Mitophagy in the Pathogenesis of Liver Diseases †


1
Department of Biochemistry & Molecular Biology and Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan 33302, Taiwan
2
Liver Research Center, Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan
3
Division of Allergy, Immunology, and Rheumatology, Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan
†
Running Title: Mitophagy and Liver Diseases.
Cells 2020, 9(4), 831; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040831
Submission received: 3 March 2020
/
Revised: 25 March 2020
/
Accepted: 27 March 2020
/
Published: 30 March 2020
(This article belongs to the Special Issue Highlights in Autophagy: From Basic Mechanisms to Human Disorder Treatments)
Abstract
:Autophagy is a catabolic process involving vacuolar sequestration of intracellular components and their targeting to lysosomes for degradation, thus supporting nutrient recycling and energy regeneration. Accumulating evidence indicates that in addition to being a bulk, nonselective degradation mechanism, autophagy may selectively eliminate damaged mitochondria to promote mitochondrial turnover, a process termed “mitophagy”. Mitophagy sequesters dysfunctional mitochondria via ubiquitination and cargo receptor recognition and has emerged as an important event in the regulation of liver physiology. Recent studies have shown that mitophagy may participate in the pathogenesis of various liver diseases, such as liver injury, liver steatosis/fatty liver disease, hepatocellular carcinoma, viral hepatitis, and hepatic fibrosis. This review summarizes the current knowledge on the molecular regulations and functions of mitophagy in liver physiology and the roles of mitophagy in the development of liver-related diseases. Furthermore, the therapeutic implications of targeting hepatic mitophagy to design a new strategy to cure liver diseases are discussed.
Keywords:
autophagy; mitophagy; liver disease; liver injury; hepatitis; steatosis; fibrosis; hepatocellular carcinoma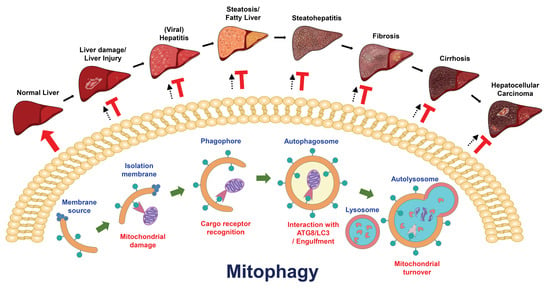
1. Introduction
Autophagy is a catabolic process that eliminates unwanted intracellular materials via lysosomal degradation to supply nutrients and energy for the maintenance of cellular homeostasis [1,2]. Autophagy is often activated in cells to counteract a variety of stresses, including nutrient starvation, organelle damage, protein misfolding and aggregation, and pathogen infection [3,4]. Loss of control of autophagy regulation is involved in the development of numerous human diseases, such as cancer, metabolic dysfunction, neurodegenerative disorders, and liver-associated diseases [1,2]. Despite its bulk and nonselective degradation mechanism, autophagy has been demonstrated to selectively eliminate damaged organelles to promote organelle regeneration, a process known as “organellophagy” [3,4,5]. Mitophagy is a specific kind of organellophagy responsible for the clearance of damaged mitochondria and thus promotes mitochondrial turnover [6,7]. Improper regulation of mitophagy is implied to participate in the pathogenesis of age-related neurodegenerative and cardiovascular diseases [8,9], metabolic syndrome [10], and tissue injury [11,12]. Very recently, mitophagy was shown to function in the maintenance of hepatic function and to protect the liver from tissue damage via the removal of damaged mitochondria. In addition, deregulation of mitophagy is also implicated in the development of liver-associated diseases, including liver injury, liver steatosis/fatty liver disease, viral hepatitis, hepatic fibrosis, and liver cancer. Thus, a comprehensive understanding of the functional roles of mitophagy will provide an opportunity to identify a new mitophagy modulation-related therapeutic target for the development of a novel and effective strategy for curing and intervening with liver disease. In this review, I summarize the current knowledge on the functional role(s) of mitophagy in the regulation of liver physiology and provide an overview of how mitophagy is altered to prevent and/or promote the development and pathogenesis of liver diseases. Furthermore, I discuss the potential implications of therapeutically targeting hepatic mitophagy for the clinical treatment of liver diseases.
2. Autophagy
2.1. Discovery of Autophagy and Identification of Autophagy-Related Genes (ATGs)
Autophagy is a “self-eating” process; its name is derived from the Greek words “auto” (self) and “phagy” (eating). During its discovery, autophagy was morphologically characterized by transmission electron microscopy (TEM)-based analysis of double- and single-membraned, vesicle-like, dense structures that sequestered mitochondria and fragments of the endoplasmic reticulum (ER) membrane in different animal tissues [13,14,15,16]. Later observations showed that these dense bodies were related to lysosomal degradation, prompting the discovery of a new cell-autonomous destruction process, which was coined “autophagy” by Christine de Duve, the 1974 Nobel Laureate in Physiology or Medicine, at the Ciba Symposium on Lysosomes in 1963 [17,18]. During the 1970s and 1980s, several stimuli, including hormones and amino acid deprivation, were demonstrated to induce autophagy [19,20,21,22,23,24]. In addition, the signal transduction pathways mediating autophagic processes and pharmacological inhibitors of autophagy, such as 3-methyladenine (3-MA), were characterized and identified [24,25,26,27,28,29,30,31]. Shortly thereafter, in the early 1990s, the concept of the double-membrane structure of autophagic vacuoles derived from the membranes of intracellular organelles, such as endosomes and peroxisomes, was developed [32,33,34,35,36,37,38,39]. The molecular mechanism responsible for autophagy regulation was first unveiled by the isolation and molecular cloning of ATGs by Yoshinori Ohsumi, the 2016 Nobel Laureate in Physiology or Medicine. Ohsumi et al. employed a genetic screen of temperature-sensitive mutants of Saccharomyces cerevisiae that were deficient in autophagic degradation and identified approximately 15 ATGs involved in the autophagic process of Saccharomyces cerevisiae [40,41,42]. Subsequently, approximately 40 ATGs with homologous functions in autophagy regulation in other eukaryotes and mammals were identified and characterized [43,44,45,46,47,48,49] and were further unified by the autophagy research community [47,48,49].
2.2. Three Major Types of Autophagy
To date, three major types of autophagy, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), have been identified [50,51]. Among these types, macroautophagy (hereafter referred to as autophagy), which involves a membrane rearrangement process to sequester cytosolic components in autophagic vacuoles and deliver them to lysosomes for degradation, is the most well characterized [51,52]. Various stresses, including nutrient starvation, accumulation of damaged organelles or aggregated proteins, and pathogen infection, have been shown to induce autophagy to eliminate harmful components in cells and maintain cellular homeostasis; thus, autophagy serves as a guardian of human health [53,54]. Accordingly, improper alteration of autophagy has been demonstrated to participate in the pathogenesis of various human diseases and biological processes, such as tumorigenesis, neurodegenerative disorders, infectious diseases, cardiovascular diseases, metabolic syndrome, and aging [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. Microautophagy is an engulfment process that randomly and/or selectively delivers intracellular materials into the lysosomal lumen for degradation through the rearrangement and invagination of the lysosomal membrane into the lumen [72,73,74]. In addition to core ATGs in the autophagic process, the endosomal sorting complexes required for transport (ESCRT) machinery was recently shown to function in the membrane protrusion and scission processes of microautophagy [75,76,77,78]. To date, the molecular process that regulates microautophagy and the physiological importance of microautophagy to human health remain unclear. CMA proceeds through a selective sequestration process involving the recognition of degradative substrates containing the pentapeptide “Lys-Phe-Glu-Arg-Gln″ (KFERQ) motif by a molecular chaperone, heat shock cognate protein of 70 kDa (HSC70), and translocation of these substrates into the lumen of lysosomes through the docking of lysosomal membrane protein 2A (LAMP2A) onto the lysosomal membrane [79,80]. CMA has been shown to be activated by various stimuli, such as nutrient deprivation, metabolic imbalance, oxidative stress, and genotoxicity [81,82,83,84,85,86], and is required for biological processes ranging from energy production, lipid metabolism, gene regulation, immune response control, and cell cycle regulation to aging [82,86,87,88,89,90,91,92,93,94,95,96,97]. Unsurprisingly, deregulation of CMA has also been suggested to contribute to the development of multiple kinds of human diseases [98,99,100,101,102,103,104,105,106,107,108,109,110].
2.3. Functional ATGs in the Regulation of Autophagic Process
2.3.1. Membrane Nucleation and Phagophore Formation
The entire process of autophagy relies on the stepwise biogenesis of vacuoles that begins with rearrangement of the membrane for nucleation of the isolation membrane (IM)/phagophore [111,112,113,114]. Different intracellular organelles, such as the ER [115,116], Golgi apparatus [117], mitochondria [118], plasma membrane [119], recycling endosome [120,121], and mitochondria-associated ER membrane [122], supply the membrane source for reconstituting the membranous structure of the IM/phagophore. The cup-shaped IM/phagophore then elongates and matures into a double-membrane autophagosome [123,124,125,126], which subsequently fuses with a lysosome, forming an autolysosome in which the enclosed materials are degraded by lysosomal proteases [125,127,128,129]. In addition, the core ATG complexes and concerted actions of signaling cascades are required for the maturation of autophagic vacuoles (Figure 1) [52,130,131]. In eukaryotic cells, nutrient deprivation often suppresses the activity of mammalian target of rapamycin (mTOR), a serine/threonine protein kinase that regulates cellular metabolism (Figure 1) [132,133]. Suppression of mTOR leads to translocation of the unc-51 like-kinase (ULK) complex, which contains ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101, from the cytoplasm to a membrane-enclosed compartment derived from the ER (Figure 1) [134,135]. Subsequently, the translocated ULK complex recruits the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex (which contains Vps34/PI3KC3, Vps15, Beclin 1, and ATG14) to the nucleated domain from the ER membrane and activates the class III PI(3)K complex via phosphorylation (Figure 1) [131,136,137,138]. In turn, activation of the class III (PI(3)K) complex leads to the generation of phosphatidylinositol-3-phosphate (PtdIns(3)P) [131,136,137], which induces the recruitment of double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI, the mammalian orthologue of ATG18) family proteins to trigger the reconstitution of an ER-organized omegasome structure (also termed the IM/phagophore) (Figure 1) [131,136,137,139,140]. Moreover, ATG9-mediated vesicle trafficking from the trans-Golgi network (TGN) to the ER and the interaction of ER membrane-bound vacuole membrane protein 1 (VMP1) with the class III PI(3)K complex supply the lipid constituents required for the formation of autophagic vacuoles [141,142,143] and promote the nucleation of the IM/phagophore (Figure 1) [144,145,146].
2.3.2. The Biogenesis of Autophagosome
Two ubiquitin-like (UBL) conjugation cascades are required for the elongation and closure of the IM/phagophore into a mature autophagosome (Figure 1) [147,148,149,150]. The enzymatic activities of ATG7 (E1) and ATG10 (E2) induce the formation of the ATG5-ATG12 conjugate, which further associates with ATG16L to form an ATG5-ATG12-ATG16L trimeric complex (Figure 1) [147,148,151,152,153]. On the other hand, the C-terminal region of ATG8 family proteins (including the microtubule-associated protein 1 light chain 3 [LC3] and gamma-aminobutyric acid receptor-associated protein [GABARAP] subfamilies) are cleaved by ATG4 family proteases to generate ATG8/LC3-I [154,155]. Subsequently, the ATG7 (E1) and ATG3 (E2) enzyme cascade promotes the lipidation (referred to as the conjugation of phosphatidylethanolamine [PE]) of ATG8/LC3-I to form lipidated ATG8/LC3, termed “ATG8-LC3-II” (or lipidated ATG8-LC3) (Figure 1) [154,155,156]. Then, lipidated ATG8/LC3 and the ATG5-ATG12-ATG16L complex cooperate to accomplish autophagosome maturation by promoting the elongation of autophagosomal membranes [157], supporting tethering and membrane fusion between autophagic vacuoles [150,158], and serving as an E3 ubiquitin ligase-like enzyme to promote the formation of lipidated ATG8/LC3 [158,159]. According to recent studies, in addition to these two UBL conjugation complexes, the ESCRT machinery also participates in the closure of IM/phagophore to form autophagosomes [160,161,162].
2.3.3. The Fusion of Autophagosome with Lysosome to Form Autolysosome
Ultimately, an autophagosome fuses with a lysosome, forming an autolysosome that contains acidic proteases to degrade the interior materials. The maturation of autolysosomes requires several biological processes, including cytoskeleton-mediated vesicle trafficking and membrane fusion processes (Figure 1) [123,126,127,129,163,164,165]. The interaction of the autophagosomal membrane-associated small GTPase Ras-related protein 7 (Rab7) with kinesin- and dynactin-linked FYVE and coiled-coil domain-containing 1 (FYCO1) and Rab-interacting lysosomal protein (RILP) allows the transport of autophagosomes along microtubules, facilitating autophagosome-lysosome fusion (Figure 1) [166,167,168,169,170]. On the other hand, histone deacetylase 6 (HDAC6)-triggered remodeling and assembly of the actin cytoskeleton also functions in autolysosome maturation [171,172]. Endosome- and lysosome-associated Rab7 also function in the fusion process between autophagosomal and lysosomal membranes by recruiting pleckstrin homology domain-containing protein family member 1 (PLEKHM1) and the homotypic fusion and protein sorting (HOPS) complex (Figure 1). The interaction between the ATG8/LC3-interacting motif within PLEKHM1 and ATG8/LC3 occurs on the autophagosomal membrane, and the binding of PLEKHM1 to HOPS and the Rab7 complex links autophagosome to the lysosome for fusion [173]. Very recently, this endosomal conversion of phosphatidylinositol-4-phosphate (PtdIns(4)P) to phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) was shown to promote the disassociation of Rab7 and the release of PLEKHM1 from endosomes for autophagosome-lysosome fusion [174]. In addition, the association of the HOPS complex with PI(3)K-associated UV radiation resistance associated (UVRAG) was shown to activate Rab7 and coordinate autophagosome-lysosome fusion (Figure 1) [175]. Moreover, the association of Rubicon and ATG14L with the Beclin 1- PI(3)K complex was indicated to regulate autolysosome maturation [137,176]. Alternatively, the protein complex containing ATG14L, syntaxin 17 (STX17), synaptosome-associated protein 29 (SNAP29), and vesicle-associated membrane protein 8 (VAMP8) was shown to promote the tethering and fusion of membranes for autophagosome-lysosome fusion (Figure 1) [177,178]. In addition to VAMP8, the preferential binding of STX17 to VAMP7A, not VAMP7B, through the regulation of divergent protein kinase domain 2A (DIPK2A) was indicated to control autophagosome-lysosome fusion [179]. Interestingly, the 20S proteasome was recently shown to degrade SNAP29 and STX17 in a ubiquitin-independent manner to control autophagosome-lysosome fusion [180]. ATG8/LC3 proteins were recently shown to play a critical role in autophagosome-lysosome fusion in addition to activating the initial maturation of autophagosomes [181]. Recent studies have demonstrated that Golgi reassembly stacking protein 2 (GORASP2) may promote autophagosome-lysosome fusion by mediating the interaction between LC3 on autophagosomes and LAMP2 on lysosomes [182]. When the autolysosome forms, the encompassed components are degraded by lysosomal proteases in low-pH compartments to support nutrient recycling and energy production.
2.3.4. The Termination of Autophagy
Information regarding the mechanism by which autophagy is terminated and lysosomes are regenerated is still limited. The current model shows that replenishment of nutrients can reactivate mTOR to suppress the induction of autophagy and, coincidently, promote autophagic lysosome reformation (ALR), which terminates the autophagic process [183]. Several other molecules, including clathrin and PtdIns(4,5)P2 [184,185], kinesin 1 [186,187,188], and Spinster [189], have been reported to function in ALR. A recent study also implied that the Cullin 3-Kelch-like protein 20 (KLHL20) E3 ubiquitin ligase promotes the degradation of ULK1 and Vps34 complexes to terminate the autophagic process [190].
3. Selective Autophagy and Organellophagy
3.1. Cargo Recognition in Selective Autophagy
In the past decade, autophagy has no longer been considered only as a nonselective, bulk degradation process. In contrast, autophagy represents a new and specific route to selectively eliminate sequestered organelles and proteins (referred to as cargoes), a process termed “selective autophagy” [3,4,5,191]. Selective autophagy was first observed in the degradation of β-granules in rat pancreatic tissue in the early 1970s [192,193], and the underlying molecular mechanism and regulatory genes were initially delineated and identified in the late 2000s [3,4,5,191]. Before being targeted for degradation by selective autophagy, cargoes are often tagged by polyubiquitination or the binding of an adaptor protein and further recognized by specific cargo receptors, which then deliver the targeted proteins for autophagy by interacting with ATG8/LC3 on the autophagosomal membrane [191,194,195,196,197,198,199]. These cargo receptors, including p62/sequestosome 1 (p62/SQSTM1), neighbor of BRCA1 gene 1 (NBR1), calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2, or NDP52), optineurin (OPTN), and Tax1-binding protein 1 (TAX1BP1) often contain LC3-interacting regions (LIRs) for binding to ATG8/LC3 family proteins and ubiquitin-associated domains for recognizing the ubiquitinated cargoes [191,196,197,199]. Recent studies have identified that in addition to LIR-containing cargo receptors, the ATG8-interacting motifs (AIMs), GABARAP-interacting motifs (GIMs), and ubiquitin-interacting motifs (UIMs) found in several ATGs and other cellular proteins may potentially control the degradation process of selective autophagy [191,198,200,201,202,203,204,205].
3.2. Quality Control of Organelle Biogenesis by Selective Autophagy
Selective autophagy has been found to play a functional role in the turnover of damaged organelles, termed “organellophagy” [3,5,206], which protects cells from the consequences of organelle damage and supports the recycling of constituents for the regeneration of the ER, mitochondria, lipid droplets (LDs), peroxisomes, ribosomes, lysosomes, and nuclei (Figure 2) [3,5,206]. To successfully eliminate harmful organelles, several cargo receptors participate in the targeting of damaged organelles for the degradation process of organellophagy [3,5,206]. During mitochondrial turnover by organellophagy (referred to as mitophagy) [6,207], mitochondrial oxidation and depolarization leads to mitochondrial damage [208,209,210,211,212,213,214,215], in turn triggering polyubiquitination of mitochondrial proteins on the outer mitochondrial membrane by the E3 ubiquitin ligase Parkin [213,214,215,216,217,218,219]. Subsequently, Calcoco2/NDP52 and OPTN are recruited for the removal of mitochondria by selective autophagy (Figure 2) [6,220]. In addition to Parkin, other mitochondrial outer membrane (MOM) proteins, such as FUN14 domain-containing 1 (FUNDC1), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L, or Nix), yeast ATG32, and autophagy/beclin-1-regulator-1 (AMBRA1), also participate in the mitophagy process in a ubiquitin-independent manner (Figure 2) [221,222,223,224,225,226]. Recently, novel cargo receptors for mitophagy, such as prohibitin 2 (PHB2), Toll-interacting protein (Tollip), nitrophenylphosphatase domain and nonneuronal SNAP25-like protein homolog (NIPSNAP) family proteins, and the adenine nucleotide translocator (ANT) complex (Figure 2) [227,228,229,230], were identified. Finally, cargo receptors for mitophagy recruit components of the core autophagy initiation complex, including ULK1, DFCP1 and WIPI/ATG18 family proteins, for autophagosome maturation near the damaged mitochondria [220,231], which involves PtdIns(4,5)P2- and F-actin-mediated disassembly of mitoaggregates [232].
For the degradation of damaged peroxisomes by organellophagy (referred to as pexophagy), two protein kinases, yeast Hrr25 and mammalian ataxia-telangiectasia-mutated (ATM), phosphorylate ATG36 in yeast and NBR1 and p62/SQSTM1 in mammals, respectively [233,234,235,236], thus promoting the ability of these mediators to regulate peroxisome turnover by pexophagy (Figure 2). Recent studies demonstrated that the ubiquitination of peroxisomal (PEX) membrane proteins, including PEX5 and the 70-kDa PEX membrane protein (PMP70), may enhance the targeting of damaged peroxisomes by cargo receptors [237,238]. The targeting of damaged ER components for organellophagy, called ER-phagy, requires the functions of several ATGs in yeast, such as ATG39, ATG11, and ATG40 [239], and cargo receptors, including ER-associated reticulon family proteins and family with sequence similarity 134, member B (FAM134B) in mammals (Figure 2) [240,241]. Intriguingly, ATG39 and ATG11 were shown to not only function in ER-phagy but also to regulate nuclear organellophagy, termed “nucleophagy″, to promote the recycling of nuclear components in yeast (Figure 2) [239,242]. Lysophagy is a form of organellophagy that degrades injured lysosomes through the interaction between galectin-3 and LC3 and the p62/SQSTM1-mediated recognition process (Figure 2) [243,244]. Analogously, the removal of other intracellular organelles, including ribosomes (ribophagy) [245,246] and LDs (lipophagy) [247,248], by organellophagy has been shown to regulate cellular homeostasis. However, the specific cargo receptors for organellophagy have not yet been comprehensively identified, the molecular mechanism underlying the regulation of organellophagy is not completely unveiled, and the physiological significance of organellophagy in human health remains to be further studied.
3.3. Elimination of Protein Aggregates and Infecting Pathogens by Selective Autophagy
In addition to mediating organelle turnover via organellophagy, selective autophagy also participates in the clearance of protein aggregates and invading pathogens (referred to as aggrephagy and xenophagy, respectively) (Figure 2). For the degradation of protein aggregates by aggrephagy, the p62/SQSTM1 and HDAC6-mediated recruitment of Lys63 (K63)-linked protein ubiquitination determines the specificity of degradative protein aggregates [171,249,250,251]. In addition, NBR1 and autophagy-linked FYVE (ALFY), two cargo receptors, associate with p62/SQSTM1 to target protein aggregates for clearance by aggrephagy (Figure 2) [252,253,254,255]. In addition to removing protein aggregates by aggrephagy, selective autophagy was also found to target specific proteins for degradation. For instance, ferritin heavy and light chains have been shown to be specifically bound by an ATG8/LC3-interacting protein, nuclear receptor coactivator 4 (NCOA4), and targeted for autophagic degradation in a process known as “ferritinophagy″, to precisely maintain the intracellular pool of iron (Figure 2) [256,257] and function in the regulation of erythropoiesis and DNA replication in blood cells [258,259]. To eliminate invading microbes by xenophagy [260,261,262], several cargo receptors, including p62/SQSTM1, NDP52, and OPTN, recognize infecting pathogens and deliver them for degradation (Figure 2) [67,263,264]. Protein kinase-mediated phosphorylation of p62/SQSTM1 and OPTN also serves as a signal for the activation of xenophagy [264,265,266,267]. In summary, selective autophagy plays a critical role in the specific degradation of harmful components to regulate organelle integrity, metabolic homeostasis, and the defense against pathogen infection.
4. Mitophagy
4.1. Mitochondrial Turnover via Mitophagy
Mitochondria are intracellular factories that not only generate energy in the form of adenosine triphosphate (ATP) via oxidative phosphorylation but also maintain cellular homeostasis by promoting the anabolism of macromolecules and catabolizing metabolic waste [268]. Mitochondria are intracellular organelles that contain the MOM, the mitochondrial inner membrane (MIM), the intermembrane space, and the matrix, which collectively regulate bioenergetics, biosynthesis, and signal transduction [269]. Mitochondria are highly dynamic organelles that undergo cycles of fusion and fission (known as mitochondrial dynamics) to regulate their reshaping, rebuilding, redistribution, and recycling of constituents, thus supporting mitochondrial mass and integrity [269,270]. To control mitochondrial quality in stressed cells, the evolutionarily conserved degradative process of mitophagy is invoked to remove damaged and dysfunctional mitochondria in order to regenerate mitochondria and preserve energy production [6,271].
Current knowledge suggests that mitophagy can be classified into three types according to its physiological roles in cells: basal mitophagy, programmed mitophagy, and stress-induced mitophagy (Figure 3). Less is known about how basal mitophagy (also termed steady-state mitophagy) is regulated in a physiological cellular context, since mitophagy is typically studied and measured under chemical-induced conditions. The observation and monitoring of basal autophagy was instituted by the establishment of mitophagy reporter mice [272,273,274], which are a useful tool for the detection of mitophagy in vivo. These studies in mitophagy reporter mice not only allow us to understand the extent to which mitophagy is differentially activated in various kinds of tissues to routinely regulate mitochondrial regeneration but also suggest that basal mitophagy occurs in tissues to support the metabolic demand [272,273,274,275]. Programmed mitophagy has been shown to be induced to regulate the determination of cell fates, such as erythrocyte differentiation [276,277], cardiomyocyte maturation [278], and stem cell pluripotency [279,280]. In addition, programmed mitophagy was also found to eliminate sperm mitochondria to avoid parental mitochondrial DNA (mtDNA) inheritance [281,282,283,284,285]. Several external stimuli, such as hypoxia, nutrient deprivation, and mitochondrial uncoupling, have been demonstrated to trigger stress-induced mitophagy to promote the clearance of dysfunctional mitochondria [209,224,286,287,288,289]. Deregulation of mitophagy impairs the new synthesis of healthy mitochondria and leads to the accumulation of defective mitochondria, which has been implicated in the pathogenesis of a wide spectrum of human diseases, such as cancer [290,291], neurodegenerative diseases [292,293], cardiovascular diseases [8], tissue injury [11,294,295], metabolic disorders [296], and autoimmune diseases [297]. Thus, modulation of mitophagy has emerged as a potential target for developing new treatment strategies for human diseases.
4.2. PINK1/Parkin-Dependent and PINK1/Parkin-Independent Mitophagy
The PTEN-induced putative kinase 1 (PINK1)-dependent protein phosphorylation and Parkin E3 ubiquitin ligase-mediated protein ubiquitination pathway is the most well-studied signaling cascade that regulates mitophagy (Figure 3) [213,214,215,216,298]. Mutations in PINK1 and Parkin have been shown to be associated with mitochondrial dysfunction in the development of neurodegenerative Parkinson’s disease [299,300,301]. Under normal conditions, translated PINK1 is immediately translocated into the IMM through MOM- and MIM-associated translocases; cleaved by protein proteases, matrix processing protease (MPP) and presenilin-associated rhomboid-like protein (PARL) [302,303,304]; and degraded by the E3 ubiquitin ligases UBR1, UBR2, and UBR4 through the N-end rule pathway [305]. When mitochondria are depolarized by external stimuli, impaired mitochondrial import interferes with proteolytic cleavage of PINK1 and suppresses the degradation of PINK [302,303]. Stabilization of PINK1 leads to its accumulation on the MOM and association with the translocase of the MOM (TOM) protein complex [306], thus triggering autophosphorylation and the phosphorylation of ubiquitin at serine (Ser)65 and of Parkin [217,218,298,307,308,309,310,311]. This phosphorylation in turn recruits the E3 ubiquitin ligase Parkin [213,214,215,216,217,218,306] and then promotes the ubiquitination of mitochondrial proteins on the MOM [213,214,215,216,219,306], leading to recognition by specific cargo receptors for the elimination of mitochondria by autophagy [6,213,214,215,216,220,298]. PINK1/Parkin-dependent mitophagy was shown to be required for mitochondrial uncoupler-induced mitophagy and programmed mitophagy [213,214,215,216,224,278,279,280,298,312,313].
In addition to PINK1/Parkin-mediated mitophagy, multiple layers of molecular mechanisms have been shown to regulate mitophagy. Several E3 ubiquitin ligases, such as mitochondrial E3 ubiquitin protein ligase 1 (MUL1) [282,314,315], SMAD ubiquitination regulatory factor 1 (SMURF1) [316], seven in absentia homolog 1 (SIAH1) [317], Glycoprotein 78 (Gp78) [318,319], Ariadne RBR E3 ubiquitin ligase homolog 1 (ARIH1) [320], and HECT, UBA and WWE domain-containing protein 1 (HUWE1) [226], may specifically ubiquitinate mitochondrial proteins to trigger mitophagy. Intriguingly, the PINK1/Parkin-dependent pathway is not required for the regulation of basal mitophagy [275,321].
4.3. Cargo Receptors and Activators of Mitophagy
Several cargo receptors have been identified to function in mitophagy regulation. ATG32, a mitochondria-anchored protein, was the first mitophagy receptor shown to interact with ATG8 and ATG11 and initiate the autophagic degradation of mitochondria to counteract oxidative stress in budding yeast (Figure 3) [287,288,322,323]. Very recently, the mammalian homolog of yeast ATG32, BCL2-like 3 (BCL2-L3), was also proven to function as a mitophagy receptor [324]. In mammals, the BCL2 family protein BNIP3L/Nix was demonstrated to function in mitochondrial turnover by interacting with ATG8/LC3 family proteins during erythrocyte maturation [222,276,277,325,326]. Similarly, BNIP3 was reported to act as a mitophagy receptor for the removal of mitochondria in cardiac myocytes by inducing mitochondrial permeability, cytochrome C release, and mitochondrial fission; interfering with mitochondrial bioenergetics; and binding to LC3 [223,327,328,329,330,331]. BNIP3 and BNIP3L/Nix are also involved in PINK1/Parkin-mediated mitophagy by suppressing the proteolytic cleavage of PINK1 and serving as ubiquitination substrates of Parkin [312,330,332].
p62/SQSTM1 was also shown to activate PINK1/Parkin-dependent mitophagy by bridging the interaction between ubiquitinated voltage-dependent anion-selective channel 1 (VDAC1) on degrading mitochondria and LC3 on autophagosomal membranes [333] and mediating the clustering of depolarized mitochondria [334,335]. In addition to p62/SQSTM1, NBR1 was indicated to be recruited to mitochondria by ubiquitinated BNIP3L/Nix to mediate PINK1/Parkin-dependent mitophagy [332]. FUNDC1 also serves as a cargo receptor for the regulation of mitochondrial dynamics and activation of mitophagy to eliminate mitochondria with hypoxia-induced dysfunction [224,336,337]. The MOM-associated mitofusin 2 (MFN2) is a mitophagy receptor for the quality control of cardiac mitochondria and metabolic maturation of the perinatal heart through phosphorylation by PINK1 and ubiquitination by Parkin [338,339]. OPTN, a causative gene of mitochondrial dysfunction-related amyotrophic lateral sclerosis (ALS) and glaucoma diseases, was shown to participate in PINK1/Parkin-mediated mitophagy by recruiting damaged mitochondria via its LIR [340,341,342]. Unlike wild-type OPTN, the ALS-associated mutant (with a mutation in the ubiquitin-associated domain [E478G]) and LIR mutant of OPTN cannot restore OPTN deficiency-impaired mitochondrial turnover, suggesting the detrimental role of OPTN deficiency in defective mitophagy-linked neurodegenerative diseases [340,342].
In addition to mitochondrial proteins located on the MOM, the MIM-associated protein PHB2 was shown to recruit autophagosomal membrane through its LIR domain for PINK/Parkin-dependent mitophagy [228,343]. A recent study showed that PHB2 can destabilize PARL and suppress its proteolytic activity, thus promoting the stability of PINK1 to activate mitophagy [344]. The Parkin-interacting protein AMBRA1 was shown to induce mitophagy by prolonging mitochondrial depolarization and triggering mitochondrial clearance—not by recruiting Parkin to damaged mitochondria [345]. In contrast, other studies indicated that AMBRA1 can drive mitophagy by a LIR motif-mediated interaction with LC3 on autophagosomes and that concerted localization of AMBRA1 is sufficient to activate mitophagy in a PINK1/Parkin-independent manner [346,347].
Recent studies have demonstrated that NIPSNAP1 and NIPSNAP2, two mitochondrial matrix proteins, can translocate to the outer surface of mitochondria and interact with ATG8/LC3 and other mitophagy receptors to play roles in the removal of depolarized mitochondria, which may prevent the occurrence of parkinsonism [230,348]. The ANT complex is another MIM-anchored protein that was recently shown to trigger mitophagy by stabilizing PINK1, and interference with ANT-mediated mitophagy may contribute to mitochondrial abnormalities during the development of cardiomyopathy [229]. The SNARE protein STX17 was similarly indicated to regulate the induction of PINK1/Parkin-dependent mitophagy by interfering with the dephosphorylation ability of a mitochondrial phosphatase, phosphoglycerate mutase family member 5 (PGAM5) [349], and by recruiting the core complex of the autophagy machinery through interaction with ATG14 [350]. Analogously, Rab GDP/GTP exchange factor 1 (RabGEF1)-directed translocation of Rab5 and Rab7 and phosphorylation of Rab7 were also shown to be required for the recruitment of ATG9 vesicles for mitophagy regulation [351,352]. Choline dehydrogenase (CHDH) was demonstrated to translocate to the MOM and form a complex with p62/SQSTM1 and LC3 to activate CCCP-induced Parkin-dependent mitophagy [353]. High-mobility group box 1 (HMGB1), a chromatin-associated protein functioning in nuclear homeostasis, has been reported to regulate mitophagy via the downstream heat shock protein beta-1 (HSPB1, also called HSP27) for mitochondrial quality control [354,355].
Although several cargo receptors are indicated to function redundantly in the induction of PINK1/Parkin-dependent mitophagy, recent studies by Youle et al. specifically clarified that NDP52 and OPTN are two detrimental receptors for PINK1/Parkin responsible for recruiting the core machineries of phagophore biogenesis and autophagosome biogenesis [220,356]. In addition, NDP52 was reported to interact with mitochondrial RNA poly(A) polymerase (MTPAP) to form a receptor complex to promote mitochondrial turnover [357]. Collectively, these studies imply that several kinds of cargo receptors function in multiple regulatory mechanisms to control mitophagy. However, new receptors and regulators of mitophagy are still being identified, and the molecular mechanisms underlying the control of mitophagy remain to be investigated and revised, particularly those controlling basal mitophagy and programmed mitophagy—two lesser-studied modes of mitophagy.
4.4. Posttranslational Modifications of Mitophagy Regulators
4.4.1. Phosphorylation and Dephosphorylation
Phosphorylation is the most well-studied posttranslational modification (PTM) of mitophagy regulators [358,359,360]. When mitochondria are depolarized and damaged, PINK1 phosphorylates Parkin at Ser65 to stimulate E3 ubiquitin ligase activity [307,308,311,361] and induces the phosphorylation of ubiquitin at Ser65 to establish a feed-forward amplification loop to both promote Parkin ubiquitination and translocation to mitochondria [217,218,307,311,362,363,364,365] and stabilize the assembly of the ubiquitin chain [366]. PINK1 also phosphorylates MFN2 (at threonine [Thr]111 and Ser442) and mitochondrial GTPase 1 (MIRO1) (at Thr298 and Thr299) to activate mitophagy [338,367,368,369]. PINK1-dependent phosphorylation is a positive regulator of mitophagy, and Tank-binding kinase 1 (TBK1) also functions in mitophagy activation by phosphorylating mitophagy receptors. TBK1 phosphorylates OPTN at Ser177 to promote its interaction with ATG8/LC3 proteins [370] and at Ser473 and Ser513 to confer it with the ability to bind to the ubiquitin chain [371,372,373]. In addition, TBK1-mediated autophosphorylation, p62/SQSTM1 phosphorylation at Ser403, Rab7 phosphorylation at Ser72, and other phosphorylation events have been shown to regulate mitophagy [266,374,375,376]. A recent study also noted that the Ca2+-binding protein TBC1 domain family member 9 (TBC1D9) may induce mitophagy by activating TBK1 [377]. Casein kinase 2 (CK2) is another kinase involved in the activation of mitophagy by phosphorylating FUNDC1 at Ser13 and phosphorylating translocase of MOM 22 (TOM22) at Ser15 and Thr43 [336,378,379]. On the other hand, adenosine monophosphate-activated protein kinase (AMPK) was demonstrated to phosphorylate ULK1 at Ser555 to promote mitophagy and cell survival under nutrient starvation conditions [380], suggesting a molecular mechanism coupling energy sensing and mitophagy. Additionally, the ULK1 complex was shown to be recruited to depolarized mitochondria [381], presumably through the interaction between FIP200 and NDP52 [220,231]. In addition, ULK1 mediates the phosphorylation of FUNDC1 at Ser17 to promote its interaction with LC3, which facilitates mitophagy induction [225]. Collectively, these studies suggest the promoting role of protein phosphorylation in mitophagy activation.
In contrast to phosphorylation, phosphatase-mediated dephosphorylation was demonstrated to antagonize mitophagy activation. The yeast protein phosphatase 2A-like protein 1 and mammalian PGAM5 were demonstrated to suppress mitophagy by dephosphorylating ATG32, FUNDC1 and dynamin-related protein 1 (DRP1) [349,379,382]. Similarly, phosphatase and tensin homolog (PTEN)-long (PTEN-L), an isoform of PTEN, was recently shown to translocate to the MOM and dephosphorylate Ser65 of ubiquitin to negatively regulate PINK1/Parkin-dependent mitophagy [383,384]. Reciprocally, the expression and mitochondrial localization of PGAM5 were recently shown to be regulated by the PHB2- PARL axis and STX17, respectively, to control mitophagy activation [344,349]. Collectively, these studies indicate that not only phosphorylation but also dephosphorylation represents an antagonistic approach to regulate mitophagy.
4.4.2. Ubiquitination and Deubiquitination
Ubiquitination is a molecular process involving a coordinated cascade of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases [385] and is reversibly regulated by deubiquitination through deubiquitinating enzymes (DUBs) [386]. Multiple types of E3 ubiquitin ligases promote the ubiquitination of mitochondrial proteins to tag damaged mitochondria in order to recruit the autophagic machinery for degradation [387]; these proteins include Parkin [365,371,388,389,390], MUL1 [282,314], SMURF1 [316], SIAH1 [317], Gp78 [318,319], ARIH1 [320], and HUWE1 [226,391]. Parkin-mediated heterotypic ubiquitin chains, such as lysine (K)6, K11, K48, and K63, on mitochondrial proteins of mitophagy are the most often decoded tags [365,371,387,388,389,390]. Several cytoplasmic and mitochondrial proteins have been shown to be ubiquitinated by Parkin during mitochondrial depolarization and to be involved in mitophagy regulation; these proteins include the MOM proteins MFN1, MFN2, TOM20, TOM70, and VDAC family proteins and the cargo receptors of selective autophagy p62/SQSTM1, NDP52, OPTN, and Tax1bp1 [365,371,388,389,390]. MUL1 participates in the ubiquitination of MFN in parallel to PINK1/Parkin-promoted mitochondrial turnover [314]. Additionally, MUL1 has been shown to ubiquitinate ULK1 to positively regulate selenite-induced mitophagy [315]. In addition, MUL1 plays a role redundant with that of Parkin in eliminating parental mitochondria for mouse embryo development [282]. The E3 ubiquitin ligase SMURF1 was reported to target damaged mitochondria for degradation, most likely through the recruitment of autophagosomal membrane to mitochondria via the membrane-binding ability of its C2 domain rather than via ubiquitin ligase activity [316]. SIAH1 was demonstrated to interact with synphilin-1 and translocate to mitochondria in order to promote mitochondrial ubiquitination to regulate PINK1/Parkin-dependent mitophagy [317]; however, the specific substrate that can be ubiquitinated by SIAH1 is unidentified. The E3 ubiquitin ligase activity of Gp78 was shown to be required for the ubiquitination of MFN1 and MFN2 to support mitochondrial turnover during calcium concentration increase-induced and PINK1-independent mitophagy [318,319]. ARIH1 is an E3 ubiquitin ligase widely expressed in numerous cancer cells that has also been demonstrated to ubiquitinate mitochondria to induce mitophagy in a PINK1- but not Parkin-dependent manner, which may contribute to chemotherapeutic resistance during cancer treatment [320]. The HUWE1 E3 ubiquitin ligase promotes PINK1/Parkin-independent mitophagy through multiple processes, including ubiquitination of mitochondria, enhancement of IkB kinase alpha (IKK-alpha)-mediated phosphorylation of AMBRA1 at Ser104, and destabilization of the mitochondrial protein myeloid cell leukemia 1 (MCL1) [226,391]. Another E3 ubiquitin ligase, membrane-associated RING finger protein 5 (MARCH5, also called MITOL), was indicated to negatively regulate hypoxia-inducing mitophagy, in contrast to activating mitophagy, by promoting the degradation of FUNDC1 [392]. In contrast, a recent study showed that MARCH5 may facilitate the ubiquitination of mitochondrial proteins, thus establishing a positive feedback loop to promote PINK1/Parkin-dependent mitophagy [393]. Collectively, these studies imply that ubiquitination serves as an accelerator of mitophagy induction.
Various DUBs, including ubiquitin-specific peptidase (USP)15 [394], USP30 [395,396,397], USP8 [398], USP14 [399], USP33 [400], and USP36 [401], have been demonstrated to regulate mitophagy. USP15 was first shown to counteract PINK1/Parkin-dependent mitophagy by diminishing Parkin-triggered mitochondrial ubiquitination [394]. The mitochondrial DUB USP30 was demonstrated to antagonize PINK1/Parkin-mediated mitochondrial turnover by removing ubiquitin from TOM20 and MIRO [395,396,397]. In addition, recent studies identified two DUBs that suppress PINK1/Parkin-dependent mitophagy: USP33, which acts by removing K6-, K11-, K48-, and K63-linked ubiquitin conjugates on Parkin; and USP36, which acts by downregulating ATG14 [400,401]. In contrast to suppressing mitophagy, USP8-mediated removal of the K6-linked ubiquitin chain from Parkin facilitates efficient recruitment of Parkin to depolarized mitochondria to induce mitophagy [398]. Similarly, USP14 was shown to promote PHB2-mediated mitochondrial membrane rupture in the cellular context of PINK1 and Parkin deficiency [399]. These studies collectively suggest that DUB-dependent deubiquitination may regulate mitophagy through multiple unrevealed and unresolved molecular mechanisms.
4.4.3. Other PTMs
Several other PTMs, such as acetylation, deacetylation, and sumoylation, have been demonstrated to potentially regulate mitophagy. The NAD-dependent protein deacetylase sirtuin-2 (SIRT2) was shown to localize to mitochondria and participate in the regulation of mitophagy via ATG5 deacetylation [402]. Intriguingly, SIRT1 plays an opposite role in mitophagy inhibition by reducing Parkin translocation to mitochondria [403]. In addition to protein deacetylation, the decrease in mitochondrial protein acetylation induced by depletion of the mitochondrial-enriched GCN5-like 1 (GCN5L1) gene was demonstrated to induce PINK1/Parkin-independent mitophagy [404]. Another acetyltransferase, the protein N-terminal acetyltransferase A (NatA), was reported to promote yeast mitophagy via ATG32 induction [405]. In addition to protein acetylation and deacetylation, sumoylation of an RNA-binding protein, fused in sarcoma/translocated in sarcoma (FUS), was recently shown to be involved in the regulation of mitophagy in glioma pathogenesis [406]. These results suggest that other PTMs may function in the regulation of mitophagy and remain to be further investigated.
5. The Regulation of Liver Function and Liver Diseases by Autophagy
Autophagy has been extensively shown to regulate live physiology in the past few decades. Hepatic autophagy was first shown to promote glycogen degradation to balance liver metabolism and protect hepatocytes from liver atrophy [407,408,409]. This regulation of hepatic metabolism by autophagic degradation is triggered by deprivation of nutrients [21,410,411] and altered levels of metabolites [410,412,413]. Similarly, autophagy also functions in the turnover of intracellular RNA and proteins [414], as well as organelles, such as Mallory-Denk bodies (MDBs) [415,416,417], ER [418,419], mitochondria [420,421,422,423], peroxisome [424,425,426,427,428,429,430,431,432] and LDs [88,89,247,248,433,434,435,436]. In addition, autophagy also participates in the protection of liver cells from damage [437,438], the balancing of the intracellular levels of amino acids and iron [439,440], and the regeneration of transplanted liver tissues [441]. Thus, these studies collectively underlie that autophagy plays a protective role in maintaining the balance of liver metabolism and in protecting liver cells from injury.
In addition to the physiological role of autophagy in liver, modulation of autophagy was shown to participate in the pathogenesis of liver diseases. Autophagy has been demonstrated to eliminate the aggregate of alpha (1)-antitrypsin Z mutant in the liver of patients with alpha (1)-antitrypsin deficiency to prevent liver against cell death [442,443,444,445]. Deregulated LDs catabolism by abnormal autophagy was also reported to be involved in the development of liver steatosis and fatty liver diseases [446,447,448,449]. Additionally, impaired autophagy was evidenced to be correlated with the pathogenesis of liver cancer. The genetic ablation of ATG5 and ATG7 has been reported to spontaneously induce the development of liver cancer through the accumulation of p62/SQSTM1-driven nuclear factor erythroid-2-related factor 2 (Nrf2) and induction of antioxidant responses [450,451]. The hyperphosphorylation of p62/SQSTM1 and downstream Nrf2-antioxidant axis was recently shown to promote metabolic reprogramming to enhance the chemoresistance of hepatocellular carcinoma (HCC) cells [452], and to enhance the diethylnitrosamine (DEN) carcinogenic activity for HCC development [453]. Moreover, autophagy has consistently been shown to be activated in liver cells infected with hepatitis viruses, including hepatitis B virus [454,455,456], and hepatitis C virus [457,458,459,460]. Based on these findings, alterations in autophagy may be involved in the development of liver diseases and represent as a specific target for the diagnosis and treatment of liver diseases.
6. The Physiological Role(s) of Mitophagy in the Liver
6.1. The Discovery of Mitochondrial Autophagy in the Liver
The liver was the first tissue leading to the discovery of mitochondrial autophagy; in the early 1960s, Ashford et al. observed mitochondria sequestered within autophagic vacuoles associated with lysosomes in glucagon-perfused rat hepatocytes [14]. Additionally, mitochondria were the most common intracellular organelles initially observed within autophagic vacuoles in the late 1950s and early 1960s [13,14,15,16]. Soon thereafter, mitochondrial autophagy was observed in rat hepatic tissues under different kinds of stimuli, including exposure to the detergent Triton WR-1339 [461], fasting [461], exposure to glucagon [20,411,462], and amino acid deprivation25. The TEM-based ultrastructural and biochemical fractionation analyses in these studies suggested that a large portion of fragmented and dysfunctional mitochondria are engulfed by autophagic vacuoles, in which lysosomal acidic proteases degrade the enclosed materials [20,461,462], suggesting that hepatic autophagy may function in mitochondrial turnover. Later, the concept of selective degradation of organelles, including mitochondria, by autophagic process, emerged from biochemical and morphological studies on liver tissues [23,411,463,464,465,466,467,468,469,470,471,472]. In addition, autophagic vacuoles in hepatic tissues were shown to contain mitochondrial enzymes [35,36,473,474,475]. These studies collectively imply that mitochondrial autophagy may promote mitochondrial degeneration in the liver.
6.2. The Functional Role(s) of Mitophagy in Liver Physiology
The procedure for the specific detection of mitophagy was initially established by immunogold TEM-mediated labeling of mitochondrial enzymes within autophagic vacuoles and fluorescent probe-based tracking of mitochondrial engulfment by autophagy [475,476]. Mitochondrial degradation by autophagy was first demonstrated in the liver tissues from patients with alpha (1)-antitrypsin (α1-AT) deficiency, a chronic liver disease (Table 1) [444,477,478], suggesting that autophagic degradation of mitochondria may be activated to counteract the mitochondrial injury induced by α1-AT deficiency. In addition, failure of autophagic mitochondrial degradation was shown to be associated with age-dependent accumulation of 8-oxo-2′-deoxyguanosine (8-OHdG) in mtDNA in the rat liver, and activated mitochondrial autophagy was demonstrated to degrade injured mitochondria and rescue older cells from 8-OHdG-mtDNA accumulation (Table 1) [423,479]. Komatsu et al. first demonstrated that ATG7 deficiency led to the accumulation of fragmented and deformed mitochondria in mouse liver tissue (Table 1) [480]. Mitochondrial degradation by autophagy was further shown to be induced by nutrient starvation in order to remove depolarized mitochondria from the mouse liver via the pre-autophagosomal structure (PAS)-mediated nucleation process [421]. Loss of BNIP3, a cargo receptor for mitophagy in the mouse liver, was reported to induce an increase in the mitochondrial mass and the accumulation of abnormal mitochondria (Table 1) [481], suggesting the functional role of BNIP3-dependent mitophagy in the maintenance of mitochondrial integrity. These studies may imply a critical role of basal mitophagy in mitochondrial quality control. On the other hand, interference with hepatic autophagy by ischemia/reperfusion (I/R) and anoxia/reoxygenation (A/R) was indicated to trigger mitochondrial dysfunction by enhancing mitochondrial permeability transition (MPT) (Table 1) [482], and aging can aggravate I/R-induced liver injury via Parkin-mediated mitophagy (Table 1) [483]. In addition, sequestration of mitochondria within autophagic vacuoles was shown to be correlated with acute liver cell damage in patients with anorexia nervosa (Table 1) [437]. Thus, these studies imply that stress-induced mitophagy is pivotal to mitochondrial homeostasis [482].
Later, mitochondrial degradation by autophagy was shown to be activated during the remodeling of primary rat hepatocytes through the MPT, suggesting that mitophagy underlies hepatic remodeling in response to stimuli (Table 1) [420]. Consistent with this observation, mitophagy was demonstrated to be triggered by ethanol in mouse liver tissues (Table 1) [446], and Parkin was shown to be required for ethanol-induced mitophagy (Table 1) [484,485,486], suggesting that mitophagy may mitigate ethanol-induced hepatotoxicity. In addition, efavirenz, the specific reverse transcriptase inhibitor used to treat HIV, was reported to induce mitophagy to rescue mitochondrial dysfunction, thus circumventing drug-induced liver damage (Table 1) [487,488]. Additionally, dynamin 1-like-dependent mitochondrial fragmentation was shown to potently activate mitophagy in liver cells to mediate cadmium-induced hepatotoxicity (Table 1) [489]. Moreover, mitophagy was indicated to be involved in the removal of dysfunctional mitochondria to overcome acetaminophen-induced liver injury (Table 1) [490,491], most likely through a PINK1/Parkin-independent pathway (Table 1) [492,493]. In addition, hepatic mitophagy was reported to be activated by AMPK signaling and to inhibit NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome activation to diminish acetaminophen-triggered hepatotoxicity (Table 1) [494,495]. Collectively, these findings indicate that mitophagy fundamentally promotes mitochondrial turnover in liver cells and acts as a guardian to prevent hepatic cells from injury.
7. Role(s) of Mitophagy in the Development of Liver Diseases
7.1. Liver Injury
The association of mitophagy and/or mitochondrial autophagy with liver injury was first discovered in earlier studies showing that α1-AT deficiency leads to mitochondrial dysfunction by interfering with autophagic degradation of mitochondria (Table 2) [444,477,478]. α1-AT is a serum glycoprotein that functions as an inhibitor of destructive neutrophil proteases [496,497]. Several naturally occurring mutants of α1-AT [498], including the S variant (in which the glutamic acid [Glu] at residue 264 [Glu264] is mutated to valine [Val]) and the Z variant (in which Glu264 is mutated to lysine [Lys]), have been identified and shown to participate in the pathogenesis of human diseases, such as chronic liver-associated diseases [499,500,501]. Homozygosity for the Z variant of α1-AT (α1-ATZ) leads to a conformational change promoting α1-AT polymerization and the formation of inclusion bodies that are retained in the ER, thus impairing α1-AT secretion into the bloodstream and causing liver damage [502,503,504]. Numerous studies have demonstrated that autophagy may function in the elimination of α1-ATZ protein aggregates to prevent hepatic injury [442,443,505,506,507]. Interference with the autophagic pathway by genetic knockout of ATGs, such as ATG5, was demonstrated to inhibit α1-ATZ degradation in yeast cells [505,508]. Reciprocally, enhancement of autophagy by rapamycin and other autophagy-enhancing drugs was proven to promote the clearance of α1-ATZ to restore α1-AT deficiency-induced liver damage [509,510,511,512]. α1-ATZ aggregation was reported to induce mitochondrial dysfunction in the liver tissues of patients with α1-AT deficiency (Table 2) [444]. Later, mitochondrial injury-associated mitophagy was shown to be induced in liver tissues of patients with α1-AT deficiency and in the livers of α1-ATZ transgenic mice (Table 2) [477,478]. In addition, defects in mitophagy were shown to enhance the MPT and induce mitochondrial dysfunction in the setting of I/R- and A/R-induced hepatic injury (Table 2) [482]. I/R-induced downregulation of SIRT1 was demonstrated to reduce MFN2 deacetylation (at the Lys655 and Lys662 residues) and impair mitochondrial autophagy, thus inducing mitochondrial dysfunction and liver injury (Table 2) [513,514]. Overexpression of SIRT1 by adenoviral gene delivery and enhancement of SIRT1 activity by a pharmacological activator, SIRT1702, were shown to restore mitochondrial autophagy and to suppress mitochondrial abnormalities and cell death after I/R (Table 2) [513]. Consistent with these studies, recent studies indicated that aging may reduce Parkin and ATG5 expression, interfering with the mitophagy process and exacerbating hepatic I/R injury (Table 2) [483], and that Parkin deficiency can aggravate hepatic I/R injury by suppressing mitophagy-mediated mitochondrial turnover (Table 2) [515]. Heme oxygenase-1 (HO-1) was recently demonstrated to restore hepatic I/R-impaired mitophagy by activating PGAM5 signaling, which protected against hepatocellular damage during I/R injury (Table 2) [516]. In contrast, microRNA 330-3p (miR-330-3p) was reported to elevate hepatic I/R injury by suppressing PGAM5-mediated mitophagy (Table 2) [517]. Moreover, mitophagy was demonstrated to remove dysfunctional mitochondria to protect liver cells from hepatotoxicity induced by drugs such as acetaminophen [490,491], efavirenz [487], and cadmium [489] (Table 2). In addition, mitophagy was proven to prevent both acute ethanol-triggered hepatotoxicity in mice (Table 2) [446,518] and polyethylenimine-induced liver damage [519]. Collectively, these studies imply that mitophagy may act to protect liver cells from different kinds of liver injury and serve as a therapeutic target for the design of a new therapy for liver injury.
7.2. Steatosis and Fatty Liver Diseases
Numerous studies have shown that autophagy may balance lipid metabolism by catabolizing LDs [88,89,248,433,436,520,521] and regulating LD biogenesis [247,522]. Thus, autophagy has been indicated to prevent the development of fatty liver, and elevation of autophagic activity is considered a strategy to be exploited for developing therapeutics to treat fatty liver disease [435,523,524,525]. The induction of mitochondrial damage in alcoholic liver disease was first indicated during the 1970s and 1980s by the increased levels of giant and rounded mitochondria in liver biopsies from patients with alcoholic liver disease and in the fatty liver tissue of ethanol-treated mice (Table 3) [526,527,528]. The relationship between mitophagy and hepatic steatosis was first observed in an earlier study showing that the expression of BNIP3, a mitophagy receptor, is induced by fasting and that loss of BNIP3 triggers hepatic steatosis and promotes its transition into steatohepatitis in mice (Table 3) [481]. Additionally, loss of BNIP3 was shown to result in reduced mitochondrial integrity and increased lipogenesis in the livers of BNIP3-null mice [481], suggesting the critical role of mitophagy in regulating normal liver metabolism and preventing hepatic steatosis. Moreover, the induction of hepatic steatosis in rats by ethanol was demonstrated to dramatically activate mitophagy by elevating PINK1 expression on mitochondria to eliminate damaged mitochondria (Table 3) [529]. In addition, ethanol-activated PINK1-dependent mitophagy was shown to be strongly correlated with the mitochondrial expression of Parkin and the level of the indicator of oxidative mtDNA damage, 8-OHdG, in the rat model of ethanol-induced hepatic steatosis (Table 3) [530,531]. The functional role of Parkin-dependent mitophagy in alcohol-induced hepatic steatosis was further evidenced by decreased mitophagy, mitochondrial respiration, and cytochrome c oxidase activity (Table 3) [484,486]. Based on these studies, mitophagy plays a protective role in combating alcohol-induced mitochondrial dysfunction, hepatic steatosis, and hepatic injury, indicating a promising opportunity to develop feasible therapeutic and preventative strategies for alcoholic fatty liver disease. In support of this idea, enhancement of mitophagy by quercetin, a naturally occurring flavonoid, was shown to alleviate ethanol-triggered mitochondrial damage [532].
Despite the role of mitophagy in alcoholic hepatic steatosis, accumulating evidence also indicates that mitophagy may be involved in the development of nonalcoholic fatty liver disease (NAFLD), which encompasses a wide spectrum of progressive diseases ranging from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH) to liver cirrhosis and, consequently, to hepatocellular carcinoma (HCC) and represents a major public health burden associated with a modern lifestyle [540]. Mitochondrial abnormalities in the liver tissues of patients with NAFLD were first demonstrated by the formation of megamitochondria containing linear crystalline inclusions (Table 3) [533]. Later, diet-induced NAFLD in mice was shown to block hepatic autophagy and lead to oxidative stress and mitochondrial dysfunction (Table 3) [534]. Additionally, diet-induced NAFLD was indicated to reduce thyroid hormone-induced mitophagy in mice, suggesting the role of insufficient mitophagy in NAFLD-induced abnormal mitochondrial homeostasis and hepatic injury (Table 3) [535]. In addition, induction of NAFLD in HepG2 cells treated with oleic acid (OA) was shown to activate mitophagy through expression of the p53-dependent damage-regulated autophagy modulator (DRAM) to promote hepatocyte apoptosis (Table 3) [536]. According to another study conducted in mice, diet-induced NAFLD suppresses the completion of mitophagy and leads to the accumulation of mitophagy intermediates, e.g., megamitochondria that contain p62/SQSTM1, Kelch-like ECH-associated protein (Keap1), and ubiquitin (Table 3) [537]. Mitochondrial stasis induced by simultaneous deletion of two dynamin-related GTPases for division (DRP1) and fusion (OPA1) in the mouse liver rescues the mitochondrial integrity and liver function by restoring Parkin-independent mitophagy mediated by p62/SQSTM1/Keap1/RING-box protein 1 (RBX1) (Table 3) [537]. Moreover, increased degradation of mitochondrial oxidative phosphorylation subunits was suggested to contribute to the suppression of mitophagy and induction of mitochondrial defects in hepatocytes from mice with NAFLD (Table 3) [538]. On the other hand, impairment of mitophagy was shown to activate the NLRP3 inflammasome to promote the progression of NAFL to NASH [539]. These studies collectively indicate the pathological role of deregulated mitophagy in the development of NAFLD and suggest the therapeutic potential of exploiting mitophagy enhancement for the design of a new curative therapy for NAFLD. Consistent with this idea, several studies have reported that restoration of mitophagy may ameliorate the progression of NAFLD [541,542,543,544].
7.3. Liver Cancer
The alteration of mitochondrial homeostasis in liver cancer was first discovered in the 1950s [545,546]. In subsequent studies, TEM ultrastructural analyses of subcellular compartments in liver specimens from patients with liver cancer and in liver tissues from mice with safrole-induced HCC further indicated that the loss of mitochondrial integrity is associated with the initiation of hepatocarcinogenesis (Table 4) [547,548]. Concanavalin A (ConA), a lectin that functions in the activation of acute hepatic inflammation, was shown to suppress hepatoma cell growth in vitro through BNIP3-mediated mitophagy and to inhibit the formation of liver tumor nodules in vivo (Table 4) [549,550]. Adriamycin, a chemotherapeutic drug commonly used to treat HCC, and curcumin, an extract of Curcuma longa, were also reported to activate mitophagy to promote the apoptosis of hepatoma and HepG2 cells (Table 4) [551,552]. Moreover, melatonin was shown to trigger mitophagy to increase the sensitivity of human HCC cells to the cytotoxic effects of sorafenib, a kinase inhibitor approved for the treatment of liver cancer (Table 4) [553]. Consistent with these studies, DRAM-mediated mitophagy was implied to promote the apoptosis of HCC cells (Table 4) [554]. According to a recent study, FUNDC1-mediated mitophagy may suppress the initial development of HCC in mice by interfering with inflammasome activation (Table 4) [555]. Although it can play a protective role in liver tumorigenesis, mitophagy could be an initiator and/or accelerator of hepatocarcinogenesis. Mitochondrial fission, which promotes cell survival via reactive oxygen species (ROS) production and is positively correlated with a poor prognosis for patients, was frequently reported to be elevated in liver specimens from patients with HCC (Table 4) [556]. Mitophagy was also shown to attenuate p53 activity to support the maintenance of hepatic cancer stem cells (CSCs), thus promoting the development of liver cancer (Table 4) [557]. When mitophagy is suppressed in cells, PINK1-mediated phosphorylation of p53 at Ser392 in the mitochondria induces nuclear translocation of p53 and prevents the expression of NANOG gene, a homeobox transcription factor that mediates the maintenance of CSC stemness and self-renewal ability, thus decreasing the hepatic CSC population (Table 4) [557]. Notably, FUNDC1 was reported to accumulate in the liver tissues of patients with HCC (Table 4) [555], suggesting that FUNDC1-induced mitophagy may contribute to the pathogenesis of late-stage HCC. To date, whether mitophagy is altered and how mitophagy participates in the development of cholangiocarcinoma remain largely unknown, although p62/SQSTM1-mediated mitophagy and regulation of mitochondrial dynamics were shown to sensitize cholangiocarcinoma cells to the cytotoxic effects of the chemotherapeutic drug cisplatin [558]. Collectively, these studies indicate that mitophagy plays a multifaceted role in preventing and/or contributing to liver cancers. However, further investigations are required to comprehensively delineate the functional role(s) of mitophagy in the progression of liver cancer. In perspective, mitophagy modulation is a valuable strategy to be exploited for the development of a new therapy for liver cancer.
7.4. Viral Hepatitis
In the past decade, autophagy has been widely demonstrated to function in the life cycles of hepatitis viruses, including hepatitis B virus (HBV) and hepatitis C virus (HCV) [457,458,459,460,559,560,561,562]. HCV infection was shown to induce autophagy to promote the replication of viral RNA [458,559,560,561,563,564], translation of incoming viral RNA [459], and assembly of infectious virions [565,566,567,568]. In addition, HCV-activated autophagy was reported to suppress the innate antiviral response [459,460,562,569], protect infected hepatocytes from cell death [570], and promote LD catabolism in infected liver cells [459,571]. HCV infection was also reported to induce PINK1/Parkin-dependent mitophagy, in which mitophagosomes contribute to mitochondrial injury associated with chronic HCV infection (Table 5) [572,573]. Moreover, HCV infection was shown to trigger DRP1 phosphorylation at Ser616 and translocation of DRP1 to mitochondria, thus inducing mitochondrial fission and mitophagy [574]. Virus-activated mitophagy was further indicated to attenuate apoptosis and promote persistent viral infection (Table 5) [574]. Consistent with this finding, the HCV nonstructural protein 5A (NS5A) was reported to disrupt mitochondrial dynamics, by concomitantly increasing ROS production and triggering mitophagy (Table 5) [575]. In contrast, the HCV core protein was demonstrated to suppress mitophagy by interacting with Parkin and interrupting its translocation to mitochondria to sustain HCV-induced mitochondrial injury in infected liver cells (Table 5) [576]. Collectively, these studies suggest that HCV may subvert the removal of damaged mitochondria by mitophagy to prevent the death of infected cells and maintain viral persistence.
The role(s) of autophagy in HBV infection were first revealed by an earlier study showing that heterozygous deletion of Beclin 1, an ATG protein in the PI(3)K complex, suppresses autophagy in liver cells, thus promoting HBV-associated premalignant lesions [578]. Later, HBV-induced autophagy mediated through an increase in PI(3)K enzyme activity was further demonstrated in an in vitro cell culture model and in liver tissues of HBV transgenic mice and was shown to promote viral DNA replication [454,455,456]. HBV x protein (HBx) and HBV small surface protein (SHBs) were shown to induce autophagy to support viral replication [579,580]. Autophagy and the autophagic machinery were indicated to participate not only in viral genome replication [454,455,456,579,580,581,582], but also in the secretion of infectious virions by infected cells [583,584], viral envelopment [580], virus-induced secretion of cytokines [585,586], the suppression of HBV-associated tumorigenesis [587], and the degradation of infecting virions [588,589]. In contrast, HBx was demonstrated to impair autophagic flux by suppressing lysosomal function to contribute to the development of HBV-associated HCC [590]. HBV has been shown to induce Parkin-mediated mitophagy though DRP1-mediated mitochondrial fission to attenuate the apoptosis of infected cells (Table 5) [577]. Additionally, thyroid hormone was reported to trigger mitophagy to protect hepatocytes from HBx-induced carcinogenesis [591]. Moreover, HBx was recently shown to enhance Parkin-dependent mitophagy through the Lon peptidase in hepatocytes under starvation conditions [592]. These studies imply the functional roles of mitophagy in HBV-host interactions.
7.5. Other Liver Diseases
An impairment of mitophagy was shown to potentially participate in carbon tetrachloride (CCl4)-induced hepatic fibrosis in the mouse liver, and melatonin was indicated to restore mitophagy and protect hepatocytes from hepatic fibrosis (Table 6) [593]. In addition, another study showed that interference with T-cell immunoglobulin domain and mucin domain-4 (TIM-4) in Kupffer cells may suppress PINK1/Parkin-dependent mitophagy to mitigate CCl4-induced hepatic fibrosis in mice (Table 6) [594]. Mitophagy was also reported to be induced by increased ROS production to promote PM2.5-induced hepatic stellate cells (HSCs) activation and hepatic fibrosis (Table 6) [595]. In contrast, inhibition of mitophagy was shown to promote inflammation in HSCs during acute liver injury (Table 6) [596]. Very recently, activation of DRP1-mediated mitochondrial fission and suppression of FUNDC1-dependent mitophagy were shown to promote DNA-dependent protein kinase catalytic subunit (DNA-PKcs)-induced alcoholic liver disease (Table 6) [597].
Mitophagy was also shown to control the fatty acids metabolism. Genetic studies in mice showed that the deletion of BNIP3 in liver triggers lipogenesis by elevating the expressions of lipogenic enzymes and decreasing the β-oxidation of fatty acids, thus increasing the mitochondrial mass and upregulating hepatic respiration [481]. On the other hand, the β-oxidation of fatty acids was shown to be enhanced through inducing the gene expression of carnitine palmitoyltransferase 1α (CPT1α), a rate-limiting enzyme required for fatty acid oxidation by thyroid hormone-activated mitophagy in liver cells [598]. Similarly, CPT1α expression was recently shown to be increased and accompanied by the increased expressions of mitophagy-related genes, such as BNIP3L and Parkin in REDD1 (regulated in development and DNA damage response-1) knockout mice with NAFLD [599]. These studies together concluded that hepatic mitophagy may integrate lipid metabolism through increasing fatty acids β-oxidation.
It was also evinced that mitophagy is regulated by hepatic insulin resistance. Insulin has been shown to interfere with autophagy by suppressing the expressions of ATGs, such as ATG12 and Vps34, in cultured hepatocytes [600]. Moreover, starvation- and glucagon-induced mitophagy are repressed by insulin resistance in cultured hepatocytes, which are induced by the long-term exposure to insulin [600]. Intriguingly, gene knockout of Parkin in mice does not increase insulin resistance and obesity, presumably due to an impairment in intestinal lipid absorption [601], and a decrease in ER stress and the activation of AMPK [602]. Loss of FUNDC1-mediated mitophagy was demonstrated to increase insulin resistance through inducing adipose tissue-associated macrophage infiltration and hyperactivation of mitogen-activated protein kinase 8 (MAPK8, also named JNK1) [603]. Thus, these studies imply that mitophagy is not only regulated by hepatic insulin resistance, mitophagy but also functions in the insulin resistance-associated metabolic disorder. Collectively, these studies indicate that mitophagy may participate in the pathogenesis of other liver diseases. However, the exact role(s) of mitophagy in the development of these liver diseases are still enigmatic and required further investigation.
8. Translation of Mitophagy to the Control of Liver Diseases
Because autophagy and mitophagy have been extensively shown to be modulated in the pathogenesis of liver diseases, the knowledge of mitophagy regulation might conceivably be translated into the clinic for the diagnosis and treatment of liver diseases. Mitophagy and mitophagy-related genes are differentially regulated in liver diseases, and thus the monitoring of changes in mitophagy and the mitophagy regulators might represent a promising strategy to diagnose liver diseases. For example, p62/SQSTM1 accumulates in the damaged mitochondria in the liver during NAFLD development [537], and the increased expression of FUNDC1 supports the tumorigenesis of HCC [555]; thus, these proteins represent potential biomarkers for diagnosing liver diseases. On the other hand, the mitophagy-associated alteration in the activation of the inflammasome and downstream interleukin secretion might represent another diagnostic marker for the early detection of NAFLD and HCC [539,555]. Notably, conclusions on the role(s) of mitophagy in liver diseases were drawn from animal experiments, rather than from clinical samples obtained from patients with liver diseases. Further investigations of the modulation of these potential mitophagy-related biomarkers in cohorts of patients with liver diseases are urgently needed to confirm the reliability and feasibility of these new methods designed for diagnosing liver diseases.
An increase in hepatic mitophagy has been shown to protect liver cells from damage and to prevent the progression of liver diseases, thus suggesting that the precise activation of mitophagy potentially represents a rational strategy to interfere with the development of liver diseases. For example, the induction of mitophagy has been widely shown to prevent liver injury caused by drugs and ethanol [446,487,489,490,491,518,519]; therefore, a study aiming to search for and identify the highly potent inducers of mitophagy for clinical used as treatments for liver injury would be valuable. Several enhancers of mitophagy that trigger or restore mitophagy have been identified, such as p62/SQSTM1-mediated inducer (PMI) [604], urolithin A [605], quercetin [532], and ivermectin [376]. However, researchers have not confirmed whether these inducers are able to be safely utilized for treating liver diseases because extensive clinical trials of these drugs in patients with liver diseases have not been successfully completed. Questions still exist regarding potential side effects of these mitophagy inducers after administration for curing liver disease. Very recently, an increase in mitophagy mediated by the induction of the MPT has been shown to unexpectedly induce hepatic cell death in response to I/R injury and to shorten the lifespan [606]. Therefore, further studies are needed to determine whether manipulations of mitophagy represent a druggable target for systematically curing liver diseases. The comprehensive knowledge of the role(s) of mitophagy in liver diseases will allow us to overcome obstacles of applying the therapeutic modulation of mitophagy in the clinic. In addition to mouse genetic models, studies of the alterations in mitophagy in liver specimen and the identification of the clinically relevant correlation between altered mitophagy and the pathogenesis of liver diseases will help us to search for more specific molecular targets, such as a particular cargo receptor or regulator of mitophagy that is dispensable in general mitophagy, and develop a new therapeutic strategy to treat liver diseases.
9. Conclusions and Perspectives
Mitophagy plays a pivotal role in the elimination of dysfunctional mitochondria to promote mitochondrial turnover and maintain mitochondrial integrity, thus physiologically regulating liver function. On the other hand, accumulating evidence implies the extensive modulation of mitophagy in various types of liver diseases. Induction of mitophagy has historically been believed to protect liver cells from damage and injury and to serve as a guardian to prevent the development of liver diseases. Accordingly, the enhancement and/or restoration of hepatic mitophagy seems to be a promising strategy to exploit for the development of new therapeutics for liver diseases. On the other hand, mitophagy is subverted to promote the pathogenesis of liver diseases, suggesting that proper suppression of mitophagy could be harnessed to alleviate the progression of liver diseases. However, the implications of manipulating hepatic mitophagy to treat liver diseases remain largely uncertain, most likely due to the functional role(s) of mitophagy in liver diseases, which are controversial and under debate. Additionally, there are large discrepancies among different studies, and most of the conclusions on mitophagy and liver diseases are experimental model- and disease stage-dependent, thus impeding the comprehensive understanding of the functions of mitophagy in liver diseases. However, further investigations are required to unveil the detailed role(s) of mitophagy in the development of liver diseases and to improve knowledge on the clinical relevance of autophagy in different stages of liver disease progression. Most importantly, innovative and integrative biomedical research approaches should be incorporated to identify molecular targets of mitophagy, such as a specific cargo receptor, that can be translated into the design of a safe, feasible, and effective clinical therapeutic strategy for liver diseases.
Author Contributions
Conceptualization, P.-Y.K.; writing—original draft preparation, P.-Y.K.; writing—review and editing, P.-Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Health Research Institute (NHRI-EX103-10322SC, NHRI-EX104-10322SC, NHRI-EX105-10322SC, and NHRI-EX106-10322SC), Miaoli; the Ministry of Science and Technology (MOST 102-2320-B-182-037-MY3, MOST 105-2628-B-182-001-MY3, and MOST108-2320-B-182-011), Taipei; and Chang Gung Memorial Hospital (CMRPD1C0211, CMRPD1D0021, CMRPD1D0022, CMRPD1D0023, CMRPD1G0281, CRRPD1F0031, CRRPD1F0032, CRRPD1F0033 and CMRPD1H0681), Taoyuan, Taiwan.
Acknowledgments
The author apologizes to colleagues whose studies could not be cited due to space constraints. The author thanks Chih-Wei Chang for the help on the preparation of figures.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Chu, C.T. Multiple pathways for mitophagy: A neurodegenerative conundrum for Parkinson’s disease. Neurosci. Lett. 2019, 697, 66–71. [Google Scholar] [CrossRef]
- Liu, L.; Liao, X.; Wu, H.; Li, Y.; Zhu, Y.; Chen, Q. Mitophagy and its contribution to metabolic and aging associated disorders. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazari, Y.; Bravo-San Pedro, J.M.; Hetz, C.; Galluzzi, L.; Kroemer, G. Autophagy in hepatic adaptation to stress. J. Hepatol. 2020, 72, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.L., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Essner, E. Cytolysomes and mitochondrial degeneration. J. Cell Biol. 1962, 15, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B. The proximal tubule cell in experimental hydronephrosis. J. Biophys. Biochem. Cytol. 1959, 6, 136–138. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [Green Version]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U. Inhibition by insulin of the physiological autophagic breakdown of cell organelles. Acta Biol. Med. Ger. 1977, 36, 1691–1694. [Google Scholar] [PubMed]
- Pfeifer, U. Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J. Cell Biol. 1978, 78, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.L.; Molnar, K.; Seglen, P.O. Inhibition of autophagic sequestration and endogenous protein degradation in isolated rat hepatocytes by methylated adenosine derivatives. FEBS Lett. 1981, 134, 194–196. [Google Scholar] [CrossRef] [Green Version]
- Blankson, H.; Holen, I.; Seglen, P.O. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp. Cell Res. 1995, 218, 522–530. [Google Scholar] [CrossRef]
- Holen, I.; Gordon, P.B.; Seglen, P.O. Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem. J. 1992, 284, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Holen, I.; Stromhaug, P.E.; Gordon, P.B.; Fengsrud, M.; Berg, T.O.; Seglen, P.O. Inhibition of autophagy and multiple steps in asialoglycoprotein endocytosis by inhibitors of tyrosine protein kinases (tyrphostins). J. Biol. Chem. 1995, 270, 12823–12831. [Google Scholar] [CrossRef] [Green Version]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, A.L.; Seglen, P.O. Inhibition of hepatocytic protein degradation by methylaminopurines and inhibitors of protein synthesis. Biochim. Biophys. Acta 1981, 676, 213–220. [Google Scholar] [CrossRef]
- Gordon, P.B.; Kovacs, A.L.; Seglen, P.O. Temperature dependence of protein degradation, autophagic sequestration and mitochondrial sugar uptake in rat hepatocytes. Biochim. Biophys. Acta 1987, 929, 128–133. [Google Scholar] [CrossRef]
- Plomp, P.J.; Wolvetang, E.J.; Groen, A.K.; Meijer, A.J.; Gordon, P.B.; Seglen, P.O. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 1987, 164, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.B.; Seglen, P.O. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 1988, 151, 40–47. [Google Scholar] [CrossRef]
- Yamamoto, A.; Masaki, R.; Tashiro, Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J. Histochem. Cytochem. 1990, 38, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S. Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur. J. Cell Biol. 1993, 61, 67–80. [Google Scholar]
- Yokota, S.; Himeno, M.; Roth, J.; Brada, D.; Kato, K. Formation of autophagosomes during degradation of excess peroxisomes induced by di-(2-ethylhexyl)phthalate treatment. II. Immunocytochemical analysis of early and late autophagosomes. Eur. J. Cell Biol. 1993, 62, 372–383. [Google Scholar]
- Yokota, S.; Himeno, M.; Kato, K. Formation of autophagosomes during degradation of excess peroxisomes induced by di-(2-ethylhexyl)-phthalate treatment. III. Fusion of early autophagosomes with lysosomal compartments. Eur. J. Cell Biol. 1995, 66, 15–24. [Google Scholar]
- Liou, W.; Geuze, H.J.; Geelen, M.J.; Slot, J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997, 136, 61–70. [Google Scholar] [CrossRef]
- Fengsrud, M.; Roos, N.; Berg, T.; Liou, W.; Slot, J.W.; Seglen, P.O. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: Effects of vinblastine and asparagine on vacuole distributions. Exp. Cell Res. 1995, 221, 504–519. [Google Scholar] [CrossRef]
- Furuno, K.; Ishikawa, T.; Akasaki, K.; Lee, S.; Nishimura, Y.; Tsuji, H.; Himeno, M.; Kato, K. Immunocytochemical study of the surrounding envelope of autophagic vacuoles in cultured rat hepatocytes. Exp. Cell Res. 1990, 189, 261–268. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Matsuura, A.; Wada, Y.; Ohsumi, Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1995, 210, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.M.; Hefner-Gravink, A.; Thumm, M.; Klionsky, D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996, 271, 17621–17624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlumpberger, M.; Schaeffeler, E.; Straub, M.; Bredschneider, M.; Wolf, D.H.; Thumm, M. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Straub, M.; Bredschneider, M.; Thumm, M. AUT3, a serine/threonine kinase gene, is essential for autophagocytosis in Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 3875–3883. [Google Scholar] [CrossRef] [Green Version]
- Lang, T.; Schaeffeler, E.; Bernreuther, D.; Bredschneider, M.; Wolf, D.H.; Thumm, M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998, 17, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sowers, J.R.; Zhang, Y. Metabolic Stress, Autophagy, and Cardiovascular Aging: From Pathophysiology to Therapeutics. Trends Endocrinol. Metab. 2018, 29, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Krick, R.; Muehe, Y.; Prick, T.; Bremer, S.; Schlotterhose, P.; Eskelinen, E.L.; Millen, J.; Goldfarb, D.S.; Thumm, M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol. Biol. Cell 2008, 19, 4492–4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, M.; Maeda, Y.; Kagohashi, Y.; Kondo, T.; Yamada, M.; Fujimoto, T.; Sakai, Y. Evidence for ESCRT- and clathrin-dependent microautophagy. J. Cell Biol. 2017, 216, 3263–3274. [Google Scholar] [CrossRef] [Green Version]
- Sattler, T.; Mayer, A. Cell-free reconstitution of microautophagic vacuole invagination and vesicle formation. J. Cell Biol. 2000, 151, 529–538. [Google Scholar] [CrossRef]
- Wang, C.W.; Miao, Y.H.; Chang, Y.S. A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J. Cell Biol. 2014, 206, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Finn, P.F.; Dice, J.F. Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem. 2005, 280, 25864–25870. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Sun, Y.; Fei, M.; Tan, C.; Wu, J.; Zheng, J.; Tang, J.; Sun, W.; Lv, Z.; Bao, J.; et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 2014, 10, 1015–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Cai, Z.; Tao, K.; Zeng, W.; Lu, F.; Yang, R.; Feng, D.; Gao, G.; Yang, Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 2016, 12, 1215–1228. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, D.H.; Yoon, S.Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Bose, J.K.; Majumder, P.; Lee, K.H.; Huang, J.T.; Huang, J.K.; Shen, C.K. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef]
- Li, Y.; Lu, L.; Luo, N.; Wang, Y.Q.; Gao, H.M. Inhibition of PI3K/AKt/mTOR signaling pathway protects against d-galactosamine/lipopolysaccharide-induced acute liver failure by chaperone-mediated autophagy in rats. Biomed. Pharmacother. 2017, 92, 544–553. [Google Scholar] [CrossRef]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G950–G963. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Walker, S.A.; Ktistakis, N.T. Autophagosome Biogenesis Machinery. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.e8. [Google Scholar] [CrossRef] [Green Version]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Formation and maturation of autophagosomes in higher eukaryotes: A social network. Curr. Opin. Cell Biol. 2018, 53, 29–36. [Google Scholar] [CrossRef]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molejon, M.I.; Ropolo, A.; Re, A.L.; Boggio, V.; Vaccaro, M.I. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci. Rep. 2013, 3, 1055. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Lo Re, A.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovanna, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.G.; Chen, Y.; Miao, G.; Zhao, H.; Qu, W.; Li, D.; Wang, Z.; Liu, N.; Li, L.; Chen, S.; et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol. Cell 2017, 67, 974–989.e6. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.W.; Sorensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Liang, X.; Hattori, T.; Tang, Z.; He, H.; Chen, H.; Liu, X.; Abraham, T.; Imamura-Kawasawa, Y.; Buchkovich, N.J.; et al. VPS37A directs ESCRT recruitment for phagophore closure. J. Cell Biol. 2019, 218, 3336–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Johansen, T. FYCO1: Linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010, 6, 550–552. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.; Toth, D.J.; Sengupta, N.; Kim, Y.J.; Balla, T. Phosphatidylinositol 4,5-bisphosphate controls Rab7 and PLEKHM1 membrane cycling during autophagosome-lysosome fusion. EMBO J. 2019, 38, e100312. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Diao, J.; Li, L.; Lai, Y.; Zhong, Q. In Vitro Reconstitution of Autophagosome-Lysosome Fusion. Methods Enzymol. 2017, 587, 365–376. [Google Scholar]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zheng, P.; Zhou, C.; Wang, X.; Ma, H.; Ma, W.; Zhou, X.; Teng, J.; Chen, J. DIPK2A promotes STX17- and VAMP7-mediated autophagosome-lysosome fusion by binding to VAMP7B. Autophagy 2019. [Google Scholar] [CrossRef]
- Njomen, E.; Tepe, J.J. Regulation of Autophagic Flux by the 20S Proteasome. Cell Chem. Biol. 2019, 26, 1283–1294.e5. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, L.; Ireland, S.C.; Ahat, E.; Li, J.; Bekier, M.E., 2nd; Zhang, Z.; Wang, Y. GORASP2/GRASP55 collaborates with the PtdIns3K UVRAG complex to facilitate autophagosome-lysosome fusion. Autophagy 2019, 15, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef]
- Du, W.; Su, Q.P.; Chen, Y.; Zhu, Y.; Jiang, D.; Rong, Y.; Zhang, S.; Zhang, Y.; Ren, H.; Zhang, C.; et al. Kinesin 1 Drives Autolysosome Tubulation. Dev. Cell 2016, 37, 326–336. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar]
- Liu, X.; Klionsky, D.J. Regulation of autophagic lysosome reformation by kinesin 1, clathrin and phosphatidylinositol-4,5-bisphosphate. Autophagy 2018, 14, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol. Cell 2016, 61, 84–97. [Google Scholar] [CrossRef]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Melmed, R.N.; Benitez, C.J.; Holt, S.J. Intermediate cells of the pancreas. 3. Selective autophagy and destruction of beta-granules in intermediate cells of the rat pancreas induced by alloxan and streptozotocin. J. Cell Sci. 1973, 13, 297–315. [Google Scholar]
- Kirkin, V. History of the Selective Autophagy Research: How Did It Begin and Where Does It Stand Today? J. Mol. Biol. 2020, 432, 3–27. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif - crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Sawa-Makarska, J.; Martens, S. Beyond Atg8 binding: The role of AIM/LIR motifs in autophagy. Autophagy 2017, 13, 978–979. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [Green Version]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Fracchiolla, D.; Sawa-Makarska, J.; Zens, B.; Ruiter, A.; Zaffagnini, G.; Brezovich, A.; Romanov, J.; Runggatscher, K.; Kraft, C.; Zagrovic, B.; et al. Mechanism of cargo-directed Atg8 conjugation during selective autophagy. Elife 2016, 5, e18544. [Google Scholar] [CrossRef]
- Marshall, R.S.; Hua, Z.; Mali, S.; McLoughlin, F.; Vierstra, R.D. ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell 2019, 177, 766–781.e24. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Di Rita, A.; Peschiaroli, A.; Pasquale, D.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef]
- Princely Abudu, Y.; Pankiv, S.; Mathai, B.J.; Hakon Lystad, A.; Bindesboll, C.; Brenne, H.B.; Yoke Wui Ng, M.; Thiede, B.; Yamamoto, A.; Mutugi Nthiga, T.; et al. NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Dev. Cell 2019, 49, 509–525.e12. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.W.; Yang, W.Y. Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 2019, 10, 969. [Google Scholar] [CrossRef] [PubMed]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Oikawa, K.; Yoshimoto, K.; Kondo, M.; Mano, S.; Yamada, K.; Hayashi, M.; Sakamoto, W.; Ohsumi, Y.; Nishimura, M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013, 25, 4967–4983. [Google Scholar] [CrossRef] [Green Version]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6, e25555. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Schiff, M.; Czymmek, K.; Talloczy, Z.; Levine, B.; Dinesh-Kumar, S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005, 121, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [Green Version]
- Verlhac, P.; Gregoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimura, R.; Tanaka, K.; Komatsu, M. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Lett. 2014, 588, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Malide, D.; Liu, J.; Rovira, I.I.; Combs, C.A.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [Green Version]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Bernstein, D. METABOLISM. Mitochondria shape cardiac metabolism. Science 2015, 350, 1162–1163. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Van den Haute, C.; Cufi, S.; Corominas-Faja, B.; Cuyas, E.; Lopez-Bonet, E.; Rodriguez-Gallego, E.; Fernandez-Arroyo, S.; Joven, J.; Baekelandt, V.; et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging 2016, 8, 1330–1352. [Google Scholar] [CrossRef] [Green Version]
- Xiang, G.; Yang, L.; Long, Q.; Chen, K.; Tang, H.; Wu, Y.; Liu, Z.; Zhou, Y.; Qi, J.; Zheng, L.; et al. BNIP3L-dependent mitophagy accounts for mitochondrial clearance during 3 factors-induced somatic cell reprogramming. Autophagy 2017, 13, 1543–1555. [Google Scholar] [CrossRef] [Green Version]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef]
- Rojansky, R.; Cha, M.Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016, 5, e17896. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, H.; Xue, D. Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans. Cell Res. 2011, 21, 1662–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Song, W.H.; Yi, Y.J.; Sutovsky, M.; Meyers, S.; Sutovsky, P. Autophagy and ubiquitin-proteasome system contribute to sperm mitophagy after mammalian fertilization. Proc. Natl. Acad. Sci. USA 2016, 113, E5261–E5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Kanki, T.; Wang, K.; Cao, Y.; Baba, M.; Klionsky, D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 2009, 17, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Kondo-Okamoto, N.; Okamoto, K. Mitochondrial degradation during starvation is selective and temporally distinct from bulk autophagy in yeast. FEBS Lett. 2013, 587, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Eid, N.; Kondo, Y. Parkin in cancer: Mitophagy-related/unrelated tasks. World J. Hepatol. 2017, 9, 349–351. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, L.E.; Moore, M.E.; Sarraf, S.A.; Pickrell, A.M. Ubiquitin and Receptor-Dependent Mitophagy Pathways and Their Implication in Neurodegeneration. J. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Kulek, A.R.; Anzell, A.; Wider, J.M.; Sanderson, T.H.; Przyklenk, K. Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells 2020, 9, 214. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Nie, Y.; Huang, X.; Zhu, Y.; Feng, B.; Tang, L.; Zheng, G. Mitophagy in Hepatic Insulin Resistance: Therapeutic Potential and Concerns. Front. Pharmacol. 2019, 10, 1193. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Piramanayagam, S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.H.; Guo, M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 2014, 3, e01958. [Google Scholar] [CrossRef]
- Li, J.; Qi, W.; Chen, G.; Feng, D.; Liu, J.; Ma, B.; Zhou, C.; Mu, C.; Zhang, W.; Chen, Q.; et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 2015, 11, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef]
- Mukherjee, R.; Chakrabarti, O. Ubiquitin-mediated regulation of the E3 ligase GP78 by MGRN1 in trans affects mitochondrial homeostasis. J. Cell Sci. 2016, 129, 757–773. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Beninca, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanki, T.; Klionsky, D.J. Atg32 is a tag for mitochondria degradation in yeast. Autophagy 2009, 5, 1201–1202. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. A landmark protein essential for mitophagy: Atg32 recruits the autophagic machinery to mitochondria. Autophagy 2009, 5, 1203–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakawa, T.; Okamoto, K.; Omiya, S.; Taneike, M.; Yamaguchi, O.; Otsu, K. A Mammalian Mitophagy Receptor, Bcl2-L-13, Recruits the ULK1 Complex to Induce Mitophagy. Cell Rep. 2019, 26, 338–345.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sandoval, H.; Wang, J. Selective mitochondrial autophagy during erythroid maturation. Autophagy 2008, 4, 926–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ney, P.A. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008, 4, 354–356. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Lee, Y.; Rikka, S.; Sayen, M.R.; Molkentin, J.D.; Gottlieb, R.A.; Gustafsson, A.B. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J. Mol. Cell Cardiol. 2010, 48, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Rikka, S.; Quinsay, M.N.; Thomas, R.L.; Kubli, D.A.; Zhang, X.; Murphy, A.N.; Gustafsson, A.B. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011, 18, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, V.; Klionsky, D.J. PHB2/prohibitin 2: An inner membrane mitophagy receptor. Cell Res. 2017, 27, 311–312. [Google Scholar] [CrossRef]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Strappazzon, F.; Caruana, I.; Meneghetti, G.; Di Rita, A.; Simula, L.; Weber, G.; Del Bufalo, F.; Dalla Valle, L.; Campello, S.; et al. Reversible induction of mitophagy by an optogenetic bimodular system. Nat. Commun. 2019, 10, 1533. [Google Scholar] [CrossRef]
- Abudu, Y.P.; Pankiv, S.; Mathai, B.J.; Lamark, T.; Johansen, T.; Simonsen, A. NIPSNAP1 and NIPSNAP2 act as “eat me” signals to allow sustained recruitment of autophagy receptors during mitophagy. Autophagy 2019, 15, 1845–1847. [Google Scholar] [CrossRef]
- Sugo, M.; Kimura, H.; Arasaki, K.; Amemiya, T.; Hirota, N.; Dohmae, N.; Imai, Y.; Inoshita, T.; Shiba-Fukushima, K.; Hattori, N.; et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018, 37, e98899. [Google Scholar] [CrossRef]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Munch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 2018, 7, e31326. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Choi, S.G.; Yoo, S.M.; Son, J.H.; Jung, Y.K. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014, 10, 1906–1920. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; Van Houten, B.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Livesey, K.M.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy 2011, 7, 1256–1258. [Google Scholar] [CrossRef] [Green Version]
- Padman, B.S.; Nguyen, T.N.; Lazarou, M. Autophagosome formation and cargo sequestration in the absence of LC3/GABARAPs. Autophagy 2017, 13, 772–774. [Google Scholar] [CrossRef] [Green Version]
- Furuya, N.; Kakuta, S.; Sumiyoshi, K.; Ando, M.; Nonaka, R.; Suzuki, A.; Kazuno, S.; Saiki, S.; Hattori, N. NDP52 interacts with mitochondrial RNA poly(A) polymerase to promote mitophagy. EMBO Rep. 2018, 19, e46363. [Google Scholar] [CrossRef]
- Wang, L.; Qi, H.; Tang, Y.; Shen, H.M. Post-translational Modifications of Key Machinery in the Control of Mitophagy. Trends Biochem. Sci. 2020, 45, 58–75. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Barini, E.; Pohjolan-Pirhonen, R.; Brooks, S.P.; Singh, F.; Burel, S.; Balk, K.; Kumar, A.; Montava-Garriga, L.; Prescott, A.R.; et al. Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 2018, 8, 180108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauer, T.; Komander, D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba-Fukushima, K.; Arano, T.; Matsumoto, G.; Inoshita, T.; Yoshida, S.; Ishihama, Y.; Ryu, K.Y.; Nukina, N.; Hattori, N.; Imai, Y. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 2014, 10, e1004861. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [Green Version]
- Wauer, T.; Swatek, K.N.; Wagstaff, J.L.; Gladkova, C.; Pruneda, J.N.; Michel, M.A.; Gersch, M.; Johnson, C.M.; Freund, S.M.; Komander, D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015, 34, 307–325. [Google Scholar] [CrossRef]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Safiulina, D.; Kuum, M.; Choubey, V.; Gogichaishvili, N.; Liiv, J.; Hickey, M.A.; Cagalinec, M.; Mandel, M.; Zeb, A.; Liiv, M.; et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019, 38, e99384. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Xu, D.; Wang, Y.; Zhou, Z.; Liu, J.; Hu, S.; Gong, Y.; Yuan, J.; Pan, L. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 2018, 14, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Pollock, S.R.; Schinlever, A.R.; Rohani, A.; Kashatus, J.A.; Kashatus, D.F. RalA and RalB relocalization to depolarized mitochondria depends on clathrin-mediated endocytosis and facilitates TBK1 activation. PLoS ONE 2019, 14, e0214764. [Google Scholar] [CrossRef]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; Kishi-Itakura, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, T.; Sano, S.; Minowa-Nozawa, A.; Toh, H.; Nakajima, S.; Murase, K.; Aikawa, C.; Nakagawa, I. TBC1D9 regulates TBK1 activation through Ca(2+) signaling in selective autophagy. Nat. Commun. 2020, 11, 770. [Google Scholar] [CrossRef]
- Kravic, B.; Harbauer, A.B.; Romanello, V.; Simeone, L.; Vogtle, F.N.; Kaiser, T.; Straubinger, M.; Huraskin, D.; Bottcher, M.; Cerqua, C.; et al. In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy 2018, 14, 311–335. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Koyama-Honda, I.; Mizushima, N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 2012, 125, 1488–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, K.; Fukuda, T.; Yamashita, S.I.; Saigusa, T.; Kurihara, Y.; Yoshida, Y.; Kirisako, H.; Nakatogawa, H.; Kanki, T. The PP2A-like Protein Phosphatase Ppg1 and the Far Complex Cooperatively Counteract CK2-Mediated Phosphorylation of Atg32 to Inhibit Mitophagy. Cell Rep. 2018, 23, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Tang, Y.; Shen, H.M. PTEN-L puts a brake on mitophagy. Autophagy 2018, 14, 2023–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Harper, N.J.; Paulo, J.A.; Li, M.; Xu, Q.; Coughlin, M.; Elledge, S.J.; Harper, J.W. Integrated proteogenetic analysis reveals the landscape of a mitochondrial-autophagosome synapse during PARK2-dependent mitophagy. Sci. Adv. 2019, 5, eaay4624. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Di Rita, A.; Peschiaroli, A.; Leoncini, P.P.; Locatelli, F.; Melino, G.; Cecconi, F. HUWE1 controls MCL1 stability to unleash AMBRA1-induced mitophagy. Cell Death Differ. 2019, 27, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Yamano, K.; Kosako, H.; Tanaka, K.; Matsuda, N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem. 2019, 294, 10300–10314. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Gersch, M.; Gladkova, C.; Schubert, A.F.; Michel, M.A.; Maslen, S.; Komander, D. Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat. Struct. Mol. Biol. 2017, 24, 920–930. [Google Scholar] [CrossRef]
- Durcan, T.M.; Tang, M.Y.; Perusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, J.; von Stockum, S.; Marchesan, E.; Caicci, F.; Ferrari, V.; Rakovic, A.; Klein, C.; Antonini, A.; Bubacco, L.; Ziviani, E. USP14 inhibition corrects an in vivo model of impaired mitophagy. EMBO Mol. Med. 2018, 10, e9014. [Google Scholar] [CrossRef]
- Niu, K.; Fang, H.; Chen, Z.; Zhu, Y.; Tan, Q.; Wei, D.; Li, Y.; Balajee, A.S.; Zhao, Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 2019. [Google Scholar] [CrossRef]
- Geisler, S.; Jager, L.; Golombek, S.; Nakanishi, E.; Hans, F.; Casadei, N.; Terradas, A.L.; Linnemann, C.; Kahle, P.J. Ubiquitin-specific protease USP36 knockdown impairs Parkin-dependent mitophagy via downregulation of Beclin-1-associated autophagy-related ATG14L. Exp. Cell Res. 2019, 384, 111641. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Di Sante, G.; Pestell, T.G.; Casimiro, M.C.; Bisetto, S.; Powell, M.J.; Lisanti, M.P.; Cordon-Cardo, C.; Castillo-Martin, M.; Bonal, D.M.; Debattisti, V.; et al. Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays PARK2 translocation to mitochondria. Am. J. Pathol. 2015, 185, 266–279. [Google Scholar] [CrossRef]
- Webster, B.R.; Scott, I.; Han, K.; Li, J.H.; Lu, Z.; Stevens, M.V.; Malide, D.; Chen, Y.; Samsel, L.; Connelly, P.S.; et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J. Cell Sci. 2013, 126, 4843–4849. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. Protein N-terminal Acetylation by the NatA Complex Is Critical for Selective Mitochondrial Degradation. J. Biol. Chem. 2015, 290, 25034–25044. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Peng, Z.; Li, P.; Fu, H.; Feng, J.; Zhang, Y.; Liu, T.; Liu, Y.; Liu, Q.; Liu, Q.; et al. lncRNA RMST Suppressed GBM Cell Mitophagy through Enhancing FUS SUMOylation. Mol. Ther. Nucleic Acids 2020, 19, 1198–1208. [Google Scholar] [CrossRef]
- Pfeifer, U. Cellular autophagy: Glycogen segregation in early stages of a partial liver atrophy. Virchows Arch. B Cell Pathol. 1970, 5, 242–253. [Google Scholar]
- Pfeifer, U. Cellular autophagy and cell atrophy in the rat liver during long-term starvation. A quantitative morphological study with regard to diurnal variations. Virchows Arch. B Cell Pathol. 1973, 12, 195–211. [Google Scholar]
- Krustev, L.P. Cell autophagy of the liver in starvation and undernutrition. Bibl. Nutr. Details 1976, 23, 145–154. [Google Scholar]
- Salas, M.; Tuchweber, B.; Kourounakis, P.; Selye, H. Temperature-dependence of stress-induced hepatic autophagy. Experientia 1977, 33, 612–614. [Google Scholar] [CrossRef]
- Schworer, C.M.; Mortimore, G.E. Glucagon-induced autophagy and proteolysis in rat liver: Mediation by selective deprivation of intracellular amino acids. Proc. Natl. Acad. Sci. USA 1979, 76, 3169–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelburne, J.D.; Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. Cyclic AMP- and dibutyryl cyclic AMP-stimulated autophagy in rat liver. Am. J. Pathol. 1973, 72, 521–540. [Google Scholar] [PubMed]
- Aguas, A.P.; Soares, J.O.; Nunes, J.F. Autophagy in mouse hepatocytes induced by lysine acetylsalicylate. Experientia 1978, 34, 1618–1619. [Google Scholar] [CrossRef] [PubMed]
- Lardeux, B.R.; Mortimore, G.E. Amino acid and hormonal control of macromolecular turnover in perfused rat liver. Evidence for selective autophagy. J. Biol. Chem. 1987, 262, 14514–14519. [Google Scholar]
- Harada, M.; Hanada, S.; Toivola, D.M.; Ghori, N.; Omary, M.B. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008, 47, 2026–2035. [Google Scholar] [CrossRef]
- Strnad, P.; Zatloukal, K.; Stumptner, C.; Kulaksiz, H.; Denk, H. Mallory-Denk-bodies: Lessons from keratin-containing hepatic inclusion bodies. Biochim. Biophys. Acta 2008, 1782, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Harada, M. Autophagy is involved in the elimination of intracellular inclusions, Mallory-Denk bodies, in hepatocytes. Med. Mol. Morphol. 2010, 43, 13–18. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Kwanten, W.J.; Vandewynckel, Y.P.; Martinet, W.; De Winter, B.Y.; Michielsen, P.P.; Van Hoof, V.O.; Driessen, A.; Timmermans, J.P.; Bedossa, P.; Van Vlierberghe, H.; et al. Hepatocellular autophagy modulates the unfolded protein response and fasting-induced steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G599–G609. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Kai, Y.; Maldonado, E.; Currin, R.T.; Lemasters, J.J. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy 2009, 5, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Lemasters, J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Cell Physiol. 2011, 300, C308–C317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, G.; Donati, A.; Taddei, M.; Bergamini, E. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy 2007, 3, 26–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamini, E.; De Tata, V.; Cubeddu, T.L.; Masiello, P.; Pollera, M. Increased degradation in rat liver induced by antilipolytic agents: A model for studying autophagy and protein degradation in liver? Exp. Mol. Pathol. 1987, 46, 114–122. [Google Scholar] [CrossRef]
- Iwata, J.; Ezaki, J.; Komatsu, M.; Yokota, S.; Ueno, T.; Tanida, I.; Chiba, T.; Tanaka, K.; Kominami, E. Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 2006, 281, 4035–4041. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Makita, T. Inhibition of peroxisomal degradation by 3-methyladenine (3MA) in primary cultures of rat hepatocytes. Anat. Rec. 1997, 247, 449–454. [Google Scholar] [CrossRef]
- Locci Cubeddu, T.; Masiello, P.; Pollera, M.; Bergamini, E. Effects of antilipolytic agents on rat liver peroxisomes and peroxisomal oxidative activities. Biochim. Biophys. Acta 1985, 839, 96–104. [Google Scholar] [CrossRef]
- Luiken, J.J.; van den Berg, M.; Heikoop, J.C.; Meijer, A.J. Autophagic degradation of peroxisomes in isolated rat hepatocytes. FEBS Lett. 1992, 304, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Sartori, C.; Stefanini, S. Selective autophagy of clofibrate-induced rat liver peroxisomes. Cytochemistry and immunocytochemistry on tissue specimens and on fractions obtained by Nycodenz density gradient centrifugation. Cell Mol. Biol. 2000, 46, 1277–1290. [Google Scholar]
- Reinke, P.; David, H.; Uerlings, I.; Decker, T. Pathology of hepatic peroxisomes in chronic hepatitis B and immunosuppression. Exp. Pathol. 1988, 34, 71–77. [Google Scholar] [CrossRef]
- Walter, K.M.; Schonenberger, M.J.; Trotzmuller, M.; Horn, M.; Elsasser, H.P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2alpha promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.; Dariush Fahimi, H. Degradation of excess peroxisomes in mammalian liver cells by autophagy and other mechanisms. Histochem. Cell Biol. 2009, 131, 455–458. [Google Scholar] [CrossRef]
- Skop, V.; Cahova, M.; Papackova, Z.; Palenickova, E.; Dankova, H.; Baranowski, M.; Zabielski, P.; Zdychova, J.; Zidkova, J.; Kazdova, L. Autophagy-lysosomal pathway is involved in lipid degradation in rat liver. Physiol. Res. 2012, 61, 287–297. [Google Scholar] [PubMed]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Cheng, J.; Fujita, A.; Tokumoto, T.; Fujimoto, T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol. Biol. Cell 2006, 17, 2674–2683. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Cazals-Hatem, D.; Moreau, R.; Francoz, C.; Feldmann, G.; Lebrec, D.; Ogier-Denis, E.; Bedossa, P.; Valla, D.; Durand, F. Acute liver cell damage in patients with anorexia nervosa: A possible role of starvation-induced hepatocyte autophagy. Gastroenterology 2008, 135, 840–848.e3. [Google Scholar] [CrossRef]
- Wang, J.H.; Ahn, I.S.; Fischer, T.D.; Byeon, J.I.; Dunn, W.A., Jr.; Behrns, K.E.; Leeuwenburgh, C.; Kim, J.S. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology 2011, 141, 2188–2199.e6. [Google Scholar] [CrossRef] [Green Version]
- Shigemitsu, K.; Tsujishita, Y.; Hara, K.; Nanahoshi, M.; Avruch, J.; Yonezawa, K. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J. Biol. Chem. 1999, 274, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Ollinger, K.; Roberg, K. Nutrient deprivation of cultured rat hepatocytes increases the desferrioxamine-available iron pool and augments the sensitivity to hydrogen peroxide. J. Biol. Chem. 1997, 272, 23707–23711. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Dono, K.; Gotoh, K.; Shibata, M.; Koike, M.; Marubashi, S.; Miyamoto, A.; Takeda, Y.; Nagano, H.; Umeshita, K.; et al. Participation of autophagy in the degeneration process of rat hepatocytes after transplantation following prolonged cold preservation. Arch. Histol. Cytol. 2005, 68, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, D.H. The role of autophagy in alpha-1-antitrypsin deficiency: A specific cellular response in genetic diseases associated with aggregation-prone proteins. Autophagy 2006, 2, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ. 2009, 16, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. Liver injury in alpha1-antitrypsin deficiency: An aggregated protein induces mitochondrial injury. J. Clin. Investig. 2002, 110, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, D.; Blomenkamp, K.; Teckman, J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007, 46, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Wang, X.; Zhou, R.; Cederbaum, A. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem. Biophys. Res. Commun. 2010, 402, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Mells, J.E.; Fu, P.P.; Saxena, N.K.; Anania, F.A. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS ONE 2011, 6, e25269. [Google Scholar] [CrossRef]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [PubMed] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [PubMed]
- Sir, D.; Ann, D.K.; Ou, J.H. Autophagy by hepatitis B virus and for hepatitis B virus. Autophagy 2010, 6, 548–549. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.K.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef] [Green Version]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, L. Cytolysomes in Metabolically Active Cells. J. Cell Biol. 1963, 18, 478–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deter, R.L. Quantitative characterization of dense body, autophagic vacuole, and acid phosphatase-bearing particle populations during the early phases of glucagon-induced autophagy in rat liver. J. Cell Biol. 1971, 48, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.; Lawson, T.A.; Abbott, P.J.; Harmon, B.; Kerr, J.F. An electron-microscope study of the mode of cell death induced by cancer-chemotherapeutic agents in populations of proliferating normal and neoplastic cells. J. Pathol. 1975, 116, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L. Analog modeling of glucagon-induced autophagy in rat liver. I. Conceptual and mathematical model of telolysosome-autophagosome-autolysosome interaction. Exp. Cell Res. 1975, 94, 122–126. [Google Scholar] [CrossRef]
- Deter, R.L. Analog modeling of glucagon-induced autophagy in rat liver. II. Evaluation of iron labeling as a means for identifying telolysosome, autophagosome and autolysosome populations. Exp. Cell Res. 1975, 94, 127–139. [Google Scholar] [CrossRef]
- Schworer, C.M.; Cox, J.R.; Mortimore, G.E. Alteration of lysosomal density by sequestered glycogen during deprivation-induced autophagy in rat liver. Biochem. Biophys. Res. Commun. 1979, 87, 163–170. [Google Scholar] [CrossRef]
- Hopgood, M.F.; Clark, M.G.; Ballard, F.J. Protein degradation in hepatocyte monolayers. Effects of glucagon, adenosine 3′:5′-cyclic monophosphate and insulin. Biochem. J. 1980, 186, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Seglen, P.O.; Gordon, P.B.; Poli, A. Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim. Biophys. Acta 1980, 630, 103–118. [Google Scholar] [CrossRef]
- Marzella, L.; Sandberg, P.O.; Glaumann, H. Autophagic degradation in rat liver after vinblastine treatment. Exp. Cell Res. 1980, 128, 291–301. [Google Scholar] [CrossRef]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Isolation of autophagic vacuoles from rat liver: Morphological and biochemical characterization. J. Cell Biol. 1982, 93, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Stromhaug, P.E.; Berg, T.O.; Fengsrud, M.; Seglen, P.O. Purification and characterization of autophagosomes from rat hepatocytes. Biochem. J. 1998, 335, 217–224. [Google Scholar] [CrossRef]
- Seglen, P.O.; Brinchmann, M.F. Purification of autophagosomes from rat hepatocytes. Autophagy 2010, 6, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.L.; Roche, E.; Knecht, E.; Grisolia, S. Differences in the half-lives of some mitochondrial rat liver enzymes may derive partially from hepatocyte heterogeneity. FEBS Lett. 1987, 224, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Jorda, A.; Perez-Pastor, E.; Portoles, M. Effect of streptozotocin-diabetes on rat liver mitochondrial adenosine triphosphatase turnover. Biochem. J. 1988, 251, 621–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, E.; Martinez-Ramon, A.; Grisolia, S. Autophagy of mitochondria in rat liver assessed by immunogold procedures. J. Histochem. Cytochem. 1988, 36, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Venezuela. Decree No. 2006. partially amending Decree No. 1906 of 30 December 1987, published in La Gaceta Oficial of the same date, 18 February 1988. Annu. Rev. Popul. Law 1988, 15, 112. [Google Scholar]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.K.; Loethen, S.; Perlmutter, D.H. Fasting in alpha1-antitrypsin deficient liver: Constitutive [correction of consultative] activation of autophagy. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1156–G1165. [Google Scholar] [CrossRef]
- Donati, A.; Taddei, M.; Cavallini, G.; Bergamini, E. Stimulation of macroautophagy can rescue older cells from 8-OHdG mtDNA accumulation: A safe and easy way to meet goals in the SENS agenda. Rejuvenat. Res. 2006, 9, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Glick, D.; Zhang, W.; Beaton, M.; Marsboom, G.; Gruber, M.; Simon, M.C.; Hart, J.; Dorn, G.W., 2nd; Brady, M.J.; Macleod, K.F. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell Biol. 2012, 32, 2570–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Nitta, T.; Mohuczy, D.; O’Malley, K.A.; Moldawer, L.L.; Dunn, W.A., Jr.; Behrns, K.E. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology 2008, 47, 1725–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ruan, D.Y.; Jia, C.C.; Zheng, J.; Wang, G.Y.; Zhao, H.; Yang, Q.; Liu, W.; Yi, S.H.; Li, H.; et al. Aging aggravates hepatic ischemia-reperfusion injury in mice by impairing mitophagy with the involvement of the EIF2alpha-parkin pathway. Aging 2018, 10, 1902–1920. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.M.; Ding, Y.; Ding, W.X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ding, W.X. Mitophagy, mitochondrial spheroids, and mitochondrial-derived vesicles in alcohol-induced liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G515. [Google Scholar] [CrossRef] [Green Version]
- Eid, N.; Ito, Y.; Otsuki, Y. Mitophagy in steatotic hepatocytes of ethanol-treated wild-type and Parkin knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G513–G514. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Gortat, A.; Blas-Garcia, A.; Esplugues, J.V. Autophagy as a rescue mechanism in efavirenz-induced mitochondrial dysfunction: A lesson from hepatic cells. Autophagy 2011, 7, 1402–1404. [Google Scholar] [CrossRef]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Gortat, A.; Blas-Garcia, A.; Esplugues, J.V. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology 2011, 54, 1009–1019. [Google Scholar] [CrossRef]
- Pi, H.; Xu, S.; Zhang, L.; Guo, P.; Li, Y.; Xie, J.; Tian, L.; He, M.; Lu, Y.; Li, M.; et al. Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy 2013, 9, 1780–1800. [Google Scholar] [CrossRef]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wu, F.; Lin, S.; Pan, X.; Jin, L.; Lu, T.; Shi, L.; Wang, Y.; Xu, A.; Li, X. Adiponectin protects against acetaminophen-induced mitochondrial dysfunction and acute liver injury by promoting autophagy in mice. J. Hepatol. 2014, 61, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.M.; Haynes, A.; Manley, S.; Li, Y.; Jaeschke, H.; Ding, W.X. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice. J. Biol. Chem. 2015, 290, 10934–10946. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ni, H.M.; Chao, X.; Ma, X.; Rodriguez, Y.A.; Chavan, H.; Wang, S.; Krishnamurthy, P.; Dobrowsky, R.; Xu, D.X.; et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol. 2019, 22, 101148. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Shan, S.; Shen, Z.; Zhang, C.; Kou, R.; Xie, K.; Song, F. Mitophagy protects against acetaminophen-induced acute liver injury in mice through inhibiting NLRP3 inflammasome activation. Biochem. Pharmacol. 2019, 169, 113643. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Mahadeva, R. Alpha1-antitrypsin polymerization and the serpinopathies: Pathobiology and prospects for therapy. J. Clin. Investig. 2002, 110, 1585–1590. [Google Scholar] [CrossRef]
- Huber, R.; Carrell, R.W. Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry 1989, 28, 8951–8966. [Google Scholar] [CrossRef]
- Teckman, J.H.; Qu, D.; Perlmutter, D.H. Molecular pathogenesis of liver disease in alpha1-antitrypsin deficiency. Hepatology 1996, 24, 1504–1516. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. alpha1-Antitrypsin deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef]
- Lomas, D.A.; Hurst, J.R.; Gooptu, B. Update on alpha-1 antitrypsin deficiency: New therapies. J. Hepatol. 2016, 65, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Rudnick, D.A.; Perlmutter, D.H. Alpha-1-antitrypsin deficiency: A new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology 2005, 42, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Sifers, R.N.; Rogers, B.B.; Hawkins, H.K.; Finegold, M.J.; Woo, S.L. Elevated synthesis of human alpha 1-antitrypsin hinders the secretion of murine alpha 1-antitrypsin from hepatocytes of transgenic mice. J. Biol. Chem. 1989, 264, 15696–15700. [Google Scholar] [PubMed]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlmutter, D.H. Alpha1-antitrypsin deficiency: Liver disease associated with retention of a mutant secretory glycoprotein in the endoplasmic reticulum. Methods Mol. Biol. 2003, 232, 39–56. [Google Scholar] [PubMed]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; Perlmutter, D.H. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G961–G974. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Lomas, D.A. Alpha1-antitrypsin deficiency and autophagy. N. Engl. J. Med. 2010, 363, 1863–1864. [Google Scholar] [CrossRef]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell 2006, 17, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. 2010, 235, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, T.; Ohsaki, Y.; Suzuki, M.; Shinohara, Y.; Tatematsu, T.; Cheng, J.; Okada, M.; Ohmiya, N.; Hirooka, Y.; Goto, H.; et al. Inhibition of Niemann-Pick-type C1-like1 by ezetimibe activates autophagy in human hepatocytes and reduces mutant alpha1-antitrypsin Z deposition. Hepatology 2014, 59, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cobanoglu, M.C.; Li, J.; Hidvegi, T.; Hale, P.; Ewing, M.; Chu, A.S.; Gong, Z.; Muzumdar, R.; Pak, S.C.; et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded alpha1-antitrypsin Z. PLoS ONE 2019, 14, e0209748. [Google Scholar]
- Biel, T.G.; Lee, S.; Flores-Toro, J.A.; Dean, J.W.; Go, K.L.; Lee, M.H.; Law, B.K.; Law, M.E.; Dunn, W.A., Jr.; Zendejas, I.; et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 2016, 23, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.K.; Lee, S.; Flores-Toro, J.U.R.Y.; Yang, M.J.; Go, K.L.; Biel, T.G.; Miney, C.E.; Pierre Louis, S.; Law, B.K.; Law, M.E.; et al. Loss of sirtuin 1 and mitofusin 2 contributes to enhanced ischemia/reperfusion injury in aged livers. Aging Cell 2018, 17, e12761. [Google Scholar] [CrossRef]
- Ning, X.J.; Yan, X.; Wang, Y.F.; Wang, R.; Fan, X.L.; Zhong, Z.B.; Ye, Q.F. Parkin deficiency elevates hepatic ischemia/reperfusion injury accompanying decreased mitochondrial autophagy, increased apoptosis, impaired DNA damage repair and altered cell cycle distribution. Mol. Med. Rep. 2018, 18, 5663–5668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.M.; Lee, S.M. Heme oxygenase-1 protects liver against ischemia/reperfusion injury via phosphoglycerate mutase family member 5-mediated mitochondrial quality control. Life Sci. 2018, 200, 94–104. [Google Scholar] [CrossRef]
- Sun, X.L.; Zhang, Y.L.; Xi, S.M.; Ma, L.J.; Li, S.P. MiR-330-3p suppresses phosphoglycerate mutase family member 5 -inducted mitophagy to alleviate hepatic ischemia-reperfusion injury. J. Cell Biochem. 2019, 120, 4255–4267. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Yin, X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Yao, L.; Song, Q.; Zhu, L.; Xia, Z.; Xia, H.; Jiang, X.; Chen, J.; Chen, H. The association of autophagy with polyethylenimine-induced cytotoxicity in nephritic and hepatic cell lines. Biomaterials 2011, 32, 8613–8625. [Google Scholar] [CrossRef]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.H.; Ke, P.Y.; Liao, C.J.; Wu, S.M.; Chi, H.C.; Tsai, C.Y.; Chen, C.Y.; Lin, Y.H.; Lin, K.H. Chromosome 19 open reading frame 80 is upregulated by thyroid hormone and modulates autophagy and lipid metabolism. Autophagy 2014, 10, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.V.; Dincsoy, H.P. The significance of giant mitochondria in liver biopsies as observed by light microscopy. Am. J. Clin. Pathol. 1982, 78, 293–298. [Google Scholar] [CrossRef]
- Bruguera, M.; Bertran, A.; Bombi, J.A.; Rodes, J. Giant mitochondria in hepatocytes: A diagnostic hint for alcoholic liver disease. Gastroenterology 1977, 73, 1383–1387. [Google Scholar] [CrossRef]
- Gordon, E.R. Mitochondrial functions in an ethanol-induced fatty liver. J. Biol. Chem. 1973, 248, 8271–8280. [Google Scholar]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef]
- Eid, N.; Ito, Y.; Horibe, A.; Otsuki, Y. Ethanol-induced mitophagy in liver is associated with activation of the PINK1-Parkin pathway triggered by oxidative DNA damage. Histol. Histopathol. 2016, 31, 1143–1159. [Google Scholar]
- Eid, N.; Ito, Y.; Horibe, A.; Otsuki, Y.; Kondo, Y. Ethanol-Induced Mitochondrial Damage in Sertoli Cells is Associated with Parkin Overexpression and Activation of Mitophagy. Cells 2019, 8, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Xu, Y.; Zhang, S.; Sun, J.; Liu, P.; Xiao, L.; Tang, Y.; Liu, L.; Yao, P. Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy. Nutrients 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.A.; Yen, P.M. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.; Liu, K.; Liu, D.; Lv, F.; Zang, Y.; Xie, F.; Yin, J.; Shi, Y.; Wang, Y.; Chen, D. Differential effects of reticulophagy and mitophagy on nonalcoholic fatty liver disease. Cell Death Dis. 2018, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic Mitochondrial Defects in a Nonalcoholic Fatty Liver Disease Mouse Model Are Associated with Increased Degradation of Oxidative Phosphorylation Subunits. Mol. Cell Proteom. 2018, 17, 2371–2386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Shao, N.; Yu, X.Y.; Ma, X.F.; Lin, W.J.; Hao, M.; Kuang, H.Y. Exenatide Delays the Progression of Nonalcoholic Fatty Liver Disease in C57BL/6 Mice, Which May Involve Inhibition of the NLRP3 Inflammasome through the Mitophagy Pathway. Gastroenterol. Res. Pract. 2018, 2018, 1864307. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Annau, E.; Manginelli, A.; Roth, A. Alteration of the mitochondrial pattern in the liver of tumor-bearing mice. Cancer Res. 1951, 11, 404–405. [Google Scholar]
- Hogeboom, G.H.; Schneider, W.C. Proteins of liver and hepatoma mitochondria. Science 1951, 113, 355–358. [Google Scholar] [CrossRef]
- Hruban, Z. Ultrastructure of hepatocellular tumors. J. Toxicol. Environ. Health 1979, 5, 403–433. [Google Scholar] [CrossRef]
- Lipsky, M.M.; Hinton, D.E.; Klaunig, J.E.; Trump, B.F. Biology of hepatocellular neoplasia in the mouse. III. Electron microscopy of safrole-induced hepatocellular adenomas and hepatocellular carcinomas. J. Natl. Cancer Inst. 1981, 67, 393–405. [Google Scholar]
- Chang, C.P.; Yang, M.C.; Liu, H.S.; Lin, Y.S.; Lei, H.Y. Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology 2007, 45, 286–296. [Google Scholar] [CrossRef]
- Lei, H.Y.; Chang, C.P. Induction of autophagy by concanavalin A and its application in anti-tumor therapy. Autophagy 2007, 3, 402–404. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Yang, Y. Alterations of cellular organelles in human liver-derived hepatoma G2 cells induced by adriamycin. Anticancer Drugs 2009, 20, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Yang, Y.; Wang, X. Curcumin enhanced adriamycin-induced human liver-derived Hepatoma G2 cell death through activation of mitochondria-mediated apoptosis and autophagy. Eur J. Pharm. Sci. 2011, 43, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Dominguez, N.; Ordonez, R.; Fernandez, A.; Mendez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Mauriz, J.L.; Gonzalez-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shi, Y.; Guo, X.H.; Ouyang, Y.B.; Wang, S.S.; Liu, D.J.; Wang, A.N.; Li, N.; Chen, D.X. Phosphorylated AKT inhibits the apoptosis induced by DRAM-mediated mitophagy in hepatocellular carcinoma by preventing the translocation of DRAM to mitochondria. Cell Death Dis. 2014, 5, e1078. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Yu, H.; Cui, N.; Kong, X.; Liu, X.; Chang, Y.; Wu, Y.; Sun, L.; Wang, G. ABT737 enhances cholangiocarcinoma sensitivity to cisplatin through regulation of mitochondrial dynamics. Exp. Cell Res. 2015, 335, 68–81. [Google Scholar] [CrossRef]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. Hepatitis C Virus Induces the Localization of Lipid Rafts to Autophagosomes for Its RNA Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog. 2017, 13, e1006609. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ou, J.J. Regulation of Apolipoprotein E Trafficking by Hepatitis C Virus-induced Autophagy. J. Virol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Elgner, F.; Jiang, B.; Himmelsbach, K.; Medvedev, R.; Ploen, D.; Hildt, E. The Autophagosomal SNARE Protein Syntaxin 17 Is an Essential Factor for the Hepatitis C Virus Life Cycle. J. Virol. 2016, 90, 5989–6000. [Google Scholar] [CrossRef] [Green Version]
- Estrabaud, E.; De Muynck, S.; Asselah, T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J. Hepatol. 2011, 55, 1150–1153. [Google Scholar] [CrossRef] [Green Version]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef] [Green Version]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Jassey, A.; Liu, C.H.; Changou, C.A.; Richardson, C.D.; Hsu, H.Y.; Lin, L.T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009, 49, 60–71. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011, 85, 6319–6333. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, J.; Liu, Y.; Zeng, X.; Wei, M.; Wu, S.; Xiong, Q.; Song, F.; Yuan, X.; Xiao, Y.; et al. Hepatitis B Virus Induces Autophagy to Promote its Replication by the Axis of miR-192-3p-XIAP Through NF kappa B Signaling. Hepatology 2019, 69, 974–992. [Google Scholar] [CrossRef]
- Chen, L.; Ming, X.; Li, W.; Bi, M.; Yan, B.; Wang, X.; Yang, P.; Yang, B. The microRNA-155 mediates hepatitis B virus replication by reinforcing SOCS1 signalling-induced autophagy. Cell Biochem. Funct. 2020. [Google Scholar] [CrossRef]
- Inoue, J.; Krueger, E.W.; Chen, J.; Cao, H.; Ninomiya, M.; McNiven, M.A. HBV secretion is regulated through the activation of endocytic and autophagic compartments mediated by Rab7 stimulation. J. Cell Sci. 2015, 128, 1696–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doring, T.; Zeyen, L.; Bartusch, C.; Prange, R. Hepatitis B Virus Subverts the Autophagy Elongation Complex Atg5-12/16L1 and Does Not Require Atg8/LC3 Lipidation for Viral Maturation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.X.; Wong, S.H.; Chan, M.T.; Yu, L.; Yu, S.S.; Wu, F.; Xiao, Z.; Wang, X.; Zhang, L.; Cheng, A.S.; et al. Autophagy Mediates HBx-Induced Nuclear Factor-kappaB Activation and Release of IL-6, IL-8, and CXCL2 in Hepatocytes. J. Cell Physiol. 2015, 230, 2382–2389. [Google Scholar] [CrossRef]
- Hu, S.; Liu, X.; Gao, Y.; Zhou, R.; Wei, M.; Dong, J.; Yan, H.; Zhao, Y. Hepatitis B Virus Inhibits Neutrophil Extracellular Trap Release by Modulating Reactive Oxygen Species Production and Autophagy. J. Immunol. 2019, 202, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.H.; Wu, S.Y.; Zuchini, R.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Lin, Y.J.; Wu, C.T.; Liu, H.S. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014, 59, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wu, C.; Wang, X.; Liu, S.; Kemper, T.; Li, F.; Squire, A.; Zhu, Y.; Zhang, J.; Chen, X.; et al. Synaptosomal-associated protein 29 is required for the autophagic degradation of hepatitis B virus. FASEB J. 2019, 33, 6023–6034. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, C.; Wang, X.; Liu, S.; Zhao, K.; Kemper, T.; Yu, H.; Li, M.; Zhang, J.; Chen, M.; et al. Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling. Autophagy 2020, 16, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014, 10, 416–430. [Google Scholar] [CrossRef]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Li, D.; Chen, Z.X.; Huang, Y.H.; Gao, W.Y.; Zheng, B.Y.; Wang, X.Z. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp. Cell Res. 2018, 368, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Hong, J.M.; Lee, S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016, 60, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, G.; Wang, J.; Deng, M.; Yuan, F.; Gong, J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020, 53, e12731. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.N.; Wang, G.H.; Zhou, F.; Hao, J.J.; Tian, L.; Guan, L.F.; Geng, X.K.; Ding, Y.C.; Wu, H.W.; Zhang, K.Z. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol. Environ. Saf. 2019, 167, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Yao, N.; Hu, C.; Wu, Y.; Guo, D.; Liu, J.; Yang, Y.; Chen, T.; Zhao, Y.; et al. Role of mitophagy regulation by ROS in hepatic stellate cells during acute liver failure. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G374–G384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, P.; Wang, J.; Toan, S.; Ren, J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal. Transduct. Target. Ther. 2019, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef] [Green Version]
- Dumas, K.; Ayachi, C.; Gilleron, J.; Lacas-Gervais, S.; Pastor, F.; Favier, F.B.; Peraldi, P.; Vaillant, N.; Yvan-Charvet, L.; Bonnafous, S.; et al. REDD1 deficiency protects against nonalcoholic hepatic steatosis induced by high-fat diet. FASEB J. 2020. [Google Scholar] [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.K.; Huckestein, B.R.; Edmunds, L.R.; Petersen, M.C.; Nasiri, A.; Butrico, G.M.; Abulizi, A.; Harmon, D.B.; Lu, C.; Mantell, B.S.; et al. Reduced intestinal lipid absorption and body weight-independent improvements in insulin sensitivity in high-fat diet-fed Park2 knockout mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E105–E116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, L.R.; Huckestein, B.R.; Kahn, M.; Zhang, D.; Chu, Y.; Zhang, Y.; Wendell, S.G.; Shulman, G.I.; Jurczak, M.J. Hepatic insulin sensitivity is improved in high-fat diet-fed Park2 knockout mice in association with increased hepatic AMPK activation and reduced steatosis. Physiol. Rep. 2019, 7, e14281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A DeltaPsim independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Felix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Zhou, B.; Kreuzer, J.; Kumsta, C.; Wu, L.; Kamer, K.J.; Cedillo, L.; Zhang, Y.; Li, S.; Kacergis, M.C.; Webster, C.M.; et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell 2019, 177, 299–314.e16. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Overview of the molecular mechanisms regulating autophagy. Autophagy is classified into three major types: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy. (A) Microautophagy is a dynamic lysosomal membrane process that directly enwraps and deliver the intracellular portions to the lumen of lysosomes for degradation. (B) CMA is a lysosomal degradation process that involves the recognition of proteins containing the KFERQ-like motif by HSC70 and transport into the lysosomal lumen via the interaction with LAMP2A. (C) Macroautophagy (referred to here as autophagy) is a degradation pathway that promotes membrane rearrangement to generate vacuoles to engulf the cytosolic components targeted for degradation within lysosome. Two distinct metabolic sensors, AMPK and the mTOR complex, control the initiation of autophagy. Nutrient starvation in cells triggers the suppression of mTOR by AMPK1and the translocation of the ULK1/2 complex (ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101) to the autophagy initiation site. Subsequently, the ULK1/2 complex leads to the recruitment and activation of the class III phosphatidylinositol-3-OH kinase (class III-PI3K complex, including Vps34/PI3KC3, Vps15, Beclin 1, and ATG14) to synthesize PtdIn(3)P. The generated PtdIn(3)P then recruits DFCP1 and WIPI family proteins to the ER-associated membrane compartment to induce the formation of the isolation membrane (IM)/phagophore. In addition to the ER, other organelles, such as the plasma membrane, mitochondria, Golgi apparatus, and recycling endosome, also supply membrane resources required for the membrane nucleation and the formation of phagophore in the initial step of autophagy. ATG9, VMP1, and coated protein complex-associated vesicles are involved in the trafficking of membrane resources for the formation of the phagophore. The expansion and elongation of the phagophore to form enclosed autophagosomes rely on two ubiquitin-like (UBL) conjugation systems. First, ATG12 is conjugated to ATG5 via ATG7 (ubiquitin activating enzyme 1, E1) and ATG10 (ubiquitin conjugation enzyme 2, E2), yielding an ATG5-ATG12 conjugate that binds to ATG16L to form an ATG5-ATG12-ATG16L complex. Second, ATG8/LC3 family proteins are post-translationally processed by a cysteine protease ATG4 to form ATG8/LC3-I. Then, ATG7 (E1) and ATG3 (E2) enzyme cascades mediate the covalent linkage of ATG8/LC3-I to PE to form the lipidated form of LC3 (ATG8/LC3-PE, also known as ATG8/LC3-II). The enclosed autophagosomes fuse with lysosomes to form mature autolysosomes, which eliminate the engulfed materials. The interactions of RAB) with FYCO1 and RILP regulate the fusion of autophagosomes with lysosomes. In addition, several protein-protein interactions and the assembly of protein complexes, including the interaction between UVRAG and RAB7, the association of PLEKHM1 with the HOPS complex, and the formation of a protein complex containing ATG14L, STX17, SNAP29, and VAMP8, also regulate autophagosome-lysosome fusion.
Figure 1.
Overview of the molecular mechanisms regulating autophagy. Autophagy is classified into three major types: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy. (A) Microautophagy is a dynamic lysosomal membrane process that directly enwraps and deliver the intracellular portions to the lumen of lysosomes for degradation. (B) CMA is a lysosomal degradation process that involves the recognition of proteins containing the KFERQ-like motif by HSC70 and transport into the lysosomal lumen via the interaction with LAMP2A. (C) Macroautophagy (referred to here as autophagy) is a degradation pathway that promotes membrane rearrangement to generate vacuoles to engulf the cytosolic components targeted for degradation within lysosome. Two distinct metabolic sensors, AMPK and the mTOR complex, control the initiation of autophagy. Nutrient starvation in cells triggers the suppression of mTOR by AMPK1and the translocation of the ULK1/2 complex (ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101) to the autophagy initiation site. Subsequently, the ULK1/2 complex leads to the recruitment and activation of the class III phosphatidylinositol-3-OH kinase (class III-PI3K complex, including Vps34/PI3KC3, Vps15, Beclin 1, and ATG14) to synthesize PtdIn(3)P. The generated PtdIn(3)P then recruits DFCP1 and WIPI family proteins to the ER-associated membrane compartment to induce the formation of the isolation membrane (IM)/phagophore. In addition to the ER, other organelles, such as the plasma membrane, mitochondria, Golgi apparatus, and recycling endosome, also supply membrane resources required for the membrane nucleation and the formation of phagophore in the initial step of autophagy. ATG9, VMP1, and coated protein complex-associated vesicles are involved in the trafficking of membrane resources for the formation of the phagophore. The expansion and elongation of the phagophore to form enclosed autophagosomes rely on two ubiquitin-like (UBL) conjugation systems. First, ATG12 is conjugated to ATG5 via ATG7 (ubiquitin activating enzyme 1, E1) and ATG10 (ubiquitin conjugation enzyme 2, E2), yielding an ATG5-ATG12 conjugate that binds to ATG16L to form an ATG5-ATG12-ATG16L complex. Second, ATG8/LC3 family proteins are post-translationally processed by a cysteine protease ATG4 to form ATG8/LC3-I. Then, ATG7 (E1) and ATG3 (E2) enzyme cascades mediate the covalent linkage of ATG8/LC3-I to PE to form the lipidated form of LC3 (ATG8/LC3-PE, also known as ATG8/LC3-II). The enclosed autophagosomes fuse with lysosomes to form mature autolysosomes, which eliminate the engulfed materials. The interactions of RAB) with FYCO1 and RILP regulate the fusion of autophagosomes with lysosomes. In addition, several protein-protein interactions and the assembly of protein complexes, including the interaction between UVRAG and RAB7, the association of PLEKHM1 with the HOPS complex, and the formation of a protein complex containing ATG14L, STX17, SNAP29, and VAMP8, also regulate autophagosome-lysosome fusion.

Figure 2.
Regulation of selective autophagy by cargo receptors. (A) Selective autophagy is an elimination process that involves the targeting of cargoes to the autophagy machinery by the specific receptor proteins that contain an ATG8/LC3-interacting regions (LIRs) for the interaction with ATG8/LC3 located on the membrane of the IM/phagophore. Another type of selective autophagy is mediated by the interactions between adaptor proteins with cargo receptors and ATG8/LC3. The IM/phagophore encloses mature autophagosomes that fuse with lysosomes to form autolysosomes, in which the engulfed cargoes are degraded. Ubiquitination of cargo and/or the binding to additional adaptor proteins mediate the recognition of cargoes and cargo receptors. (B) Selective autophagy participates in the degradation of damaged organelles and aggregated proteins. Several mammalian and yeast cargo receptors have been identified to eliminate the dysfunctional mitochondria, damaged lysosomes, injured peroxisomes, stressed ER and infecting pathogens by selective autophagy (mitophagy, lysophagy, pexophagy, ER-phagy, and xenophagy, respectively). Moreover, other forms of cargoes, including LDs (lipophagy), ferritin (ferritinophagy), nucleus (nucleophagy), ribosome (ribophagy), protein aggregate (aggrephagy), are degraded by selective autophagy through a process mediated by identified and unknown cargo receptors. The yeast and mammalian cargo receptors responsible for each type of selective autophagy are listed in the table.
Figure 2.
Regulation of selective autophagy by cargo receptors. (A) Selective autophagy is an elimination process that involves the targeting of cargoes to the autophagy machinery by the specific receptor proteins that contain an ATG8/LC3-interacting regions (LIRs) for the interaction with ATG8/LC3 located on the membrane of the IM/phagophore. Another type of selective autophagy is mediated by the interactions between adaptor proteins with cargo receptors and ATG8/LC3. The IM/phagophore encloses mature autophagosomes that fuse with lysosomes to form autolysosomes, in which the engulfed cargoes are degraded. Ubiquitination of cargo and/or the binding to additional adaptor proteins mediate the recognition of cargoes and cargo receptors. (B) Selective autophagy participates in the degradation of damaged organelles and aggregated proteins. Several mammalian and yeast cargo receptors have been identified to eliminate the dysfunctional mitochondria, damaged lysosomes, injured peroxisomes, stressed ER and infecting pathogens by selective autophagy (mitophagy, lysophagy, pexophagy, ER-phagy, and xenophagy, respectively). Moreover, other forms of cargoes, including LDs (lipophagy), ferritin (ferritinophagy), nucleus (nucleophagy), ribosome (ribophagy), protein aggregate (aggrephagy), are degraded by selective autophagy through a process mediated by identified and unknown cargo receptors. The yeast and mammalian cargo receptors responsible for each type of selective autophagy are listed in the table.

Figure 3.
Different types of mitophagy in yeast and mammals. In yeast cells, the mitochondrial outer membrane-associated protein ATG32 represents a receptor involved in mitophagy that interacts with ATG8 located on IM/phagophore through the WXXL-like motif. Another adaptor protein, ATG11, may mediate the interaction between ATG32 and ATG8 to facilitate mitophagy initiation. At least three types of mitophagy have been in mammals, including basal mitophagy, programmed mitophagy, and stress-induced mitophagy. Basal mitophagy has been shown to be activated in the tissues with large metabolic demands, but the specific receptor for basal mitophagy remains unclear. The cargo receptors of programmed mitophagy, including BNIP3 and BNIP3L/Nix, function in mitochondrial turnover for the maturation of erythrocytes and cardiomyocytes, and elimination of the parental mitochondrial genome during fertilization. Stress-induced mitophagy is classified into PINK1/Parkin-dependent mitophagy and PINK1/Parkin-independent mitophagy. For the activation of PINK1/Parkin-dependent mitophagy, mitochondrial depolarization triggers the suppression PINK1 degradation and leads to PINK1 accumulation on the MOM of damaged mitochondria. In turn, PINK1 phosphorylates ubiquitin and Parkin at serine residue 65 and then promotes the mitochondrial translocation of Parkin and mitochondrial ubiquitination by Parkin. On the other hand, several ubiquitin E3 ligases, including MUL1, SMURF1, SIAH1, GP78, ARIH1, and HUWE1, may promote the ubiquitination of mitochondrial proteins to activate PINK1/Parkin-independent mitophagy. Several non-mitochondrial cargo receptors, including p62/SQSTM1, Calcoco2 (also known as NDP52), OPTN, NBR1, and TAX1BP1, are responsible for PINK1/Parkin-dependent mitophagy. The phosphorylation of cargo receptors by tank-binding kinase 1, such as OPTN and p62/SQSTM1, facilitate the recognition process of mitophagy. Several MOM- and MIM-associated proteins, such as BNIP3, BNIP3L/Nix, FUNDC1, BCL2-L3, PHB2, ANT complex, and NIPSNAP1 and 2 have been identified as cargo receptors for mitophagy that directly bind to ATG8/LC3 located on the IM/phagophore.
Figure 3.
Different types of mitophagy in yeast and mammals. In yeast cells, the mitochondrial outer membrane-associated protein ATG32 represents a receptor involved in mitophagy that interacts with ATG8 located on IM/phagophore through the WXXL-like motif. Another adaptor protein, ATG11, may mediate the interaction between ATG32 and ATG8 to facilitate mitophagy initiation. At least three types of mitophagy have been in mammals, including basal mitophagy, programmed mitophagy, and stress-induced mitophagy. Basal mitophagy has been shown to be activated in the tissues with large metabolic demands, but the specific receptor for basal mitophagy remains unclear. The cargo receptors of programmed mitophagy, including BNIP3 and BNIP3L/Nix, function in mitochondrial turnover for the maturation of erythrocytes and cardiomyocytes, and elimination of the parental mitochondrial genome during fertilization. Stress-induced mitophagy is classified into PINK1/Parkin-dependent mitophagy and PINK1/Parkin-independent mitophagy. For the activation of PINK1/Parkin-dependent mitophagy, mitochondrial depolarization triggers the suppression PINK1 degradation and leads to PINK1 accumulation on the MOM of damaged mitochondria. In turn, PINK1 phosphorylates ubiquitin and Parkin at serine residue 65 and then promotes the mitochondrial translocation of Parkin and mitochondrial ubiquitination by Parkin. On the other hand, several ubiquitin E3 ligases, including MUL1, SMURF1, SIAH1, GP78, ARIH1, and HUWE1, may promote the ubiquitination of mitochondrial proteins to activate PINK1/Parkin-independent mitophagy. Several non-mitochondrial cargo receptors, including p62/SQSTM1, Calcoco2 (also known as NDP52), OPTN, NBR1, and TAX1BP1, are responsible for PINK1/Parkin-dependent mitophagy. The phosphorylation of cargo receptors by tank-binding kinase 1, such as OPTN and p62/SQSTM1, facilitate the recognition process of mitophagy. Several MOM- and MIM-associated proteins, such as BNIP3, BNIP3L/Nix, FUNDC1, BCL2-L3, PHB2, ANT complex, and NIPSNAP1 and 2 have been identified as cargo receptors for mitophagy that directly bind to ATG8/LC3 located on the IM/phagophore.


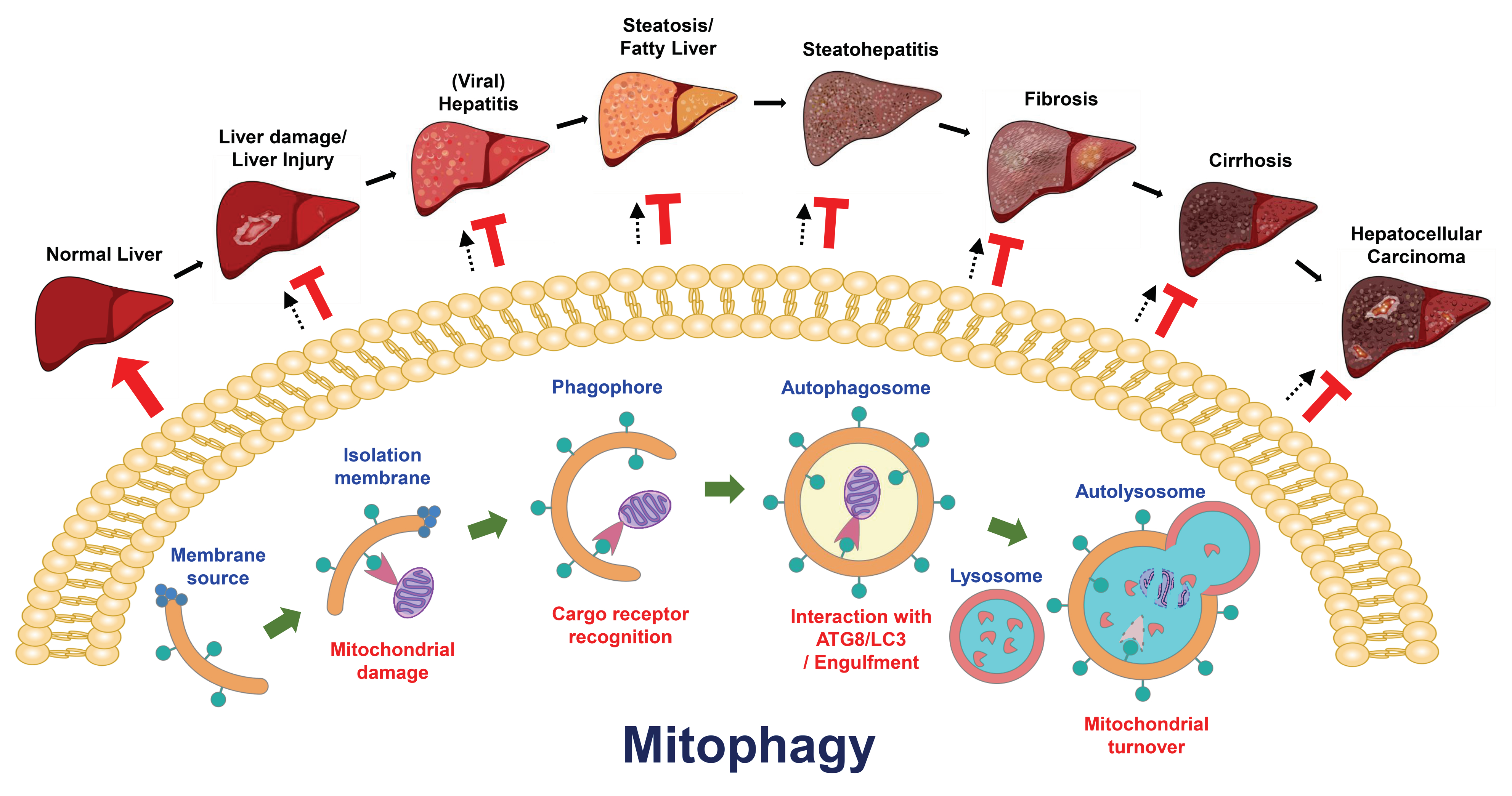{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the roles of mitophagy in liver physiology.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| 1. Liver specimens from patients with an alpha (1)-antitrypsin (α1-AT) deficiency 2. Liver tissues of α1-AT Z variant (α1-ATZ) transgenic mice | Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of human patients and α1-ATZ transgenic mice | The sequestration of deformed mitochondria associated with α1-AT deficiency-mediated chronic liver diseases | [444,477,478] |
| Rat liver tissues (ageing) | 1. Age-dependent accumulation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the mitochondrial DNA (mtDNA) in the liver tissues of aged mice 2. Age-dependent decrease in cytochrome C oxidase activity in the liver tissues of aged mice | Age-dependent loss of mitophagy activity | [423,479] |
| Liver tissues of wild type and ATG7 knockout mice | Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of ATG7 knockout mice | Degradation of damaged mitochondria by mitophagy | [480] |
| Liver tissues of GFP-LC3 transgenic mice | 1. Sequestration of GFP-LC3-labeled mitochondria in the liver tissues of nutrient-starved GFP-LC3 transgenic mice 2. Engulfment of GFP-LC3-labeled mtDNA in the liver tissues of nutrient-starved GFP-LC3 transgenic mice | Degradation of dysfunctional mitochondria by starvation-induced mitophagy | [421] |
| Liver tissues and primary hepatocytes from wild type and BNIP3 knockout mice | 1. Immunofluorescence staining for Hsp60, a mitochondrial matrix protein, was observed in primary hepatocytes isolated from wild type and BNIP3-null mice 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in primary hepatocytes isolated from wild type and BNIP3-null mice | Reduced mitochondrial turnover and increased mitochondrial mass following the loss of BNIP3-dependent mitophagy | [481] |
| Rat liver tissues (ischemia/reperfusion (I/R) and anoxia/reoxygenation (A/R) treatments) | Sequestration of GFP-LC3-labeled mitochondria in the liver tissues of I/R- and A/R-treated mice | 1. Degradation of dysfunctional mitochondria by mitophagy 2. Ageing aggravated the I/R-induced impairment in Parkin-dependent mitophagy | [482,483] |
| Liver specimens from patient with acute liver damage induced by anorexia nervosa | Electron micrographs showed autophagic vacuoles that engulfed mitochondria in liver tissues from human patients | Degradation of damaged mitochondria by starvation-induced mitophagy | [437] |
| Primary rat hepatocytes | 1. Electron micrographs showed autophagic vacuoles in the dedifferentiation-induced remodeling of rat hepatocytes 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in the dedifferentiation-induced remodeling of rat hepatocytes | Remodeling of hepatocytes by mitophagy | [420] |
| 1. Liver tissues from wild type and GFP-LC3 transgenic mice (ethanol treatment) 2. Isolated primary mouse hepatocytes (ethanol treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of ethanol-treated mice 2. Engulfment of MitoTracker-labeled mitochondria by GFP-LC3-labeled autophagic vacuoles | 1. Degradation of damaged mitochondria by ethanol-induced mitophagy 2. Protection against ethanol-induced liver injury by Parkin-dependent mitophagy | [446,484,485,486] |
| 1. Hepatocytes, Hep3B cell line 2. Primary human hepatocytes (treated with the antiretroviral drug efavirenz) | 1. Electron micrographs showed autophagic vacuoles that sequestered enlarged mitochondria in the efavirenz-treated liver cells 2. Immunofluorescence staining of MitoTracker-labeled mitochondria and LysoTracker-labeled lysosomes in the efavirenz-treated liver cells | 1. Degradation of damaged mitochondria by efavirenz-induced mitophagy 2. Mitophagy protected against efavirenz-induced hepatic injury | [487,488] |
| The human normal liver cell line, L02 (cadmium treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the cadmium-treated liver cells 2. Immunofluorescence staining revealed the co-localization of TOM20-labeled mitochondria with GFP-LC3 in the cadmium-treated liver cells 3. Immunofluorescence staining of MitoTracker- and chloromethyl-X-rosamine (CMXRos)-labeled mitochondria in the cadmium-treated liver cells | 1. Degradation of damaged mitochondria by cadmium-induced mitophagy 2. Mitophagy protected against cadmium-induced hepatotoxicity | [489] |
| Liver tissues from wild type and GFP-LC3 transgenic mice (acetaminophen treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the acetaminophen-treated mouse hepatocytes 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in the acetaminophen-treated mouse hepatocytes 3. Degradation of mitochondrial proteins in the acetaminophen-treated mouse hepatocytes | 1. Degradation of damaged mitochondria by acetaminophen-induced PINK1/Parkin-dependent mitophagy 2. Protection against acetaminophen-induced liver injury by PINK1/Parkin-dependent mitophagy 3. Activation of acetaminophen-induced mitophagy through AMPK activation and the suppression of inflammasome activation | [489,490,491,492,493,494,495] |
Table 2.
Summary of the roles of mitophagy in liver injury.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| 1. Liver specimens from patients with an α1-AT deficiency 2. Liver tissues from α1-ATZ transgenic mice | Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of human patients and α1-ATZ transgenic mice | Sequestration of deformed mitochondria that is associated with α1-AT deficiency-related chronic liver diseases | [444,477,478] |
| Rat liver tissues liver (I/R and A/R treatments) | Sequestration of GFP-LC3-labeled mitochondria in the liver tissues of I/R- and A/R-treated mice | 1. Degradation of dysfunctional mitochondria by mitophagy 2. Ageing aggravated the I/R-induced impairment in Parkin-dependent mitophagy 3. Protection against I/R-induced liver injury by Parkin-dependent mitophagy | [482,483] |
| 1. Liver specimens from human patients 2. Primary mouse hepatocytes 3. Liver tissues of I/R-treated wild type and Sirtuin 1 (SIRT1) knockout mice | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of patients 2. Immunofluorescence staining for tetramethylrhodamine methyl ester (TMRM)-labeled mitochondria in the I/R-treated hepatocytes 3. Degradation of mitochondrial proteins in the I/R-treated hepatocytes 4. Engulfment of MitoTracker-labeled mitochondria by GFP-LC3-labeled autophagic vacuoles in the I/R-treated hepatocytes | Protection against I/R-induced hepatic injury by SIRT1- and Parkin-dependent mitophagy | [483,513,514,515] |
| Liver tissues of I/R-treated mice | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of I/R-treated mice 2. Degradation of mitochondrial proteins in the I/R-treated hepatocytes 3. Quantification of the mtDNA copy number in the I/R-treated hepatocytes | Protection against I/R-induced liver injury by heme oxygenase-1 (HO-1)-induced mitophagy through phosphoglycerate mutase 5 (PGAM5) | [516] |
| 1. Liver tissues of I/R-treated mice 2. The human normal liver cell line, L02 | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of I/R-treated mice 2. Immunofluorescence staining of 5,5′,6,6′-tetrachloro-1,1′,3,3′ –tetraethyl-benzimidazolylcarbocyanine iodide (JC1)-labeled mitochondria in the I/R-treated hepatocytes | Mitophagy induced by the downregulation of microRNA330-3p protected against I/R-induced liver injury by increasing PGAM5 expression | [517] |
| 1. Hepatocytes, Hep3B cell line 2. Primary human hepatocytes (treated with the antiretroviral drug efavirenz) | 1. Electron micrographs showed autophagic vacuoles that sequestered mitochondria in the efavirenz-treated hepatocytes 2. Immunofluorescence staining of MitoTracker-labeled mitochondria and LysoTracker-labeled lysosomes in the efavirenz-treated hepatocytes | 1. Degradation of damaged mitochondria by efavirenz-induced mitophagy 2. Mitophagy protected against efavirenz-induced hepatic injury | [487,488] |
| The human normal liver cell line, L02 (cadmium treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the cadmium-treated liver cells 2. Immunofluorescence staining revealed the co-localization of TOM20-labeled mitochondria with GFP-LC3 in the cadmium-treated liver cells 3. Immunofluorescence staining of MitoTracker- and chloromethyl-X-rosamine (CMXRos)-labeled mitochondria in the cadmium-treated liver cells | 1. Degradation of damaged mitochondria by cadmium-induced mitophagy 2. Mitophagy protected against cadmium-induced hepatotoxicity | [489] |
| Liver tissues from wild type and GFP-LC3 transgenic mice (acetaminophen treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the acetaminophen-treated mouse hepatocytes 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in the acetaminophen-treated mouse hepatocytes 3. Degradation of mitochondrial proteins in the acetaminophen-treated mouse hepatocytes | 1. Degradation of damaged mitochondria by acetaminophen-induced PINK1/Parkin-dependent mitophagy 2. Protection against acetaminophen-induced liver injury by PINK1/Parkin-dependent mitophagy 3. Activation of acetaminophen-induced mitophagy through AMPK activation and the suppression of inflammasome activation | [489,490,491,492,493,494,495] |
| 1. Liver tissues from wild type and GFP-LC3 transgenic mice 2. Isolated primary mouse hepatocytes | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of ethanol-treated mice 2. Engulfment of MitoTracker-labeled mitochondria by GFP-LC3-labeled autophagic vacuoles | 1. Degradation of damaged mitochondria by ethanol-induced mitophagy 2. Protection against ethanol-induced liver injury by Parkin-dependent mitophagy | [446,484,485,486] |
Table 3.
Summary of the roles of mitophagy in steatosis and fatty liver diseases.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| Liver tissues and primary hepatocytes from wild type and BNIP3 knockout mice | 1. Immunofluorescence staining for Hsp60, a mitochondrial matrix protein, in the primary hepatocytes isolated from wild type and BNIP3-null mice 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in the primary hepatocytes isolated from wild type and BNIP3-null mice | Reduced mitochondrial turnover and increased mitochondrial mass induced by a deficiency in BNIP3-dependent mitophagy | [481] |
| Liver specimens from patients with alcoholic and nonalcoholic liver diseases (NAFLD) | Electron micrograph of giant mitochondria in the liver specimens of patients with alcoholic and nonalcoholic fatty liver diseases | Mitochondrial dysfunction in the development and pathogenesis of alcoholic and nonalcoholic fatty liver diseases | [526,527,528] |
| Rat liver tissues (ethanol treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the ethanol-treated rat liver tissues 2. Immunogold labelling of PINK1 within autophagic vacuoles in which mitochondria were sequestered | 1. Degradation of damaged mitochondria by ethanol-induced mitophagy 2. Protection against ethanol-induced fatty liver by PINK1-dependent mitophagy | [529] |
| Rat liver tissues (ethanol treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the ethanol-treated rat liver tissues 2. Immunofluorescence staining revealed the co-localization of Parkin and LC3 with mitochondrial and lysosomal markers in the ethanol-treated rat liver tissues 3. Immunogold labelling of Parkin and PINK1 within autophagic vacuoles revealed the sequestration of damaged mitochondria in the ethanol-treated rat liver tissues | 1. Degradation of damaged mitochondria by ethanol-induced mitophagy 2. Protection against ethanol-induced fatty liver by PINK1/Parkin-dependent mitophagy | [530,531] |
| 1. Liver tissues from wild type and GFP-LC3 transgenic mice 2. Isolated primary mouse hepatocytes | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of ethanol-treated mice 2. Engulfment of MitoTracker-labeled mitochondria by GFP-LC3-labeled autophagic vacuoles | 1. Degradation of damaged mitochondria by ethanol-induced mitophagy 2. Protection against ethanol-induced liver injury by Parkin-dependent mitophagy | [446,484,485,486] |
| Liver specimens from patients with nonalcoholic liver diseases | Electron micrograph of megamitochondria containing linear crystalline inclusions | Mitochondrial dysfunction in the development and pathogenesis of NAFLD | [533] |
| Liver tissues from wild type and acyl-CoA:lysocardiolipin acyltransferase-1 (ALCAT1) knockout mice (fed a high-fat diet (HFD)) | 1. Electron micrographs showed autophagic vacuoles that contained mitochondria in the liver tissues of HFD-fed mice 2. Immunofluorescence staining of MitoTracker-labeled mitochondria in the liver tissues of HFD-fed mice 3. Immunofluorescence staining revealed the co-localization of mitochondria and LC3 with mitochondrial and lysosomal markers in the liver tissues of HFD-fed mice | 1. Degradation of damaged mitochondria by HFD-induced mitophagy 2. Protection against HFD-induced NAFLD by Parkin-dependent mitophagy | [534] |
| 1. Human hepatoma cell line, HepG2 cells 2. Mouse liver tissues (fed a methionine and choline-deficient diet (MCD)) | 1. Electron micrographs showed autophagic vacuoles that contained mitochondria in the thyroid hormone-treated HepG2 cells 2. Immunofluorescence staining for mRFP-EGFP-labeled mitochondria in the thyroid hormone-treated HepG2 cells | 1.Induction of mitophagy by thyroid hormone 2. Suppression of NAFLD development by thyroid hormone-induced mitophagy | [535] |
| Human hepatoma cell line, HepG2 cells (oleic acid (OA) treatment) | 1. Immunofluorescence staining revealed the co-localization of MitoTracker-labeled mitochondria and LysoTracker-labeled lysosomes in the OA-treated HepG2 cells 2. Degradation of mitochondrial proteins in the OA-treated HepG2 cells | Activation of DRAM-mediated mitophagy in the progression of NAFLD | [536] |
| Liver tissues from wild type mice and liver-specific dynamin-related protein 1 (DRP1) knockout mice, optic atrophy protein 1 (OPA1) knockout mice and DRP1/OPA1 double knockout mice (MCD feeding) | 1. Immunofluorescence staining revealed anti-pyruvate dehydrogenase E1 (PDH1) antibody-labeled mitochondria in mouse liver tissues 2. Electron micrographs showed depolarized mitochondria in the liver tissues of DRP1, OPA1, and DRP1/OPA1 knockout mice | 1. Inhibition of mitophagy during the progression of NAFLD 2. Ubiquitination of mitochondria p62/SQSTM1/Kelch-like ECH-associated protein (KEAP1)/RING-box protein 1 (RBX1) in Parkin-independent mitophagy 3. Induced megamitochondria by impairing Parkin-independent mitophagy in fatty liver | [537] |
| Liver tissues from wild type and low-density lipoprotein cholesterol receptor (LDLR) knockout mice (Western diet (WD) feeding) | Degradation of mitochondrial proteins in the liver tissues of wild type and LDLR knockout mice | 1. Reduced protein stabilities of oxidative phosphorylation subunits in WD-induced NAFLD mice 2. Increased protein degradation of mitochondrial proteins by activated mitophagy | [538] |
| 1. Mouse liver tissues (fed a high-fat/calorie diet (HFCD)) 2. Rat primary hepatocyte (Palmitic acid, PA) | 1. Immunofluorescence staining revealed the co-localization of LC3B and cytochrome C oxidase subunit IV in the liver tissues of HFCD-fed mice 2. Induced expression of PINK1 and Parkin in the liver tissues of HFCD-fed mice 3. Immunofluorescence staining revealed the co-localization of LC3 and MitoTracker-labeled mitochondria in PA-treated rat hepatocytes 4. Degradation of TOM20 in PA-treated rat hepatocytes | 1. Impaired mitophagy in mice with HFCD-induced NAFLD 2. Induction of inflammasome activation by the suppression of mitophagy in NAFLD mice and primary hepatocytes | [539] |
Table 4.
Summary of the roles of mitophagy in liver cancer.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| 1. The BALB/c hepatoma cell line ML-1 2. Liver tissues from NOD/LtSz-PrkdcJ (SCID) mice (treated with concanavalin A (ConA)) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the ConA-treated ML-1 cells 2. Induction of BNIP3 expression in the ConA-treated ML-1 cells 3. Immunofluorescence staining revealed the co-localization of MitoTracker-labeled mitochondria and LysoTracker-labeled lysosomes | 1. Activation of BNIP3-dependent mitophagy by ConA treatment 2. Suppression of hepatoma cell growth by ConA-induced mitophagy 3. Inhibition of liver tumor nodule formation by ConA-induced mitophagy | [549,550] |
| Human hepatoma cell line, HepG2 cells (adriamycin treatment) | 1. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the adriamycin-treated HepG2 cells 2. Immunofluorescence staining of dsRed2-labeled mitochondria and JC1-labeled mitochondria in the adriamycin-treated HepG2 cells | Induction of adriamycin-induced cell apoptosis of human hepatoma cells | [551] |
| Human hepatoma cell line, HepG2 cells (adriamycin and curcumin treatment) | Immunofluorescence staining of dsRed2-labeled mitochondria and JC1-labeled mitochondria in the adriamycin- and curcumin-treated HepG2 cells | Enhancement of adriamycin-induced cell apoptosis of human hepatoma cells by curcumin | [552] |
| 1. Human hepatoma cell line, HepG2 cells 2. Human hepatoma cell line, Hep3B cells 3. Human hepatoma cell line, Huh7 cells (melatonin and sorafenib treatment) | 1. Immunofluorescence staining for anti-TOM20 antibody-labeled mitochondria and anti-LAMP2 antibody-labeled lysosomes in the melatonin- and sorafenib-treated hepatoma cells 2. Degradation of HSP60 in the melatonin- and sorafenib-treated hepatoma cells 3. Decreased mtDNA copy number in the melatonin- and sorafenib-treated hepatoma cells | The cytotoxicity of sorafenib was increased in human hepatoma cells by melatonin-induced mitophagy | [553] |
| 1. Human normal liver cell line, 7702 2. Human hepatoma cell line, HepG2 cells 3. Human hepatoma cell line, Hep3B cells 4. Human hepatoma cell line, Huh7 cells | 1. Immunofluorescence staining revealed the co-localization of anti-HSP60 antibody-labeled mitochondria and GFP-LC3-labeled autophagic vacuoles in the nutrient-starved hepatoma cells 2. Mitochondrial translocation of DRAM in the nutrient-starved hepatoma cells | 1. The apoptosis of human hepatoma cells was induced by activating DRAM-mediated mitophagy 2. Inhibition of mitophagy-triggered cell apoptosis through the inhibition of the mitochondrial translocation of DRAM | [554] |
| 1. Liver tissues from patients with HCC 2. Liver tissues from wild type and FUNDC1 knockout mice, and liver-specific FUNDC1 knockout mice | 1. Degradation of mitochondrial proteins in the liver tumor tissues of patients with HCC 2. Decreased mtDNA copy number in the liver tumor tissues from patients with HCC 3. Induction of FUNDC1 expression in the liver tumor tissues from patients with HCC 4. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in the liver tissues of diethylnitrosamine (DEN)-treated mice 5. Increased the formation of mito-Keima-labeled dot (Red+/Green-) in the isolated hepatocytes treated by DEN | 1. Suppression of the initial development of HCC by FUNDC1-mediated mitophagy through a reduction in inflammasome activation 2. Benefit to tumor growth at the late stage of HCC development by FUNDC1-mediated mitophagy | [555] |
| 1. Liver tissues of HCC patients 2. Human HCC cell lines, Bel7402 and SMMC7721 3. The liver tissues of mouse xenograft models | 1. Electron micrograph of deformed mitochondria in the tumorous liver tissues of HCC patients 2. Downregulation of dynamin-1-like protein (DNM1L) expression and upregulation of mitofusin 1 (MFN1) expression in the tumorous liver tissues of HCC patients 3. Co-localization of GFP-LC3B-labeled autophagic vacuoles with MitoTracker red-labeled mitochondria in DNM1L-knockdown Bel7402 and SMMC7721 cells | 1. Promotion of HCC cell survival by elevated DNM1L and downregulated MFN1 2. Contribution to poor prognosis of HCC patients by the elevated expressional ratio of DNM1L to MFN1 3. Suppression of tumor growth by inhibition of mitochondrial fission | [556] |
| 1. Human hepatoma cell line, HepG2 cells 2. Human hepatoma cell line, Hep3B cells 3. Human hepatoma cell line, Huh7 cells | 1. Immunofluorescence staining revealed the co-localization of nti-TOM20 antibody-labeled mitochondria and phospho-p53 (at serine 392) 2. Phosphorylation of p53 at serine 392 by PINK1 3. Mitochondrial translocation of p53 and p53 phosphorylation in the mitochondria | 1. Maintenance of the stemness of cancer stem cells (CSCs) by NANOG gene expression induced by the activation of mitophagy 2. Reduced NANOG gene expression by PINK1-dependent phosphorylation of p53 at serine 392 | [557] |
Table 5.
Summary of the roles of mitophagy in viral hepatitis.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| Human hepatoma, Huh7.5.1 cells (hepatitis C virus (HCV) infection) | 1. Translocation of Parkin to the mitochondria in HCV-infected cells 2. Mitochondrial ubiquitination was induced by HCV infection 3. Immunofluorescence staining revealed the co-localization of anti-TOM20 antibody-labeled mitochondria, GFP-LC3-labeled autophagic vacuoles, and Parkin in HCV-infected cells 4. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in HCV-infected cells | 1. Promotion of virus replication by PINK1/Parkin-dependent mitophagy 2. HCV-induced mitophagy protected infected cells from apoptosis 3. Establishment of viral persistence by HCV-activated mitophagy | [572,574] |
| Human hepatoma, Huh7.5.1 cells (transfected with HCV NS5A) | 1. Immunofluorescence staining for MitoTracker deep red-labeled depolarized mitochondria in HCV NS5A-transfected cells 2. Translocation of Parkin to the mitochondria in HCV NS5A-transfected cells | 1. Activation of mitophagy by HCV NS5A through increased ROS production 2. Degradation of depolarized mitochondria by HCV NS5A | [575] |
| 1. Human hepatoma, Huh7 cells (HCV infection) 2. Liver tissues of HCV transgenic mice | 1. Decrease in the carbonyl cyanide m-chlorophenyl hydrazone (CCCP)-induced co-localization of anti-TOM20 antibody-labeled mitochondria with Parkin in HCV-infected cells 2. Inhibition of the CCCP-induced mitochondrial translocation of Parkin in HCV-infected cells 3. Interaction of the HCV core protein with Parkin 4. Electron micrographs showed autophagic vacuoles that engulfed mitochondria in CCCP-treated cells, and the change was decreased by HCV infection | A sustained HCV infection induced mitochondrial injury by suppressing mitophagy | [576] |
| Human hepatoma Huh7 cells (transfection of the HBV viral genome) | 1. Translocation of Parkin to the mitochondria in HBV-transfected cells 2. Mitochondrial ubiquitination was induced by HBV transfection 3. Immunofluorescence staining revealed the co-localization of anti-TOM20 antibody-labeled mitochondria, GFP-LC3-labeled autophagic vacuoles, and Parkin in HBV-transfected cells | 1. Protection of infected cells from apoptosis by HBV-induced PINK1/Parkin-mediated mitophagy 2. Establishment of viral persistence by HBV-activated PINK1/Parkin-mediated mitophagy | [577] |
| Human hepatoma cell lines, HepG2 cells, HepG2.2.15 cells, and SMMC-7721 cells (transfection of HBx) | 1. Immunofluorescence staining revealed the co-localization of mitochondria, LC3-labeled autophagic vacuoles, and HBx in HBx-transfected cells 2. Induced PINK1 and Parkin expression in HBx-transfected cells 3. Translocation of Parkin to the mitochondria in HBx-transfected cells | Increase in nutrient starvation-induced PINK1/Parkin-dependent mitophagy by HBx | [577] |
Table 6.
Summary of the roles of mitophagy in other liver diseases.
| Experimental Model | Characteristics of Mitophagy | Function of Mitophagy | References |
|---|---|---|---|
| Rat liver tissues (CCl4 treatment) | 1. Electron micrographs showed dysfunctional mitochondria in the liver tissues from CCl4-treated mice 2. Decreased PINK1 and Parkin expression in the liver tissues from CCl4-treated mice | 1. Impairment in mitophagy in mice with CCl4-induced liver fibrosis 2. Protection against liver fibrosis by melatonin-induced activation of mitophagy | [593] |
| Mouse liver tissues (CCl4 treatment) | 1. Electron micrographs showed dysfunctional mitochondria in the Kupffer cells from CCl4-treated mice 2. Induced expression of PINK1 and Parkin in the Kupffer cells from CCl4-treated mice 3. Increased mitoSOX-labeled mitochondrial ROS levels | 1. Activation of PINK1/Parkin-dependent mitophagy in Kupffer cells by CCl4-induced liver fibrosis 2. Suppression of PINK1/Parkin-dependent mitophagy in Kupffer cells by T-cell immunoglobulin domain and mucin domain-4 (TIM-4) 3. Mitigation of liver fibrosis by TIM4 interference in Kupffer cells | [594] |
| 1. Human hepatic stellate cell line, LX-2 cells 2. Primary HSCs | 1. Degradation of mitochondrial proteins in the PM2.5-treated HSCs 2. Induced expression of PINK1 and Parkin the PM2.5-treated HSCs 3, Mitochondrial translocation of Parkin in the PM2.5-treated HSCs | 1. Activation of HSCs and induction of liver fibrosis by PM2.5 2. Induction of mitochondrial damage by PM2.5 through ROS production 3. Induction of PINK1/Parkin-dependent mitophagy 4. Alleviation of PM2.5-induced liver fibrosis through the inhibition of mitophagy | [595] |
| 1. Liver specimens from patients with acute liver failure 2. Mouse liver tissues (lipopolysaccharide (LPS) treatment) 3. Human hepatic stellate cell line, LX-2 cells (H2O2, LPS, N-acetyl-L-cysteine [NAC], carbonyl cyanide-p-trifluoromethoxyphenylhydrazone [FCCP], or oligomycin treatment) | 1. Inhibition of PINK1 expression and upregulation of TOM40 in the HSC model of H2O2-induced acute liver failure 2. Inhibition of PINK1 expression and upregulation of TOM40 in HSCs from mice with LPS-induced acute liver failure | 1. Inhibition of mitophagy by ROS in the HSC model of acute liver failure 2. Promotion of inflammasome activation by impairing mitophagy in the HSC model of acute liver failure | [596] |
| Hepatocytes isolated from wild type mice, orphan nuclear receptor subfamily 4 group A member 1 (NR4A1) knockout mice, and liver-specific DNA-dependent protein kinase catalytic subunit (DNA-PKcs) knockout mice (ethanol treatment) | 1. Electron micrographs showed damaged mitochondria in the ethanol-treated hepatocytes 2. Immunofluorescence staining for MitoTracker-labeled mitochondria in the ethanol-treated hepatocytes 3. Degradation of mitochondrial proteins in the ethanol-treated hepatocytes | 1. The NR4A1/DNA-PKcs/p53 axis enhanced the pathogenesis of alcohol-related liver disease (ARLD) 2. ARLD pathogenesis was induced by activating DRP1-related mitochondrial fission and restricting FUNDC1-required mitophagy. | [597] |
| 1. Liver tissues from wild type and BNIP3 knockout mice 2. Primary hepatocytes isolated from wild type and BNIP3-null mice | 1. Immunofluorescence staining for HSP60 2. Degradation of mitochondrial proteins | 1. Increase in lipogenesis via the upregulation of lipogenic enzymes through defective mitophagy 2. Suppression of the β-oxidation of fatty acids by interference with mitophagy | [481] |
| 1. Liver tissues from wild type and thyroid hormone receptor knockout mice 2. HepG2 cells | 1. Electron micrographs showed damaged mitochondria 2. Degradation of mitochondrial proteins | Increased β-oxidation of fatty acids through inducing gene expression CPT1α by thyroid hormone-activated mitophagy | [598] |
| Liver tissues from wild type and REDD1 knockout mice (HFD treatment) | 1. Electron micrographs showed damaged mitochondria 2. Degradation of mitochondrial proteins | Increased CPT1α, BNIIP3 and Parkin expression in the livers of HFD-fed REDD1 KO mice | [599] |
| 1. Liver tissues from wild type mice 2. Primary mouse hepatocytes | Co-localization of GFP-LC3 and mitochondria | Suppression of mitophagy by insulin resistance | [600] |
| 1. Liver tissues from wild type and Parkin knockout mice 2. Primary mouse hepatocytes (HFD treatment) | Degradation of mitochondrial proteins | No significant changes in obesity and insulin resistance were observed in response to an impairment in Parkin-dependent mitophagy | [601,602] |
| 1. Liver tissues from wild type and FUNDC1 knockout mice 2. Primary mouse hepatocytes (HFD treatment) | 1. Electron micrographs showed damaged mitochondria 2. Degradation of mitochondrial proteins 3. Change in the fluorescence intensity of the mt-Keima reporter | Induction of adipose tissue-associated macrophage infiltration and hyperactivation of MAPK8 (also named JNK1) by the loss of FUNDC1-mediated mitochondrial turnover | [603] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ke, P.-Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040831
AMA Style
Ke P-Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells. 2020; 9(4):831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040831
Chicago/Turabian StyleKe, Po-Yuan. 2020. "Mitophagy in the Pathogenesis of Liver Diseases" Cells 9, no. 4: 831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040831
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.