miR-17-92 cluster (also known as MIR17HG), in MYC-driven leukemia [74,75] and the inhibition of this specific microRNA endows leukemic cells with a pro-apoptotic phenotype [75], making microRNA networks an alternative entry point for interfering with MYC function in the B-cell precursor ALL. The regulation of MYC in BCR-ABL1-rearranged leukemia is depicted in Figure 2.
2.2. B lymphoblastic Leukemia with the t(v;11) MLL Rearrangement
Translocations in the histone methyltransferase MLL gene are the most common chromosomal alteration in infant leukemia and, in general, exhibit a very poor prognosis, such that the disease is an extremely lethal malignancy in infants [76,77,78]. As in other types of B-cell leukemia, the MYC gene is not commonly involved in chromosomal translocations, although rare individual cases with t(8;22) have been reported [77]. Recently, a revised characterization of the RS4;11 leukemic cell line has demonstrated the presence of i(8q), resulting in MYC duplication, which confers a selective growth advantage in vitro [79]. In B-ALL, this alteration is considered a secondary hit that contributes to disease progression. Patients with the MLL-AF4 fusion protein have a strongly enriched MYC gene signature compared with AML patients [80,81].
Consequently, MLL-fusion proteins activate the expression of the MYC oncogene in pre-B and pro-B-cell leukemia [82,83]. For instance, MLL protein can prompt additional activity by fusing to USP2 deubiquitinating protein, leading to the enrichment of USP2 activity on MDM2, ending with the enhanced degradation of p53, which, in turn, activates MYC expression [84,85].
The binding of MLL-rearranged proteins to the regulatory regions of target genes depends on the presence of chromatin adaptors that comprise the super-elongation complex (SEC). The bromodomain and extra-terminal domain (BET) family of proteins (BRD2/3/4) are part of this complex and contribute to the induction of MYC [86,87,88]. Initial evidence showed that suppression of BRD4 induces potent cell growth arrest and cell senescence, combined with MYC downregulation, meaning that the BET family is a promising therapeutic target [86,87,88,89].
For instance, a small molecule inhibitor of BET (iBET-151) prevents the recruitment of BET proteins to chromatin by inhibiting the transcription of key targets, such as BCL2 and MYC [87]. In parallel, JQ1 (a potent inhibitor of BRD4) greatly reduces MYC expression and activity, jointly with a large set of its target genes [88]. Moreover, JQ1 has also been tested in patient-derived xenograft from ALL patients, confirming its ability to inhibit MYC expression [90]. BET proteins are involved in maintaining aberrantly altered chromatin states in ALL. At this point, BRD4 cooperates with MYC in recruiting TEFb complexes to initiate transcriptional elongation at active promoters, while the transcriptional regulator HEXIM1 counteracts BRD4 and MYC role by inactivating the complex. Therefore, tight regulation of BRD4, MYC, and HEXIM1 is required for proper elongation [40,91,92]. A novel oral BRD2/3/4 inhibitor (OTX015) has been shown to reduce MYC expression and to increase HEXIM1 levels in MLL-rearranged leukemia [89]. Apart from OTX015, the specific BRD4 inhibitor CPI-0610 has been selected for phase I clinical trials in ALL patients [93].
Histone deacetylases (HDACs) have a differential expression pattern in ALL patients with MLL rearrangement and are commonly overexpressed. For instance, HDAC9 is associated with an adverse prognosis, whereas SIRT1 is involved in drug resistance through its regulation of the acetylation of the TP53, MYC, and NF-κβ genes. Consequently, HDAC inhibitors (HDACis) have emerged as potential therapeutic options in treating hematological malignancies [94,95]. However, conceptualizing the role of HDACs in leukemia as inducers of malignant transformation would be a too simplistic view, given that some histone deacetylases, such as HDAC7, carry out an opposite function. As reported by Barneda-Zahonero et al., HDAC7 is involved in the repressive transcriptional machinery of MYC and, therefore, it is often reduced in different types of leukemia and lymphoma, including MLL-rearranged malignancies [96].
In this sense, newly developed compounds that selectively inhibit specific HDAC subtypes are gaining relevance in the treatment of hematological malignancies [97]. For instance, class I/IIb-selective HDACi purinostat has demonstrated a direct effect on MYC downregulation [56], while other selective drugs (mocetinostat, entinostat) are already undergoing clinical trials for diverse hematological malignancies [97]. The use of combinatorial therapies merging selective HDACi and classical treatments emerges as a promising therapeutic option, and it is tempting to speculate that its ability to induce apoptosis resides, at least partially, in MYC negative regulation [97].
The relevance of the MYC oncogene in hematopoiesis is restricted to its functions in aberrantly proliferating B-cell precursors and in the normal hematopoietic stem cell hierarchy. The expression of MYC throughout this process is controlled by a super-enhancer region located 1.7 Mb downstream of the gene [98]. This super-enhancer, known as the “blood enhancer cluster” (BENC), is comprised of several selectively active modules that recruit a wide range of transcription factors due to the increased chromatin accessibility. This differential access to regulatory regions has also been reported in murine models of MLL-AF9-driven leukemia, indicating that MYC hyperactivation during leukemia can be driven by BENC-unbalanced modulation [99,100]. BENC deletion entails a drastic depletion of B lymphocytes during normal development, as well as an improved prognosis in MLL-AF9+ leukemia [100]. Regarding the regulation of MYC at the promoter level, we strongly consider that recently developed techniques for 3D chromatin architecture analysis will improve our knowledge about the coordination of transcriptional machinery, chromatin accessibility, and 3D structure. Not in vain, this novel methodology has already conferred a new dimension to the study of B cell development at different stages [101].
2.3. B Lymphoblastic Leukemia with the t(12;21) ETV6/RUNX1 Rearrangement
The t(12;21) translocation, which involves the ETV6 and RUNX1 (also known as AML1) genes, is the most frequent lesion in childhood B-ALL (20–30% of cases), at early diagnosis and remission [102,103]. The N-terminal region of ETV6 displays weak homology with the bHLH region of MYC protein [104]. This homology enables the induction of the targets of these factors through protein-protein interaction, enhancing MYC oncogenic function [103]. Apart from its characteristic fusion to RUNX1 protein, ETV6 also forms fusion proteins with PAX5, which is a key inducer of B-cell-specific genes (such as CD19 and CD79A) [104,105]. The combination of PAX5 activity with ETV6-mediated MYC targets induction establishes the ETV6/PAX5 fusion protein as a powerful mediator of ALL progression.
Alterations affecting the MYC gene itself should be highlighted as examples of chromosomal aberrations. For instance, a double MYC gene translocation t(8;14)t(8;9) was reported in a B-ALL patient with ETV6 amplification [106]. Copy number variation (CNV) was reported in a substantial 65% of relapsing ETV6/RUNX1-positive ALL patients, including MYC expression gain at chromosome 8 (q23.1-24.1) in 10% of cases [107].
An indirect pathway of MYC activation in ETV6/RUNX1-rearranged leukemia is mediated by the GTP-binding protein RAC1, a pivotal modulator of hematopoiesis [108], that increases the phosphorylation levels of STAT3 [109]. ETV6/RUNX1 protein enhances the activity of RAC1, increasing MYC expression, induced by the phosphorylation of STAT3 [110]. Specific STAT3 inhibitors revert MYC induction by blocking cell proliferation and promoting apoptosis in pro-B-ALL cells [110].
Additionally, the ETV6/RUNX1 fusion gene can be stabilized at the mRNA level by the RNA-binding protein IGF2BP1, which is overexpressed in this type of leukemia [111]. IGF2BP1 leads to an eventual increase of MYC, linked to aberrant leukemogenesis in ETV6/RUNX1-mediated ALL [111,112]. Finally, and similarly to the mechanism reported for BCR-ABL1-rearranged leukemia, MYC is also stabilized at the protein level through aberrantly altered phosphorylation at the Thr58 and Ser62 residues [69].
2.4. B Lymphoblastic Leukemia with other Chromosomal Rearrangements
Expression of B220 and CD43 determines the transition of pro-B into pre-B lymphocytes [113]. Leukemia derived from this developmental stage usually displays TCF3/PBX1 chromosomal rearrangement, which is commonly found in leukemia derived from pre-B lymphocytes (in more than 90% of cases) [114]. Survival of TCF3/PBX1+ cells critically depends on the activity of the pre-BCR [52,53]. Immunoglobulin µ (Igµ) heavy-chain knockdown impairs the proper assembly of pre-BCR and blocks signal transduction through the Igα-Igβ heterodimer [115]. Igµ downregulation in TCF3/PBX1-rearranged cell lines significantly suppresses MYC expression at the mRNA and protein levels. MYC is regulated by the pre-BCR in a FOXO-dependent manner since the forced expression of a constitutive form of FOXO1 reverts the blockade of pre-BCR signaling, and partially restores MYC expression [53].
Under physiological conditions, MYC mRNA is modulated by miR-24, which is able to bind at its 3′-UTR region to reduce MYC levels, thereby controlling cell-cycle progression [116]. miR-24 is frequently downregulated in TCF3/PBX1+ pre-B-ALL, concomitantly with other miRNAs involved in proliferation and apoptosis modulation in various cancers (e.g., miR-126 and miR-365) [117]. Surprisingly, the restoration of miR-24 expression in TCF3-rearranged leukemic cell lines neither affects the expression of some of its targets nor alters the frequency of apoptotic cells, suggesting that MYC is regulated by a combination of mechanisms in this type of leukemia [117].
Despite not being the most frequent alteration, the IGH gene (located at chromosome 5) can also be translocated to chromosomes 14 or 12, as is the case for the NALM-6 cell line, which harbors the t(5;12) translocation. This cell line was recently used to identify the transcriptional cofactor apoptosis antagonizing transcription factor (AATF) as being a direct target of MYC since it features canonical binding motifs at the promoter region [118]. AATF promotes cell-cycle progression by inhibiting TP53 expression and mediating the response to DNA damage [119]. It is of particular note that, when inhibiting the expression of MYC, there is a drastic downregulation of MLL gene expression, a previously described key mediator of pediatric leukemia. This downregulation can be counteracted by the exogenous induction of AATF. Therefore, AATF mediates a positive feedback loop between MYC and MLL gene in pro-B-ALL [118].
3. MYC Role in Lymphomagenesis
In hematopoietic malignancies, genomic abnormalities involving the MYC gene are almost always found in B cell lymphomas, but rarely in T cell lymphomas. 30% of all lymphoid neoplasms are B cell non-Hodgkin lymphomas. These can be classified further as Burkitt lymphoma (BL), diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), and plasmablastic lymphoma (PBL), among others.
3.1. MYC in Burkitt Lymphomas
BL arises mostly in children and young adults and has an extremely high proliferation rate. Endemic, sporadic, and immunodeficiency-associated BLs are distinguished as clinical variants in the World Health Organization (WHO) classification. BL has a mature B cell phenotype with expression of GC/post-GC markers such as immunoglobulin M (IgM), CD10, and is typically negative for BCL2 [120].
The genetic hallmark that characterizes BL is the rearrangement of MYC with one of the IG gene loci. Translocation triggers constitutive MYC hypermutation of the translocated gene in germinal centers [121], subjected to AID-dependent SHM [41], which is susceptible to generating MYC variants and increasing its oncogenic potential [122]. Specifically, an MYC translocation to the IG heavy chain gene locus 14q32 (80% of the cases), or to the IGκ or IGλ light chain genes at 2p12 or 22q11 (10%) are the main rearrangement sites [123,124]. Most mutations of the rearranged MYC gene are point SNPs or deletions in the 3’ border of the first exon and the first intron [125,126], altering the coding sequence but permitting its transcription from the translocated chromosome. In consequence, there is an MYC expression that terminates inhibiting cell differentiation and inducing proliferation, probably keeping the cells in a hyperproliferative state.
In a gene expression profile study of human samples of BL (and DLBCL), three main cytogenetic groups within the mature aggressive B cell lymphomas were distinguished: MYC-simple, with IG-MYC fusions and a low chromosomal complexity score, no IGH-BCL2 fusions, and no BCL6 breakpoints, and with a favorable prognosis; MYC-complex, including IG/MYC-rearranged BLs with highly complex karyotypes, non-IG/MYC-rearranged cases, and all IGH/BCL2 fusions and/or BCL6 breakpoints, or any combination of these; and MYC-negative, comprising lymphomas with unaltered MYC [127].
Inhibitor of DNA binding (ID) proteins, such as ID3, bind E-proteins such as TCF3 via HLH common motifs, preventing the binding of the latter to DNA. Schmitz et al. shed light on some oncogenic pathways, suggesting that MYC translocation is insufficient to induce BL [128]. The next-generation sequencing (NGS) study performed by Love et al. identified MYC and ID3 as the genes most frequently mutated in BL [128,129].
In the setting of deregulated MYC, samples with ID3 mutations show a higher level of expression of known MYC target genes, and give rise to increased G1-to-S-phase cell-cycle progression in BL, suggesting a role for ID3 as a tumor suppressor in this type of lymphoma [129]. The high level of ID3 expression in BL might be because the ID3 locus itself is a direct target of MYC [130] and due to BCR triggering, as described using an Id3-/- mouse model [131]. Functional analyses suggested that ID3-inactivating mutations and TCF3-activating mutations (by blocking ID3 binding sites) lead to the activation of a TCF3-dependent transcriptional program that consequently promotes tonic BCR signaling [132]. TCF3 would repress PTPN6, which encodes SHP-1, a BCR-attenuating factor that acts by dephosphorylating the ITAM motifs of the CD79A and CD79B signaling subunits of the BCR [39]. In addition, BCR can activate both MYC and ID3 by a sequential process in which MYC rapidly upregulates its expression. Later, upon MYC downregulation, levels of ID3 increase [131]. This effect may be produced by direct BCR activation, or through an indirect effect of MYC, highlighting the existence of a positive feedback loop between BCR, ID3, and MYC regulation.
Tonic BCR activation requires PI3K signaling in mature B cells to maintain its continuity [132], and the pro-survival pathway cooperates with MYC in BL [133]. MYC deregulation induces the expression of the MIR17HG, a microRNA host gene amplified in ~10% of BL cases [134]. Particularly, miR-19 is the key oncogenic component of the cluster, which antagonizes PTEN and, consequently, activates the AKT-mTOR pathway, the consequence of which is exacerbated cell survival in MYC-driven lymphomagenesis [128,135,136]. See [137] for an extensive review of the involvement of MYC and miRNAs in lymphomagenesis.
BCR-induced PI3K pathway activation in BL contrasts with the absence of NF-κβ survival pathway signaling in these tumors [138]. Reinforcing this, the study by Klapproth et al. in Myc transgenic mice showed that constitutive NF-κβ activity is incompatible with the development of the MYC-induced lymphomas [139]. The resting state of the NF-κβ apoptotic pathway confers a selective advantage on MYC-driven oncogenic cells.
3.2. MYC in Diffuse Large B Cell Lymphoma
DLBCL accounts for approximately 40% of all non-Hodgkin lymphomas [120]. Two major subtypes can be identified: germinal center B-cell-like (GCB), which has a gene expression profile similar to that of the GC B cell; and activated B cell-like (ABC), which has a worse outcome because it expresses genes present in activated peripheral B cells [120]. The principal translocated sites in DLBCL are a rearrangement of BCL6 (30% of cases) and t(14;18)(q32;q21) with BCL2 rearrangement to the IGH gene locus (20–30% of cases) [140,141]. Globally BCL2 is rearranged in 30% of cases in the GCB-DLBCL subgroup and in <5% of ABC-DLBCL cases [140,142,143].
Considering MYC specifically, its protein expression is detected in ~40% of diagnoses, but its rearrangement is found in only around 10% of them, suggesting that alternative mechanisms may be associated with MYC deregulation [143,144,145,146]. With regard to translocations, similar to what is observed in BLs, the IG genes are the most frequent MYC partners, the latter being most commonly fused to IGH or to non-IG genes such as BCL6, BCL2, PAX5, or IKAROS, which appear as translocation partners in 35–50% of MYC-rearranged DLBCLs [145,146,147]. In addition to MYC translocations, DLBCLs are characterized by the presence of MYC amplification and gains, and increased copy numbers of MYC are associated with higher levels of mRNA and protein, resulting in a very poor prognosis [148,149]. However, careful examination of Cosmic, a public catalog of somatic mutations in cancer, revealed that SNPs in MYC sequence could be detected in a significant fraction of DLBCLs, as reported in BL. According to this data and as described before, polymorphisms in MYC sequence do not impair its transcription, permitting in consequence, a dysregulated gene expression that could maintain the cell in a hyperproliferative state that, in the end, will endow cells with increased aggressiveness.
MYC rearrangements are often involved in complex karyotypes and are frequently associated with other oncogenic abnormalities. Lymphomas that carry MYC and either a BCL2 or a BCL6 translocation (a double-hit lymphoma, DHL) or all three rearrangements (a triple-hit lymphoma, THL) are included in the current WHO classification as a new entity termed “High-grade B cell lymphomas with MYC and BCL2 and BCL6 rearrangements” [150]. Molecularly, DHL with MYC and BCL2 rearrangements present a TP53 mutation, inhibiting TP53-mediated apoptosis at a higher frequency than DHL, including MYC and BCL6 alterations, which account for 35% and 6% of cases, respectively [151,152]. Therefore, in the first group, upregulated MYC expression promotes proliferation and disables the capacity to induce apoptosis, while BCL2 expression fosters cell survival. Together their co-expression confers an aggressive proliferating phenotype on these DHLs.
MYC overexpression is a reliable biomarker for predicting therapeutic response, since its expression is a poor prognostic factor in DLBCL [153] but, beyond the aforementioned rearrangements, the mechanisms underlying its overexpression are still unknown. The stability of MYC is regulated by GSK-3β, which phosphorylates MYC at Thr58 and induces its degradation via the ubiquitin-proteasome pathway [154]. Wang et al. demonstrated that BCR stimulation could activate downstream PI3K signaling, phosphorylating GSK-3β at Ser9, and abolishing its ability to induce MYC degradation in DLBCL [155]. Moreover, the PI3K pathway inhibitory elements such as PTEN are frequently lost in GCB-DLBCL [156], while BCR mutations also result in its constitutive activation [157], leading to MYC dysregulation in DLBCL.
Finally, the upregulation of MYC expression in DLBCL promotes BCR signaling by inducing the MIR17HG cluster, employing a mechanism similar to that described above in the section on BL [158,159]. Taken together, these data suggest that a positive feedback loop operates in the BCR-PI3K-MYC signaling axis in DLBCL.
3.3. MYC In Plasmablastic Lymphoma
PBL is an aggressive, high-grade lymphoma that is most commonly diagnosed in patients with HIV infection or an immunocompromised phenotype [120]. The cell of origin in PBL is thought to be the plasmablast, an activated B cell that has undergone SHM and CSR, and that expresses cell surface markers such as CD138, CD38, MUM1, and Ig, similar to a plasma cell [120]. Signaling pathways leading to plasma cell differentiation involve gene silencing of PAX5 and BCL6 through BLIMP1 [160,161], which also represses MYC expression through promoter binding [42]. Recurrent somatic mutations in PRDM1 (the gene encoding BLIMP1) occur in 50% of cases, where they affect the regulation of diverse targets, such as MYC [162]. Moreover, MYC and BLIMP1 proteins were found to be co-expressed in 80% of diagnoses [162]. These findings are firm evidence that PRDM1 contributes to the oncogenicity of dysregulated MYC.
Although MYC rearrangement is the genetic hallmark of BL and is characteristic of an aggressive subset of DLBCL, it is also a common finding in PBL, along with MYC gains [163,164]. In PBL, MYC rearrangements have been found in ~50% of cases, and the IG genes are the most frequent partners (~85%), with t(8;14) MYC/IGH being the commonest fusion product [163]. Gene expression analysis of PBL revealed MYC overexpression at mRNA and protein levels [165]. MYC overexpression facilitates PBL cell apoptosis escape through cell-cycle dysregulation, and jointly with loss of TP53 [166]. Together, these two processes enhance the aggressiveness of PBL.
In the absence of translocations, the mechanisms of MYC dysregulation are poorly understood, suggesting that MYC may be activated by other mutated genes. Rearrangements of BCL2, BCL6, MALT1, and PAX5, which are common in BL and DLBCL, are not detected in PBL. Conversely, gains of these loci are frequent in PBL, where 30% of cases display amplification of three or more of them [163]. Paradoxically, Ouansafi et al. demonstrated in a single case report the concomitant presence of BCL2 and MYC translocation in a rare case of FL-to-PBL transformation [167].
3.4. MYC in Other Non-Hodgkin B Cell Lymphomas
FL is an indolent non-Hodgkin lymphoma that transforms into a high-grade lymphoma, mostly DLBCL, in about one-third of patients. The genetic hallmark of FL is t(14;18)(q32;q21), which brings about BCL2/IGH fusion protein [168]. Low-grade lymphomas containing a BCL2 rearrangement need subsequent secondary genetic hits for the disease to evolve. The genetic alteration of MYC may suffice as this secondary alteration, leading to the transformation into a high-grade B cell lymphoma [169]. Actually, the majority of FLs express MYC, but only in a small fraction of the cells (<25%) [146].
Pasqualucci et al. investigated the genetic drivers of transformed follicular lymphoma (t-FL) and determined that there is a common mutated precursor that experiences distinct genetic events that are specifically associated with alterations deregulating cell-cycle progression and DNA damage [46], evidence that matches perfectly with MYC oncogene among others. t-FL to DLBCL progression occurs in 30% of the cases, mainly among GCB-DLBCL patients [170,171]. t-FL oncogenic mechanisms are characterized by the presence of a proliferation signature, together with recurrent oncogenic transformations such as TP53 mutation, CDKN2A loss, and c-REL amplification [171], giving rise to a proliferative phenotype in which MYC could be involved. In fact, genetic lesions deregulating MYC are the second most common tFL-specific lesion (including translocations, point mutations, and CNVs) [46]. Alternative pathways involving MYC and its targets could help distinguish between two types of morphologically similar lymphomas, such as tFL-derived DLBCL and de novo DLBCL, this signature being more enriched in de novo cases than in transformed ones [172].
As reported by Martinez-Climent et al., when examining gene expression changes in t-FL, a considerable number of MYC target genes are differentially expressed, although the MYC gene locus remains unaltered in terms of copy number [173]. Consequently, MYC genetic abnormalities are not the driving mutations of FL transformation and may only serve as a surrogate for the entire proliferation signature.
MCL is generally an aggressive malignancy, but it is thought in some cases to remain latently quarrelsome in an indolent phase. It is characterized by t(11;14)(q13;q32), juxtaposing IGH, and CCND1, resulting in CCND1 overexpression, which drives the cells through the G1/S transition [174]. Interestingly, a partnership between CCND1 and MYC has been reported in the oncogenic transformation of B cells to MCL [175]. The coexistence of MYC and CCND1/IGH rearrangements [176] is commonly found in double-hit (DH)-MCL [177], which is associated with a high-risk prognostic index. As reported for FL, most of the MCL cases display an intense MYC expression, but the percentage of positive cells is frequently low (<25%) [146].
Aggressive MCL subset variants can be divided between the blastoid variants (resembling lymphoblast cells) and pleomorphic variants (DLBCL-like cells). MCL is also characterized by large numbers of secondary gains and losses of genes that are mainly involved in cell-cycle regulation, response to DNA damage, and survival [178]. Regarding the blastoid variant, MYC alterations such as the rearrangement involving IGH in t(8;14), disruption of the MYC locus in t(2;8), and gains in add(8)(q24) have been described [179,180]. These aberrations, along with a high level of TP53 expression, are features associated with MCL aggressiveness [181].
CDK4 and CDK6 are catalytic subunits of the cyclin D family that govern G1-to-S-phase progression; p16INK4a and all members of the INK4 family act as their negative regulators by specifically binding to them [182]. CDK4 mutations abolish the binding motif of the INK4 family, thereby functioning as an oncogene capable of directing proliferation [183]. Surprisingly, cyclin D1/CDK4 and p16INK4a complexes are known to be upstream regulators of MYC [184], while CDK4 has been identified as a target of MYC [185]. In the context of MYC dysregulation, CDK4-INK4 imbalance plays a role in lymphomagenesis [183].
The MYC-driven gene expression network is maintained through the stability of the MYC protein, which itself is sustained in MCL by MALT1 [186]. Constitutively activated MALT1 expression, together with BCL10, is orchestrated by the activation of the BCR, which recruits CARD11 scaffold protein and ultimately results in a BCR-driven CARD11-BCL10-MALT1 (CBM) complex. CBM subsequently activates the NF-κβ pathway [187] and, combined with MYC stabilization by MALT1, drives lymphomagenesis progression.
Table 1 summarizes the essential information about the aberrant activation of MYC in leukemia and lymphoma disorders (Chapters 2 and 3).
4. Concluding Remarks
The appropriate path that lymphoid progenitors should ideally follow on their way towards fully differentiated B cells is constantly under threat of being wrecked by the alteration of regulatory mechanisms. Beyond its widely known function as an oncogene, MYC also plays an essential role at different steps of B-cell differentiation, and its deregulation is one of the main hazards that can disrupt the process. As described in this review and in a physiological context, MYC is strongly expressed on the way to producing mature B lymphocytes, whereas its transient downregulation is required at some specific points. However, MYC basal levels are maintained and are not completely switched off at any point before the late memory and plasmatic B cell stages, demonstrating that only tight regulation of MYC levels ensures that the B lymphocytes achieve their correct fate.
When the expression of MYC protein is aberrantly altered, the risk of developing a hematological malignancy, such as leukemia or lymphoma, increases substantially through the acquisition of an uncontrolled proliferative rate and a blockade of differentiation. Remarkably, the alterations that trigger MYC overexpression differ between leukemia and lymphoma cells. In fact, leukemic cells have low rates of MYC mutations and a low frequency of chromosomal translocations involving the MYC gene, whereas the aforementioned genetic alterations are a hallmark in some types of non-Hodgkin B cell lymphoma. Nevertheless, and even when not altered at the genetic level, the expression of MYC is usually disrupted in the commonest types of leukemia, where it is activated by several pathways, as well as at the post-transcriptional level. Most types of lymphoma present high levels of MYC expression that are not always correlated with a mutated MYC gene, opening the door to speculation that, as in leukemia, multiple pathways may act to facilitate its dysregulation.
In terms of therapeutic perspectives, the possibilities for interfering with MYC activity are still to be adequately explored. However, the identification of regulatory cascades and other mechanisms that trigger its induction opens a wide range of possibilities for indirectly impairing MYC function. As reported here, we believe that the disruption of altered epigenetic regulation with HDAC inhibitors, the blockade of microRNAs function, or the use of BET inhibitors that obstruct scaffold transcriptional activating machinery, are but a few examples of the promising therapeutic strategies that will lead to an improved prognosis of hematological disorders, mostly mediated by the maintenance of MYC at physiological levels.
Author Contributions
O.d.B. and A.M. did the bibliographic research and wrote the manuscript. M.P. conceived the manuscript, supervised the work, and provided critical comments on the review. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was funded by grants to M.P. by the Spanish Ministry of Science, Innovation, and Universities (SAF2017-87990-R and EUR2019-103835) and elaborated at the Josep Carreras Leukaemia Research Institute (IJC, Badalona, Barcelona). O.d.B. is funded by a Juan de la Cierva - Formación fellowship from the Spanish Ministry of Science, Innovation, and Universities (FJCI-2017-32430). A.M. is funded by the Spanish Ministry of Science, Innovation and Universities, which is part of the Agencia Estatal de Investigación (AEI), through grant PRE2018-083183 (co-funded by the European Social Fund.). We thank the CERCA Programme/Generalitat de Catalunya and the Josep Carreras Foundation for institutional support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adolfsson, J.; Månsson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.-J.; Thoren, L.A.M.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Melnick, A. Mechanisms of action of BCL6 during germinal center B cell development. Sci. China Life Sci. 2015, 58, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, F.; Cole, M. Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nature 1986, 320, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Sheiness, D.; Bishop, J.M. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Varmus, H. The molecular genetics of cellular oncogenes. Annu. Rev. Genet. 1984, 18, 553–612. [Google Scholar] [CrossRef]
- Langdon, W.Y.; Harris, A.W.; Cory, S.; Adams, J.M. The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell 1986, 47, 11–18. [Google Scholar] [CrossRef]
- Delgado, M.D.; León, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reavie, L.; Della Gatta, G.; Crusio, K.; Aranda-Orgilles, B.; Buckley, S.M.; Thompson, B.; Lee, E.; Gao, J.; Bredemeyer, A.L.; Helmink, B.A.; et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat. Immunol. 2010, 11, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, H.; Braren, R.; Fong, S.Y.; Trumpp, A.; Carlson, T.R.; Wang, R.A. C-Myc in the hematopoietic lineage is crucial for its angiogenic function in the mouse embryo. Development 2008, 135, 2467–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc Is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.K.; Zimmerman, K.; Yancopoulos, G.D. Transcriptional down-regulation of N-myc expression during B-cell development. Mol. Cell Biol. 1992, 12, 1578–1584. [Google Scholar] [CrossRef] [Green Version]
- Klemsz, M.J.; Justement, L.B.; Palmer, E.; Cambier, J. Induction of c-fos and c-myc expression during B cell activation by IL-4 and immunoglobulin binding ligands. J. Immunol. 1989, 143, 1032–1039. [Google Scholar]
- Larsson, L.G.; Schena, M.; Carlsson, M.; Sallstrom, J.; Nilsson, K. Expression of the c-myc protein is down-regulated at the terminal stages during in vitro differentiation of B-type chronic lymphocytic leukemia cells. Blood 1991, 77, 1025–1032. [Google Scholar] [CrossRef]
- Huang, C.Y.; Bredemeyer, A.L.; Walker, L.M.; Bassing, C.H.; Sleckman, B.P. Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur. J. Immunol. 2008, 38, 342–349. [Google Scholar] [CrossRef]
- Habib, T.; Park, H.; Tsang, M.; De Alborán, I.M.; Nicks, A.; Wilson, L.; Knoepfler, P.S.; Andrews, S.; Rawlings, D.J.; Eisenman, R.N.; et al. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J. Cell Biol. 2007, 179, 717–731. [Google Scholar] [CrossRef] [Green Version]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemotopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Morrow, M.A.; Lee, G.; Gillis, S.; Yancopoulos, G.D.; Alt, F.W. Interleukin-7 induces N-myc and c-myc expression in normal precursor B lymphocytes. Genes Dev. 1992, 6, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Alboran, I.M.; O’Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of c-Myc function in normal cells via conditional gene-targeted mutation. Immunity 2001, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Dalla-Favera, R.; Nussenzweig, M.C. c-MYC is required for germinal center selection and cyclic re-entry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Ci, W.; Polo, J.M.; Cerchietti, L.; Shaknovich, R.; Wang, L.; Shao, N.Y.; Ye, K.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D.; et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 2009, 113, 5536–5548. [Google Scholar] [CrossRef] [Green Version]
- Basso, K.; Saito, M.; Sumazin, P.; Margolin, A.A.; Wang, K.; Lim, W.K.; Kitagawa, Y.; Schneider, C.; Alvarez, M.J.; Califano, A.; et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood 2010, 115, 975–984. [Google Scholar] [CrossRef] [Green Version]
- Nahar, R.; Ramezani-Rad, P.; Mossner, M.; Duy, C.; Cerchietti, L.; Geng, H.; Dovat, S.; Jumaa, H.; Ye, B.H.; Melnick, A.; et al. Pre-B cell receptor-mediated activation of BCL6 induces pre-B cell quiescence through transcriptional repression of MYC. Blood 2011, 118, 4174–4178. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Mohn, S.E.; Weinmann, A.S. Molecular mechanisms that control the expression and activity of Bcl-6 in TH 1 cells to regulate flexibility with a TFH -like gene profile. Nat. Immunol. 2013, 13, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Gao, J.; Basso, K.; Kitagawa, Y.; Smith, P.M.; Bhagat, G.; Pernis, A.; Pasqualucci, L.; Dalla-Favera, R. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 2007, 12, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Weisel, F.; Shlomchik, M.J. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity 2018, 25, 1032–1057. [Google Scholar] [CrossRef] [Green Version]
- Ersching, J.; Efeyan, A.; Mesin, L.; Jacobsen, J.T.; Pasqual, G.; Grabiner, B.C.; Dominguez-Sola, D.; Sabatini, D.M.; Victora, G.D. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity 2017, 46, 1045–1058.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Roger, I.; Kim, S.H.; Griffiths, B.; Sewing, A.; Land, H. Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 1999, 18, 5310–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, C.; Thieke, K.; Maier, A.; Saffrich, R.; Hanley-Hyde, J.; Ansorge, W.; Reed, S.; Sicinski, P.; Bartek, J.; Eilers, M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999, 18, 5321–5333. [Google Scholar] [CrossRef] [PubMed]
- Cato, M.H.; Chintalapati, S.K.; Yau, I.W.; Omori, S.A.; Rickert, R.C. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol. Cell. Biol. 2011, 31, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Peled, J.U.; Yu, J.J.; Venkatesh, J.; Bi, E.; Ding, B.B.; Krupski-Downs, M.; Shaknovich, R.; Sicinski, P.; Diamond, B.; Scharff, M.D.; et al. Requirement for cyclin D3 in germinal center formation and function. Cell Res. 2010, 20, 631–646. [Google Scholar] [CrossRef]
- Calado, D.P.; Sasaki, Y.; Godinho, S.A.; Pellerin, A.; Sleckman, B.P.; Alborán, I.M.D.; Janz, M.; Rodig, S.; Rajewsky, K. MYC is essential for the formation and maintenance of germinal centers. Nat. Immunol. 2012, 13, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef] [Green Version]
- Phan, R.T.; Saito, M.; Basso, K.; Niu, H.; Dalla-Favera, R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 2005, 6, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef] [Green Version]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. C-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Ramiro, A.R.; Jankovic, M.; Eisenreich, T.; Difilippantonio, S.; Chen-Kiang, S.; Muramatsu, M.; Honjo, T.; Nussenzweig, A.; Nussenzweig, M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004, 118, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wong, K.K.; Calame, K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 1997, 276, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L.; Callahan, W.; Witte, O.N. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992, 70, 901–910. [Google Scholar] [CrossRef]
- Advani, A.S.; Pendergast, A.M. Bcr-Abl variants: Biological and clinical aspects. Leuk. Res. 2002, 26, 713–720. [Google Scholar] [CrossRef]
- Afar, D.E.H.; Goga, A.; McLaughlin, J.; Witte, O.N.; Sawyers, C.L. Differential complementation of Bcr-Abl point mutants with c-Myc. Science 1994, 264, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbò, F.; et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Feldhahn, N.; Henke, N.; Melchior, K.; Duy, C.; Soh, B.N.; Klein, F.; Von Levetzow, G.; Giebel, B.; Li, A.; Hofmann, W.K.; et al. Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells. J. Exp. Med. 2007, 204, 1157–1166. [Google Scholar] [CrossRef] [Green Version]
- Dorsett, Y.; Robbiani, D.F.; Jankovic, M.; Reina-San-Martin, B.; Eisenreich, T.R.; Nussenzweig, M.C. A role for AID in chromosome translocations between c-myc and the IgH variable region. J. Exp. Med. 2007, 204, 2225–2232. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.J.; Wu, X.; Duan, Y.; wang, Y.M.; Shan, B.; Kong, J.X.; Ma, X.B.; Bao, Y.X. AID expression is correlated with Bcr-Abl expression in CML-LBC and can be down-regulated by As2O3 and/or imatinib. Leuk. Res. 2011, 35, 1355–1359. [Google Scholar] [CrossRef]
- Tomita, N.; Tokunaka, M.; Nakamura, N.; Takeuchi, K.; Koike, J.; Motomura, S.; Miyamoto, K.; Kikuchi, A.; Hyo, R.; Yakushijin, Y.; et al. Clinicopathological features of lymphoma/leukemia patients carrying both BCL2 and MYC translocations. Haematologica 2009, 94, 935–943. [Google Scholar] [CrossRef]
- Xu, N.; Li, Y.L.; Zhou, X.; Cao, R.; Li, H.; Lu, Q.S.; Li, L.; Lu, Z.Y.; Huang, J.X.; Sun, J.; et al. CDKN2 gene deletion as poor prognosis predictor involved in the progression of adult B-lineage acute lymphoblastic leukemia patients. J. Cancer 2015, 6, 1114–1120. [Google Scholar] [CrossRef] [Green Version]
- Eswaran, J.; Sinclair, P.; Heidenreich, O.; Irving, J.; Russell, L.J.; Hall, A.; Calado, D.P.; Harrison, C.J.; Vormoor, J. The pre-B-cell receptor checkpoint in acute lymphoblastic leukaemia. Leukemia 2015, 29, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Köhrer, S.; Havranek, O.; Seyfried, F.; Hurtz, C.; Coffey, G.P.; Kim, E.; Ten Hacken, E.; Jäger, U.; Vanura, K.; O’Brien, S.; et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016, 30, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Powell, J.A.; Tong, D.; Pitson, S.M.; Bradstock, K.F.; Bendall, L.J. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing myc expression. Cancer Res. 2014, 74, 2803–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X.; Xiang, W.; He, L.; Tang, M.; Wang, F.; Wang, T.; Yang, Z.; Yi, Y.; Wang, H.; et al. Development of purine-based hydroxamic acid derivatives: Potent histone deacetylase inhibitors with marked in vitro and in vivo antitumor activities. J. Med. Chem. 2016, 59, 5488–5504. [Google Scholar] [CrossRef]
- Yang, L.; Qiu, Q.; Tang, M.; Wang, F.; Yi, Y.; Yi, D.; Yang, Z.; Zhu, Z.; Zheng, S.; Yang, J.; et al. Purinostat mesylate is a uniquely potent and selective inhibitor of HDACs for the treatment of BCR-ABL –induced B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2019, 25, 7527–7539. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.S.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K inhibitor CUDC-907 down regulates MYC and suppresses growth of MYC-dependent cancers. Mol. Cancer Ther. 2017, 16, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Luong-Gardiol, N.; Siddiqui, I.; Pizzitola, I.; Jeevan-Raj, B.; Charmoy, M.; Huang, Y.; Irmisch, A.; Curtet, S.; Angelov, G.S.; Danilo, M.; et al. γ-catenin-dependent signals maintain BCR-ABL1 + B cell acute lymphoblastic leukemia. Cancer Cell 2019, 35, 649–663.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef]
- Xie, S.; Lin, H.; Sun, T.; Arlinghaus, R.B. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 2002, 21, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Li, L.S.; Kopp, N.; Montero, J.; Chapuy, B.; Yoda, A.; Christie, A.L.; Liu, H.; Christodoulou, A.; van Bodegom, D.; et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell 2015, 28, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Krysov, S.; Dias, S.; Paterson, A.; Mockridge, C.I.; Potter, K.N.; Smith, K.A.; Ashton-Key, M.; Stevenson, F.K.; Packham, G. Surface IgM stimulation induces MEK1/2-dependent MYC expression in chronic lymphocytic leukemia cells. Blood 2012, 119, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Yeomans, A.; Thirdborough, S.M.; Valle-Argos, B.; Linley, A.; Krysov, S.; Hidalgo, M.S.; Leonard, E.; Ishfaq, M.; Wagner, S.D.; Willis, A.E.; et al. Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood 2016, 127, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Cencic, R.; Mills, J.R.; Robert, F.; Pelletier, J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008, 68, 5326–5334. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Malempati, S.; Tibbitts, D.; Cunningham, M.; Akkari, Y.; Olson, S.; Fan, G.; Sears, R.C. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 2006, 20, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [CrossRef] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mraz, M.; Kipps, T.J. MicroRNAs and B cell receptor signaling in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1836–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Deutzmann, A.; Choi, P.S.; Fan, A.C.; Felsher, D.W. BIM mediates oncogene inactivation-induced apoptosis in multiple transgenic mouse models of acute lymphoblastic leukemia. Oncotarget 2016, 7, 26926–26934. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, T.; Brady, H.J.M. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol. Dis. 2008, 40, 192–199. [Google Scholar] [CrossRef]
- Meeker, N.D.; Cherry, A.M.; Bangs, C.D.; Frazer, J.K. A pediatric B lineage leukemia with coincident MYC and MLL translocations. J. Pediatr. Hematol. Oncol. 2011, 33, 158–160. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Ragusa, D.; Makarov, E.M.; Britten, O.; Moralli, D.; Green, C.M.; Tosi, S. The RS4;11 cell line as a model for leukaemia with t(4;11)(q21;q23): Revised characterisation of cytogenetic features. Cancer Rep. 2019, 1–11, e1207. [Google Scholar] [CrossRef] [Green Version]
- Hyrenius-Wittsten, A.; Pilheden, M.; Sturesson, H.; Hansson, J.; Walsh, M.P.; Song, G.; Kazi, J.U.; Liu, J.; Ramakrishan, R.; Garcia-Ruiz, C.; et al. De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Neff, T.; Sinha, A.U.; Kluk, M.J.; Zhu, N.; Khattab, M.H.; Stein, L.; Xie, H.; Orkin, S.H.; Armstrong, S.A. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 5028–5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiner, S.; Birke, M.; García-Cuéllar, M.P.; Zilles, O.; Greil, J.; Slany, R.K. MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res. 2001, 61, 6480–6486. [Google Scholar] [PubMed]
- Jiang, X.; Huang, H.; Li, Z.; Li, Y.; Wang, X.; Gurbuxani, S.; Chen, P.; He, C.; You, D.; Zhang, S.; et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 2012, 22, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, J.J.; Coulson, J.M.; Clague, M.J.; Urbé, S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010, 62, 140–157. [Google Scholar] [CrossRef]
- Meyer, C.; Lopes, B.A.; Caye-Eude, A.; Cavé, H.; Arfeuille, C.; Cuccuini, W.; Sutton, R.; Venn, N.C.; Oh, S.H.; Tsaur, G.; et al. Human MLL/KMT2A gene exhibits a second breakpoint cluster region for recurrent MLL–USP2 fusions. Leukemia 2019, 33, 2306–2340. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [Green Version]
- Coudé, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef] [Green Version]
- McCalmont, H.; Li, K.L.; Jones, L.; Toubia, J.; Bray, S.C.; Casolari, D.A.; Mayoh, C.; Samaraweera, S.E.; Lewis, I.D.; Prinjha, R.K.; et al. Efficacy of combined CDK9/BET inhibition in preclinical models of MLL-rearranged acute leukemia. Blood Adv. 2020, 4, 296–300. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Chao, S.H.; Lane, D.P. HEXIM1 and the control of transcription elongation: From cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 2007, 6, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Osdal, T.; Ho, Y.; Chun, S.; McDonald, T.; Agarwal, P.; Lin, A.; Chu, S.; Qi, J.; Li, L.; et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell 2014, 15, 431–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-García, N.; Malatesta, R.; Estella, C.; Pérez-Jaume, S.; Esperanza-Cebollada, E.; Torrebadell, M.; Català, A.; Gassiot, S.; Berrueco, R.; Ruiz-Llobet, A.; et al. Paediatric patients with acute leukaemia and KMT2A (MLL) rearrangement show a distinctive expression pattern of histone deacetylases. Br. J. Haematol. 2018, 182, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barneda-Zahonero, B.; Collazo, O.; Azagra, A.; Fernández-Duran, I.; Serra-Musach, J.; Islam, A.B.M.M.K.; Vega-García, N.; Malatesta, R.; Camós, M.; Gómez, A.; et al. The transcriptional repressor HDAC7 promotes apoptosis and c-Myc downregulation in particular types of leukemia and lymphoma. Cell Death Dis. 2015, 6, e1635. [Google Scholar] [CrossRef] [Green Version]
- Haery, L.; Thompson, R.C.; Gilmore, T.D. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 2015, 6, 184–213. [Google Scholar]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef]
- Azagra, A.; Marina-Zárate, E.; Ramiro, A.R.; Javierre, B.M.; Parra, M. From loops to looks: Transcription factors and chromatin organization shaping terminal B cell differentiation. Trends Immunol. 2020, 41, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Borkhardt, A.; Cazzaniga, G.; Viehmann, S.; Valsecchi, M.G.; Ludwig, W.D.; Burci, L.; Mangioni, S.; Schrappe, M.; Riehm, H.; Lampert, F.; et al. Incidence and clinical relevance of TEL/AML1 fusion genes in children with acute lymphoblastic leukemia enrolled in the German and Italian Multicenter Therapy Trials. Blood 1997, 90, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Masera, G. Molecular pathogenesis of childhood acute lymphoblastic leukemia. Haematologica 1998, 83, 651–659. [Google Scholar] [PubMed]
- Cazzaniga, G.; Daniotti, M.; Tosi, S.; Giudici, G.; Aloisi, A.; Pogliani, E.; Kearney, L.; Biondi, A. The paired box domain gene PAX5 is fused to ETV6/TEL in an acute lymphoblastic leukemia case. Cancer Res. 2001, 61, 4666–4670. [Google Scholar]
- Nutt, S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. J. Immunol. 1999, 401, 556–562. [Google Scholar]
- Chae, H.; Kim, M.; Lim, J.; Kim, Y.; Han, K.; Lee, S. B lymphoblastic leukemia with ETV6 amplification. Cancer Genet. Cytogenet. 2010, 203, 284–287. [Google Scholar] [CrossRef]
- Bokemeyer, A.; Eckert, C.; Meyr, F.; Koerner, G.; von Stackelberg, A.; Ullmann, R.; Türkmen, S.; Henze, G.; Seeger, K. Copy number genome alterations are associated with treatment response and outcome in relapsed childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Haematologica 2014, 99, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Cancelas, J.A.; Williams, D.A. Rho GTPases in hematopoietic stem cell functions. Curr. Opin. Hematol. 2009, 16, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Raptis, L.; Arulanandam, R.; Geletu, M.; Turkson, J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp. Cell Res. 2011, 317, 1787–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangolini, M.; De Boer, J.; Walf-Vorderwülbecke, V.; Pieters, R.; Den Boer, M.L.; Williams, O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12;21) acute lymphoblastic leukemia. Blood 2013, 122, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Stoskus, M.; Vaitkeviciene, G.; Eidukaite, A.; Griskevicius, L. ETV6/RUNX1 transcript is a target of RNA-binding protein IGF2BP1 in t(12;21)(p13;q22)-positive acute lymphoblastic leukemia. Blood Cells Mol. Dis. 2016, 57, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, N.; Hüttelmaier, S. IGF2BP1: A post-transcriptional “driver” of tumor cell migration. Cell Adhes. Migr. 2012, 6, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.R. B-cell commitment: deciding on the players. Curr. Opin. Immunol. 2003, 15, 158–165. [Google Scholar] [CrossRef]
- Geng, H.; Hurtz, C.; Lenz, K.B.; Chen, Z.; Baumjohann, D.; Thompson, S.; Goloviznina, N.A.; Chen, W.-Y.; Huan, J.; LaTocha, D.; et al. Self-enforcing feedback activation between BCL6 and Pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 2015, 27, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankovich, A.J.; Raunser, S.; Juo, Z.S.; Walz, T.; Davis, M.M.; Garcia, K.C. Structural insight into pre-B cell receptor function. Science 2007, 316, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microrna recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef] [Green Version]
- Akbari Moqadam, F.; Boer, J.M.; Lange-Turenhout, E.A.M.; Pieters, R.; Den Boer, M.L. Altered expression of miR-24, miR-126 and miR-365 does not affect viability of childhood TCF3-rearranged leukemia cells. Leukemia 2014, 28, 1008–1014. [Google Scholar] [CrossRef]
- Folgiero, V.; Sorino, C.; Pallocca, M.; De Nicola, F.; Goeman, F.; Bertaina, V.; Strocchio, L.; Romania, P.; Pitisci, A.; Iezzi, S.; et al. Che-1 is targeted by c-Myc to sustain proliferation in pre-B-cell acute lymphoblastic leukemia. EMBO Rep. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Bruno, T.; De Nicola, F.; Iezzi, S.; Lecis, D.; D’Angelo, C.; Di Padova, M.; Corbi, N.; Dimiziani, L.; Zannini, L.; Jekimovs, C.; et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 2006, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Bemark, M.; Neuberger, M.S. The c-MYC allele that is translocated into the IgH locus undergoes constitutive hypermutation in a Burkitt’s lymphoma line. Oncogene 2000, 19, 3404–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegmiller, A.C.; Garcia, R.; Huang, R.; Maleki, A.; Karandikar, N.J.; Chen, W. Simple karyotype and bcl-6 expression predict a diagnosis of Burkitt lymphoma and better survival in IG-MYC rearranged high-grade B-cell lymphomas. Mod. Pathol. 2010, 23, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Cory, S. Activation of cellular oncogenes in hemopoietic cells by chromosome translocation. Adv. Cancer Res. 1986, 47, 189–234. [Google Scholar]
- Cesarman, E.; Dalla-Favera, R.; Bentley, D.; Groudine, M. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science 1987, 238, 1272–1275. [Google Scholar] [CrossRef]
- Rabbits, T.H.; Forster, A.; Hamlyn, P.; Baer, R. Effect of somatic mutation within translocated c-myc genes in Burkitt’s lymphoma. Nature 1984, 309, 592–597. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.E.; Bernd, H.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of burkitt’s lymphoma from transcriptional and genomic profiling michael. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Seitz, V.; Butzhammer, P.; Hirsch, B.; Hecht, J.; Gütgemann, I.; Ehlers, A.; Lenze, D.; Oker, E.; Sommerfeld, A.; von der Wall, E.; et al. Deep sequencing of MYC DNA-Binding sites in Burkitt lymphoma. PLoS ONE 2011, 6, e26837. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Sato, S.; Frederick, J.P.; Sun, X.-H.; Zhuang, Y. Impaired immune responses and B-cell proliferation in mice lacking the Id3 gene. Mol. Cell. Biol. 1999, 19, 5969–5980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Köchert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffman, J.D.; Lorimer, P.D.; Rodic, V.; Jahromi, M.S.; Downie, J.M.; Bayerl, M.G.; Sanmann, J.N.; Althof, P.A.; Sanger, W.G.; Barnette, P.; et al. Genome wide copy number analysis of paediatric Burkitt lymphoma using formalin-fixed tissues reveals a subset with gain of chromosome 13q and corresponding miRNA over expression. Br. J. Haematol. 2011, 155, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009, 23, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Robaina, M.C.; Mazzoccoli, L.; Klumb, C.E. Germinal centre B cell functions and lymphomagenesis: Circuits involving MYC and MicroRNAs. Cells 2019, 8, 1365. [Google Scholar] [CrossRef] [Green Version]
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of burkitt’s lymphoma. N. Engl. J. Med. 2006, 354, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Klapproth, K.; Sander, S.; Marinkovic, D.; Baumann, B.; Wirth, T.; Dc, W.; Klapproth, K.; Sander, S.; Marinkovic, D.; Baumann, B.; et al. The IKK2/NF-κ B pathway suppresses MYC-induced lymphomagenesis. Blood 2011, 114, 2448–2458. [Google Scholar] [CrossRef] [Green Version]
- Ngan, B.Y.; Chen-Levy, Z.; Weiss, L.M.; Warnke, R.A.; Cleart, M.L. Expression in non-hodgkin’s lymphoma of the bcl-2 protein associated with the t(14;18) chromosomal translocation. N. Engl. J. Med. 1988, 318, 1638–1644. [Google Scholar] [CrossRef]
- Lo Coco, F.; Ye, B.H.; Lista, F.; Corradini, P.; Offit, K.; Knowles, D.M.; Chaganti, R.S.K.; Dalla-Favera, R. Rearrangements of the BCL6 gene in diffuse large cell non-Hodgkin’s lymphoma. Blood 1994, 83, 1757–1759. [Google Scholar] [CrossRef] [Green Version]
- Gascoyne, R.D. Pathologic prognostic factors in diffuse aggressive non-Hodgkin’s lymphoma. Hematol. Oncol. Clin. N. Am. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Kramer, M.H.H.; Hermans, J.; Wijburg, E.; Philippo, K.; Geelen, E.; van Krieken, J.H.J.M.; de Jong, D.; Maartense, E.; Schuuring, E.; Kluin, P.M. Clinical relevance of BCL2, BCL6, and MYC rearrangements in diffuse large B-cell lymphoma. Blood 1998, 92, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Akasaka, H.; Ueda, C.; Yonetani, N.; Maesako, Y.; Shimizu, A.; Yamabe, H.; Fukuhara, S.; Uchiyama, T.; Ohno, H. Molecular and clinical features of non-burkitt’s, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J. Clin. Oncol. 2000, 18, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, C.; Ohshima, K.; Suzumiya, J.; Kanda, M.; Tsuchiya, T.; Tamura, K.; Kikuchi, M. Rearrangements of bcl-1, bcl-2, bcl-6, and c-myc in diffuse large B-cell lymphomas. Leuk. Lymphoma 2001, 42, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, K.M.; Bangs, C.D.; Bacchi, C.E.; Molina-Kirsch, H.; Cherry, A.; Natkunam, Y. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am. J. Surg. Pathol. 2015, 39, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Bastard, C.; Maingonnat, C.; Jardin, F.; Maisonneuve, C.; Courel, M.-N.; Ruminy, P.; Picquenot, J.M.; Tilly, H. Mapping of MYC breakpoints in 8q24 rearrangements involving non-immunoglobulin partners in B-cell lymphomas. Leukemia 2007, 21, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasik, C.J.; Nitta, H.; Zhang, W.; Mosher, C.H.; Cook, J.R.; Tubbs, R.R.; Unger, J.M.; Brooks, T.A.; Persky, D.O.; Wilkinson, S.T.; et al. Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica 2010, 95, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Valera, A.; Lopez-Guillermo, A.; Cardesa-Salzmann, T.; Climent, F.; Gonzalez-Barca, E.; Mercadal, S.; Espinosa, I.; Novelli, S.; Briones, J.; Mate, J.L.; et al. MYC protein expression and genetic alterations have prognostic impact in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Haematologica 2013, 98, 1554–1562. [Google Scholar] [CrossRef]
- Grimm, K.E.; O’Malley, D.P. Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues. Ann. Diagn. Pathol. 2019, 38, 6–10. [Google Scholar] [CrossRef]
- Gebauer, N.; Bernard, V.; Gebauer, W.; Thorns, C.; Feller, A.C.; Merz, H. TP53 mutations are frequent events in double-hit B-cell lymphomas with MYC and BCL2 but not MYC and BCL6 translocations. Leuk. Lymphoma 2015, 56, 179–185. [Google Scholar] [CrossRef]
- Aukema, S.M.; Kreuz, M.; Kohler, C.W.; Rosolowski, M.; Hasenclever, D.; Hummel, M.; Küppers, R.; Lenze, D.; Ott, G.; Pott, C.; et al. Biological characterization of adult MYC-translocation-positive mature B-cell lymphomas other than molecular Burkitt lymphoma. Haematologica 2014, 99, 726–735. [Google Scholar] [CrossRef]
- Cook, J.R.; Goldman, B.; Tubbs, R.R.; Rimsza, L.; Leblanc, M.; Stiff, P.; Fisher, R. Clinical significance of MYC expression and/or “High-grade” morphology in non-burkitt, diffuse aggressive B-cell lymphomas: A SWOG s9704 correlative study. Am. J. Surg. Pathol. 2014, 38, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Kamasani, U.; Prendergast, G.C. RhoB facilitates c-Myc turnover by supporting efficient nuclear accumulation of GSK-3. Oncogene 2006, 25, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.G.; Liu, Z.B.; Jiang, X.N.; Lee, J.; Zhou, X.Y.; Li, X.Q. MYC protein dysregulation is driven by BCR-PI3K signalling in diffuse large B-cell lymphoma. Histopathology 2017, 71, 778–785. [Google Scholar] [CrossRef]
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Ju, H.; Kim, D.H.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Ko, Y.H. CD79B and MYD88 mutations in diffuse large B-cell lymphoma. Hum. Pathol. 2014, 45, 556–564. [Google Scholar] [CrossRef]
- Tagawa, H.; Karube, K.; Tsuzuki, S.; Ohshima, K.; Seto, M. Synergistic action of the microRNA-17 polycistron and Myc in aggressive cancer development. Cancer Sci. 2007, 98, 1482–1490. [Google Scholar] [CrossRef]
- Psathas, J.N.; Doonan, P.J.; Raman, P.; Freedman, B.D.; Minn, A.J.; Thomas-Tikhonenko, A. Lymphoid neoplasia: The Myc-miR-17-92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: A novel lymphomagenic feed-forward loop. Blood 2013, 122, 4220–4229. [Google Scholar] [CrossRef] [Green Version]
- Shapiro-Shelef, M.; Lin, K.I.; McHeyzer-Williams, L.J.; Liao, J.; McHeyzer-Williams, M.G.; Calame, K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 2003, 19, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Lin, K.I.; Kuo, T.C.; Yu, X.; Hurt, E.M.; Rosenwald, A.; Giltnane, J.M.; Yang, L.; Zhao, H.; Calame, K.; et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002, 17, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Montes-Moreno, S.; Martinez-Magunacelaya, N.; Zecchini-Barrese, T.; De Villambrosía, S.G.; Linares, E.; Ranchal, T.; Rodriguez-Pinilla, M.; Batlle, A.; Cereceda-Company, L.; Revert-Arce, J.B.; et al. Plasmablastic lymphoma phenotype is determined by genetic alterations in MYC and PRDM1. Mod. Pathol. 2017, 30, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Valera, A.; Balagué, O.; Colomo, L.; Martínez, A.; Delabie, J.; Taddesse-Heath, L.; Jaffe, E.S.; Campo, E. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am. J. Surg. Pathol. 2010, 34, 1686–1694. [Google Scholar] [CrossRef] [Green Version]
- Taddesse-Heath, L.; Meloni-Ehrig, A.; Scheerle, J.; Kelly, J.C.; Jaffe, E.S. Plasmablastic lymphoma with MYC translocation: evidence for a common pathway in the generation of plasmablastic features. Mod. Pathol. 2010, 23, 991–999. [Google Scholar] [CrossRef]
- Chapman, J.; Gentles, A.J.; Sujoy, V.; Vega, F.; Dumur, C.I.; Blevins, T.L.; Bernal-Mizrachi, L.; Mosunjac, M.; Pimentel, A.; Zhu, D.; et al. Gene expression analysis of plasmablastic lymphoma identifies downregulation of B-cell receptor signaling and additional unique transcriptional programs. Leukemia 2015, 29, 2270–2273. [Google Scholar] [CrossRef]
- Simonitsch-Klupp, I.; Hauser, I.; Ott, G.; Drach, J.; Ackermann, J.; Kaufmann, J.; Weltermann, A.; Greinix, H.T.; Skrabs, C.; Dittrich, C.; et al. Diffuse large B-cell lymphomas with plasmablastic/plasmacytoid features are associated with TP53 deletions and poor clinical outcome. Leukemia 2004, 18, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Ouansafi, I.; He, B.; Fraser, C.; Nie, K.; Mathew, S.; Bhanji, R.; Hoda, R.; Arabadjief, M.; Knowles, D.; Cerutti, A.; et al. Transformation of follicular lymphoma to plasmablastic lymphoma with c-myc gene rearrangement. Am. J. Clin. Pathol. 2010, 134, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Lin, P.; Fayad, L.E.; Lennon, P.A.; Miranda, R.N.; Yin, C.C.; Lin, E.; Medeiros, L.J. B-cell lymphomas with MYC/8q24 rearrangements and IGH@BCL2/t(14;18) (q32;q21): An aggressive disease with heterogeneous histology, germinal center B-cell immunophenotype and poor outcome. Mod. Pathol. 2012, 25, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Jaffe, E.S.; Longo, D.L.; Raffeld, M. MYC rearrangements in histologically progressed follicular lymphomas. Blood 1992, 80, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Al-Tourah, A.J.; Gill, K.K.; Chhanabhai, M.; Hoskins, P.J.; Klasa, R.J.; Savage, K.J.; Sehn, L.H.; Shenkier, T.N.; Gascoyne, R.D.; Connors, J.M. Population-based analysis of incidence and outcome of transformed non-hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 5165–5169. [Google Scholar] [CrossRef]
- Davies, A.J.; Rosenwald, A.; Wright, G.; Lee, A.; Last, K.W.; Weisenburger, D.D.; Chan, W.C.; Delabie, J.; Braziel, R.M.; Campo, E.; et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br. J. Haematol. 2007, 136, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Lossos, I.S.; Alizadeh, A.A.; Diehn, M.; Warnke, R.; Thorstenson, Y.; Oefner, P.J.; Brown, P.O.; Botstein, D.; Levy, R. Transformation of follicular lymphoma to diffuse large-cell lymphoma: Alternative patterns with increased or decreased expression of c-myc and its regulated genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8886–8891. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Climent, J.A.; Alizadeh, A.A.; Segraves, R.; Blesa, D.; Rubio-Moscardo, F.; Albertson, D.G.; Garcia-Conde, J.; Dyer, M.J.S.; Levy, R.; Pinkel, D.; et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood 2003, 101, 3109–3117. [Google Scholar] [CrossRef]
- Lukas, J.; Jadayel, D.; Bartkova, J.; Nacheva, E.; Dyer, M.J.; Strauss, M.; Bartek, J. BCL-1/cyclin D1 oncoprotein oscillates and subverts the G1 phase control in B-cell neoplasms carrying the t(11;14) translocation. Oncogene 1994, 9, 2159–2167. [Google Scholar]
- Lovec, H.; Grzeschiczek, A.; Kowalski, M.B.; Möröy, T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. 1994, 13, 3487–3495. [Google Scholar] [CrossRef]
- Sander, S.; Bullinger, L.; Leupolt, E.; Benner, A.; Kienle, D.; Katzenberger, T.; Kalla, J.; Ott, G.; Müller-Hermelink, H.K.; Barth, T.F.E.; et al. Genomic aberrations in mantle cell lymphoma detected by interphase fluorescence in situ hybridization. Incidence and clinicopathological correlations. Haematologica 2008, 93, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Aukema, S.M.; Siebert, R.; Schuuring, E.; Van Imhoff, G.W.; Kluin-nelemans, H.C.; Boerma, E.; Kluin, P.M. Review article Double-hit B-cell lymphomas. Hematology 2011, 117, 2319–2331. [Google Scholar]
- Royo, C.; Salaverria, I.; Hartmann, E.M.; Rosenwald, A.; Campo, E.; Beà, S. The complex landscape of genetic alterations in mantle cell lymphoma. Semin. Cancer Biol. 2011, 21, 322–334. [Google Scholar] [CrossRef]
- Hao, S.; Sanger, W.; Onciu, M.; Lai, R.; Schlette, E.J.; Medeiros, L.J. Mantle cell lymphoma with 8q24 chromosomal abnormalities: A report of 5 cases with blastoid features. Mod. Pathol. 2002, 15, 1266–1272. [Google Scholar] [CrossRef]
- Hu, Z.; Medeiros, L.J.; Chen, Z.; Chen, W.; Li, S.; Konoplev, S.N.; Lu, X.; Pham, L.V.; Young, K.H.; Wang, W.; et al. Mantle cell lymphoma with MYC rearrangement: A report of 17 patients. Am. J. Surg. Pathol. 2017, 41, 216–224. [Google Scholar] [CrossRef]
- Sander, B.; Quintanilla-Martinez, L.; Ott, G.; Xerri, L.; Kuzu, I.; Chan, J.K.C.; Swerdlow, S.H.; Campo, E. Mantle cell lymphoma—A spectrum from indolent to aggressive disease. Virchows Arch. 2016, 468, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Peters, G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim. Biophys. Acta Rev. Cancer 1998, 1378, F115–F177. [Google Scholar] [CrossRef]
- Vincent-Fabert, C.; Fiancette, R.; Rouaud, P.; Baudet, C.; Truffinet, V.; Magnone, V.; Guillaudeau, A.; Cogné, M.; Dubus, P.; Denizot, Y. A defect of the INK4-Cdk4 checkpoint and Myc collaborate in blastoid mantle cell lymphomalike lymphoma formation in mice. Am. J. Pathol. 2012, 180, 1688–1701. [Google Scholar] [CrossRef]
- Haas, K.; Staller, P.; Geisen, C.; Bartek, J.; Eilers, M.; Möröy, T. Mutual requirement of CDK4 and Myc in malignant transformation: Evidence for cyclin D1/CDK4 and p16(INK4A) as upstream regulators of Myc. Oncogene 1997, 15, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; Grau, M.; Juilland, M.; Klener, P.; Höring, E.; Molinsky, J.; Schimmack, G.; Aukema, S.M.; Hoster, E.; Vogt, N.; et al. B-cell receptor–driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood 2017, 129, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Saba, N.S.; Liu, D.; Herman, S.E.M.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood 2016, 128, 82–92. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Expression and role of MYC in B lymphocyte differentiation. Schematic representation of the participation of the MYC protein throughout B-cell differentiation in the bone marrow and germinal center (GC). The percentages shown refer to the population of MYC+, BCL6+/− cells in the total number of B cells present in the GC. The blue-colored line at the top of the Figure indicates the evolution of MYC expression, where darker blue indicates steps that require higher MYC levels.
Figure 1.
Expression and role of MYC in B lymphocyte differentiation. Schematic representation of the participation of the MYC protein throughout B-cell differentiation in the bone marrow and germinal center (GC). The percentages shown refer to the population of MYC+, BCL6+/− cells in the total number of B cells present in the GC. The blue-colored line at the top of the Figure indicates the evolution of MYC expression, where darker blue indicates steps that require higher MYC levels.

Figure 2.
Activating mechanisms of c-MYC in leukemia with the BCR-ABL1 rearrangement. A summary of the different transduction signaling pathways that trigger the activation of MYC promoter in BCR-ABL1-rearranged leukemia. Apart from direct transcriptional activation pathways, marked in green, alternative mechanisms that induce c-MYC are depicted in black and highlighted in black squares. Dashed arrows indicate the translocation of proteins between the nucleus and the cytoplasm.
Figure 2.
Activating mechanisms of c-MYC in leukemia with the BCR-ABL1 rearrangement. A summary of the different transduction signaling pathways that trigger the activation of MYC promoter in BCR-ABL1-rearranged leukemia. Apart from direct transcriptional activation pathways, marked in green, alternative mechanisms that induce c-MYC are depicted in black and highlighted in black squares. Dashed arrows indicate the translocation of proteins between the nucleus and the cytoplasm.


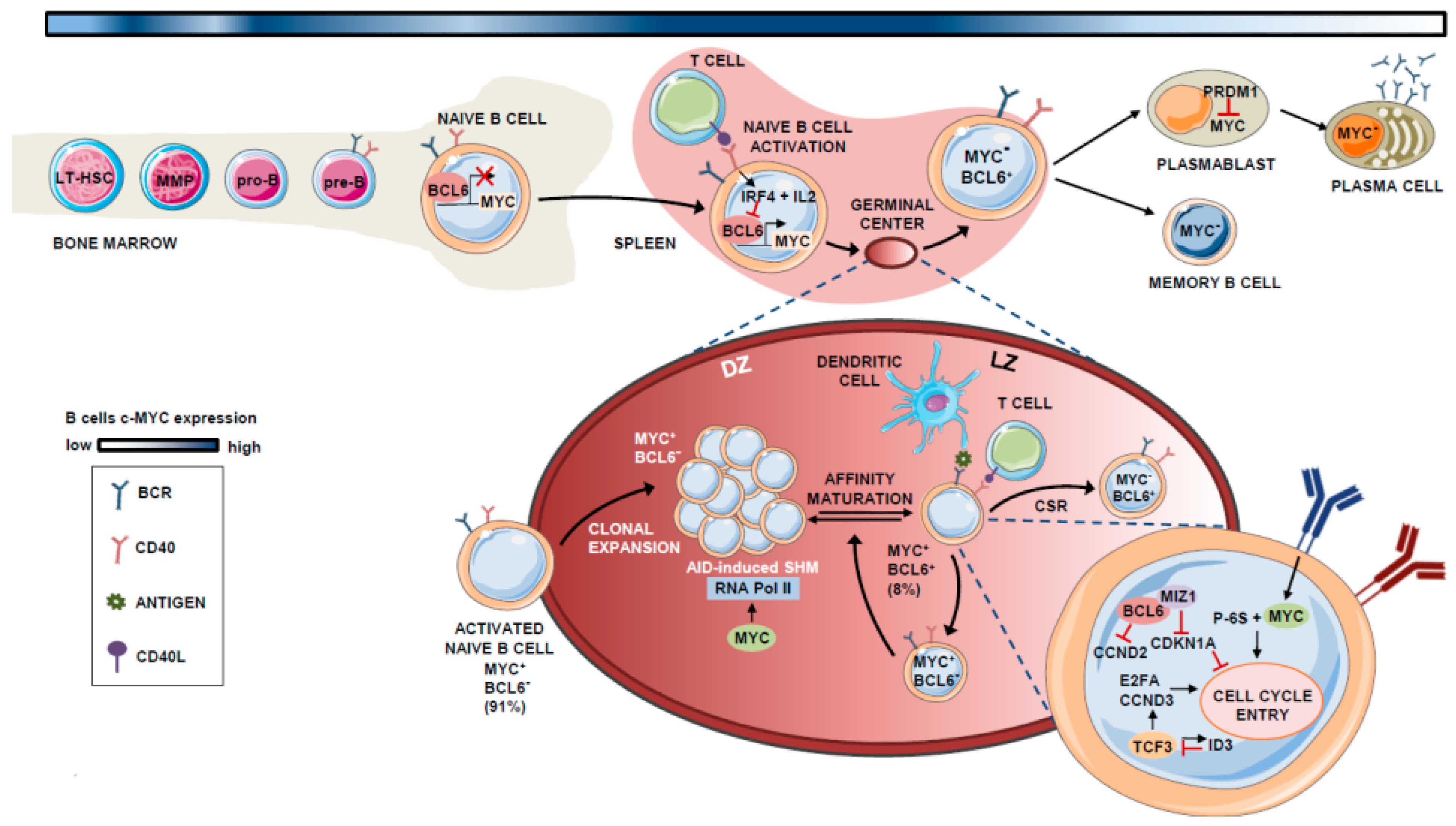{kind=link}
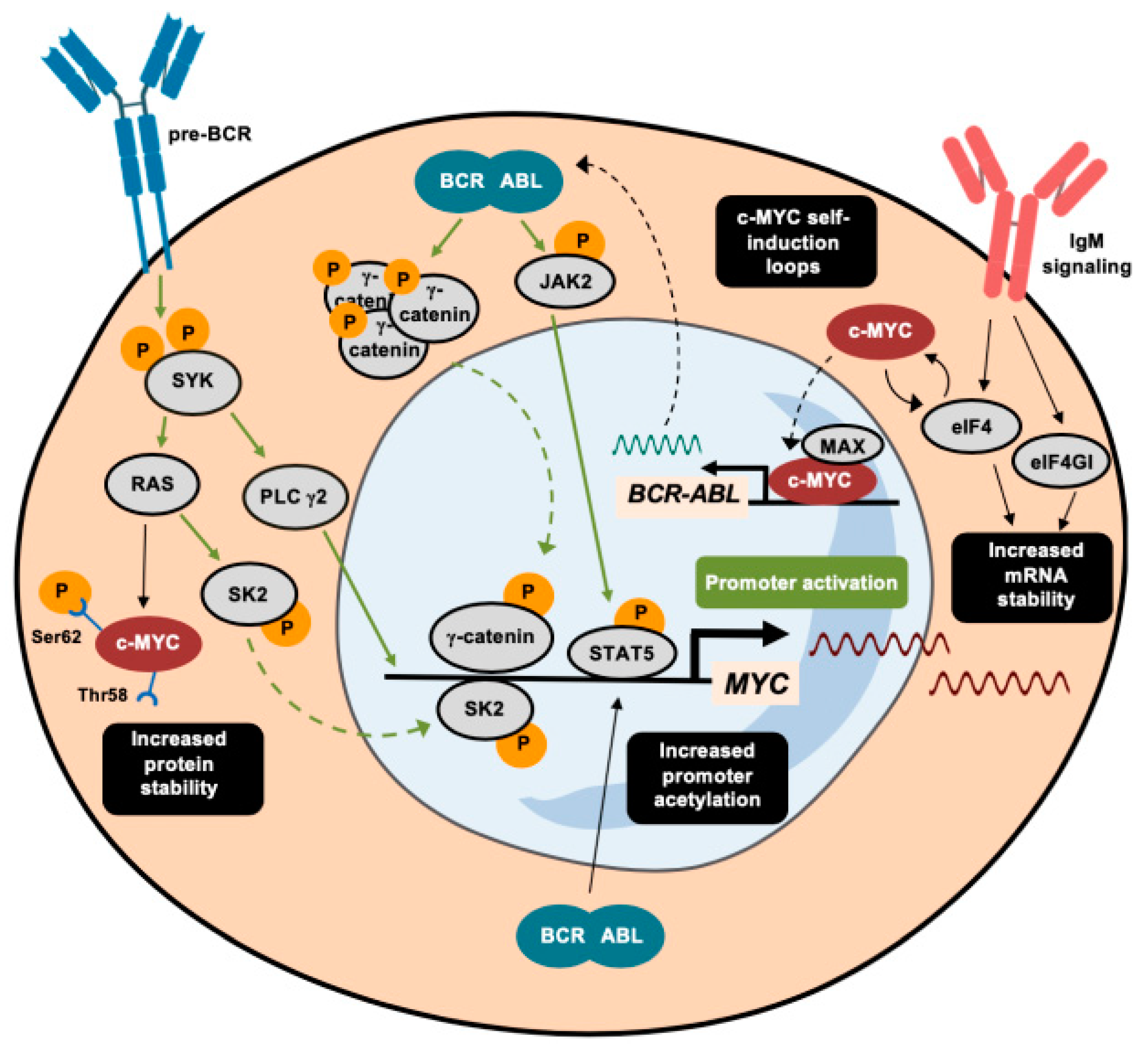{kind=link}
Table 1.
MYC alterations in leukemia and lymphoma. Summary of the gene alterations, chromosomal translocations, regulatory pathways, and post-transcriptional modifications involved in MYC activation that are included in this review, assigned to their corresponding subtype of leukemia and lymphoma.
Table 1.
MYC alterations in leukemia and lymphoma. Summary of the gene alterations, chromosomal translocations, regulatory pathways, and post-transcriptional modifications involved in MYC activation that are included in this review, assigned to their corresponding subtype of leukemia and lymphoma.
| Gene Alterations (Mutations, CNV) | Involvement of Translocations | Activating Pathways | Myc Stabilization at Post-Transcriptional Level | ||
|---|---|---|---|---|---|
| LEUKEMIA | t(9;22) BCR/ABL1 rearrangement | Almost negligible mutation rate [46] | MYC/IGH translocation is uncommon [50]. Higher frequency in CDKN2 WT patients [51] | Induced by aberrant AID [47], pre-BCR/SYK signaling [53], Wnt [58] and JAK/STAT [60,61] pathways | Translation rate controlled by EIFs [62,63,64,65], prevention of proteasomal degradation [67,68,69], induction mediated by MIR17HG cluster [74,75] |
| t(v;11) KMT2A rearrangement | i(8q) MYC duplication in RS4;11 cell line [79] | MYC is not commonly involved in translocations. Only rare cases with t(8;22) reported [77] | Indirect activation through TP53 inhibition [84,85], direct binding of MLL-fusion proteins and BET adaptors [86,87,88], regulation by BENC super-enhancer region [99,100] | ||
| t(12;21) ETV6/RUNX1 rearrangement | MYC gene expression gain 8(q23.1-24.1). CNVs reported in 65% of cases [107] | Double MYC gene translocation reported [106] | ETV6 also fuses to PAX5 and induces MYC expression [104]. Also triggered by RAC1-STAT3 [110] | Stabilized at mRNA level by IGF2BP1 [111] and at the protein level by prevention of proteasomal degradation [69] | |
| LYMPHOMA | Burkitt lymphoma | Point SNPs and deletions in the 3’border [125,126] | MYC/IGH t(8;14q32) in 80% of the cases and MYC/IGκ or Igλ t(8;2p12) or t(8;22q11) in 10% of the diagnoses [123,124] | BCR-induced PI3K pathway in cooperation with TCF3, ID3 and SHP-1 [129,130,131,132,133,134] | |
| Diffuse large B cell Lymphoma | Point SNPs, amplifications, gains and increased copy numbers of MYC [148,149] | Found in 10% of the cases, being MYC/IGL the most common [144], but also to BCL6, BCL2, PAX5, IKAROS [145,146,147]. Often participate in complex karyotypes and associatedo a second hit such as MYC/BCL2 and MYC/BCL6 [151,152] | Mutations in the BCR or PI3K pathway inhibitory elements [156,157] | GSK-3β phosphorylation abolishing MYC degradation [155] MYC upregulation promotes BCR signaling by induction of MIR17HG cluster [158,159] | |
| Plasmablastic lymphoma | Gains [163,164] and somatic mutations in MYC inhibitor PRDM1 [42,162] | Observed in 50% of the cases, being MYC/IGH the most common [163] | |||
| Follicular lymphoma | Remains unaltered in terms of copy number [173] | Second most common tFL-specific lesion [46] | Proliferation signature with oncogenic transformations such as TP53 mutations, CDKN2A loss and c-REL amplification [171] | ||
| Mantle cell lymphoma | Disruption of MYC locus in t(2;8) and MYC gains at (8)(q24) [179,180] | Coexistence of CCND1/IGH and MYC rearrangements [176,177], and also MYC/IGH t(8;14) [179,180] | CCND1/CDK4 and p16INK4a imbalance [183,184,185] | BCR-driven CARD11-BCL10-MALT1 complex together with Nf-κβ pathway orchestrates MYC stability [186,187] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
de Barrios, O.; Meler, A.; Parra, M. MYC’s Fine Line Between B Cell Development and Malignancy. Cells 2020, 9, 523. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020523
AMA Style
de Barrios O, Meler A, Parra M. MYC’s Fine Line Between B Cell Development and Malignancy. Cells. 2020; 9(2):523. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020523
Chicago/Turabian Stylede Barrios, Oriol, Ainara Meler, and Maribel Parra. 2020. "MYC’s Fine Line Between B Cell Development and Malignancy" Cells 9, no. 2: 523. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020523
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.