
Transcriptomic Analysis of Seasonal Gene Expression and Regulation during Xylem Development in “Shanxin” Hybrid Poplar (Populus davidiana × Populus bolleana)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Plant Materials
2.3. Anatomical Observation of Developing Xylem
2.4. Library Construction and Transcriptome Sequencing
2.5. Transcriptome Assembly and Annotation
2.6. Identification of DEGs and GO Enrichment Analysis
2.7. KEGG Pathway Enrichment Analysis
3. Results
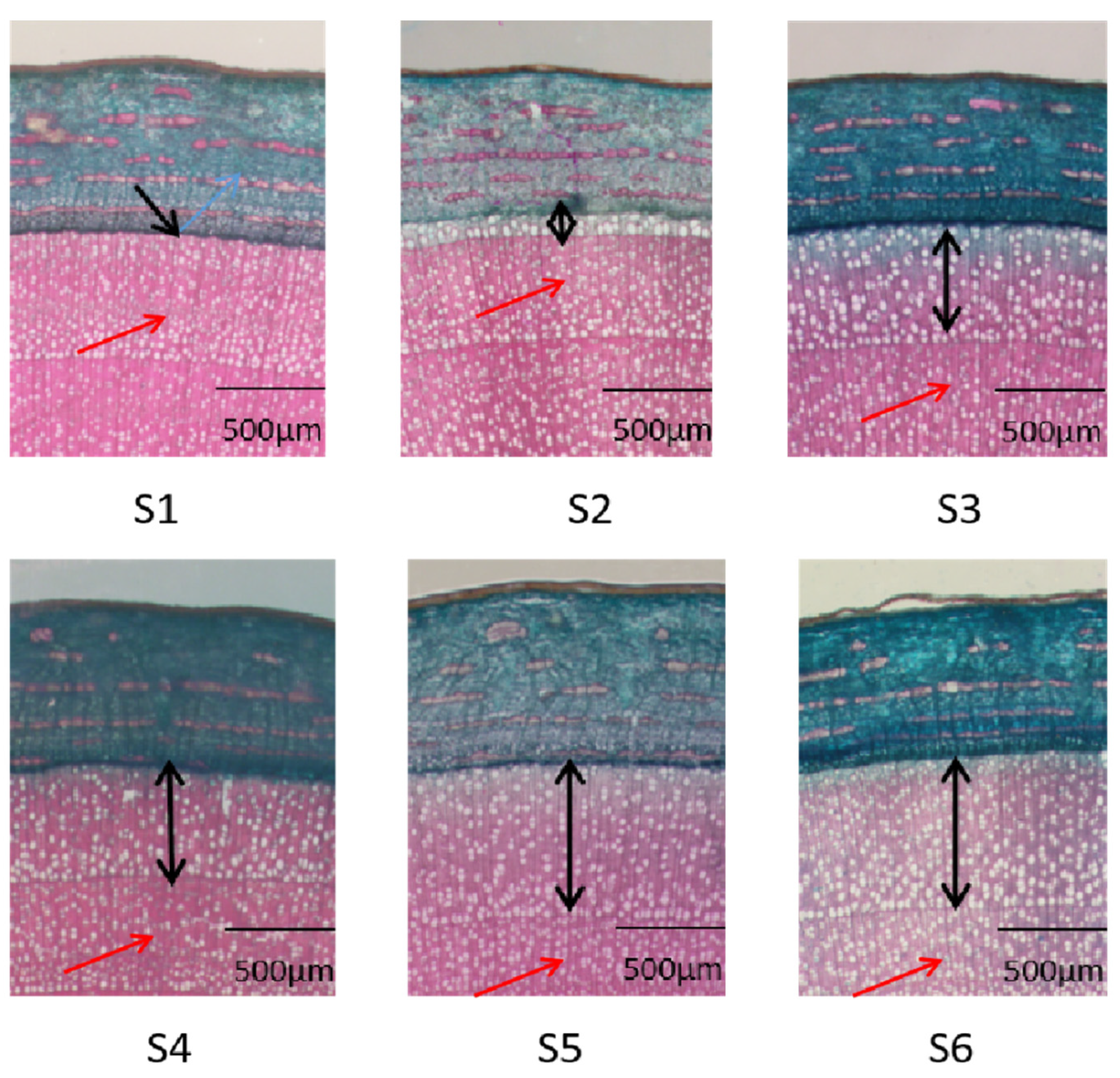
3.1. Histological Observations of Xylem Development
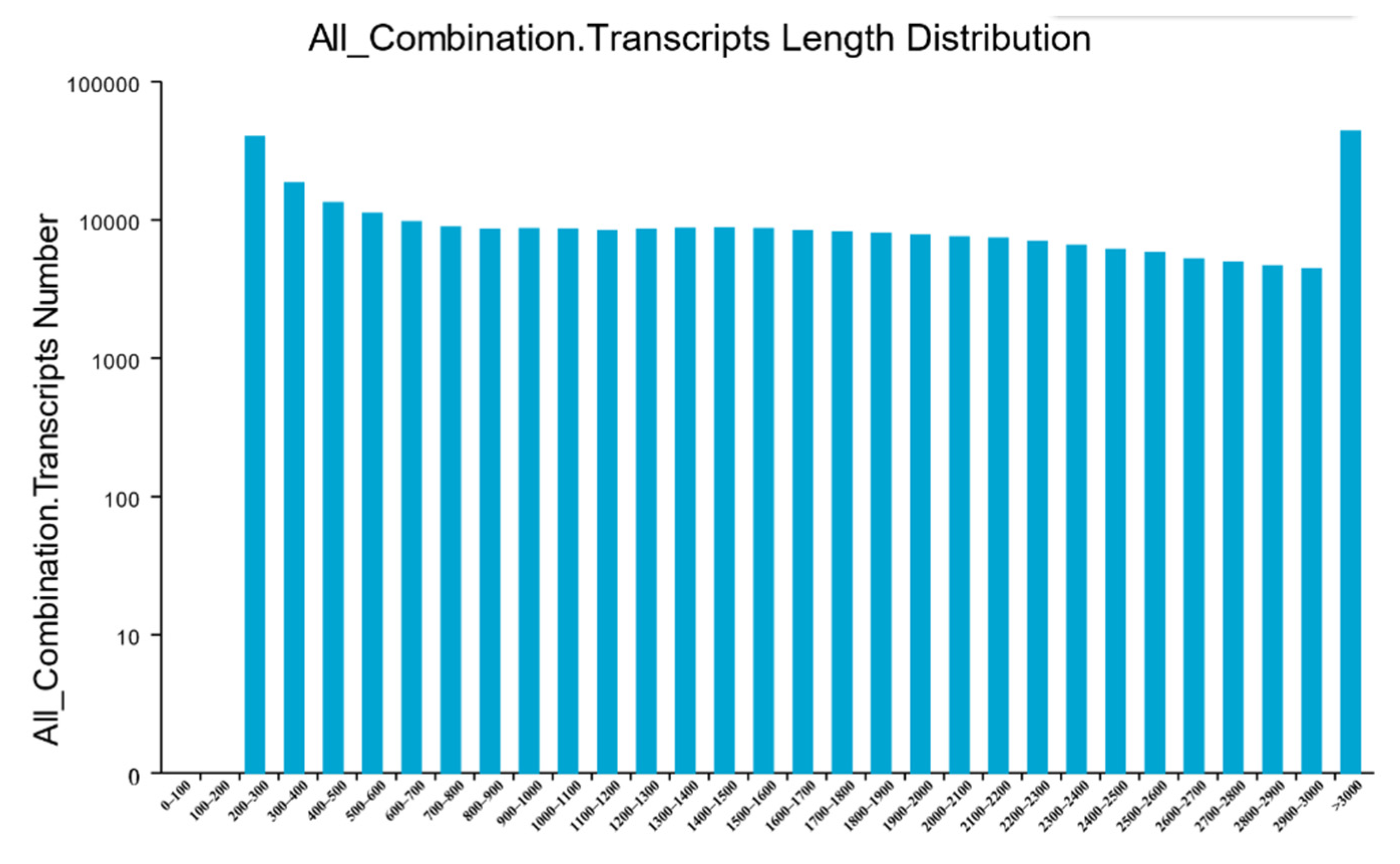
3.2. RNA Sequencing and De Novo Transcriptome Assembly
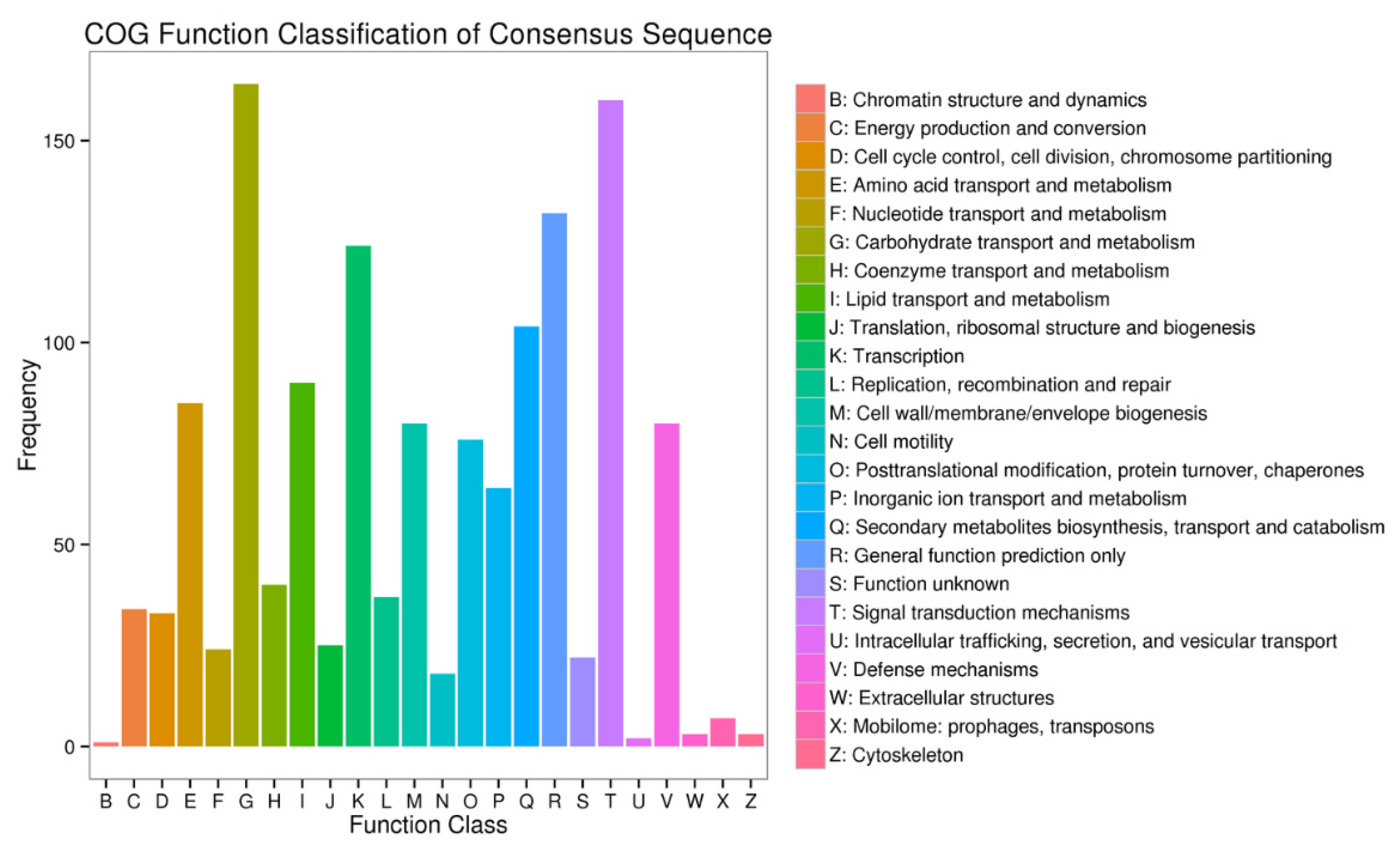
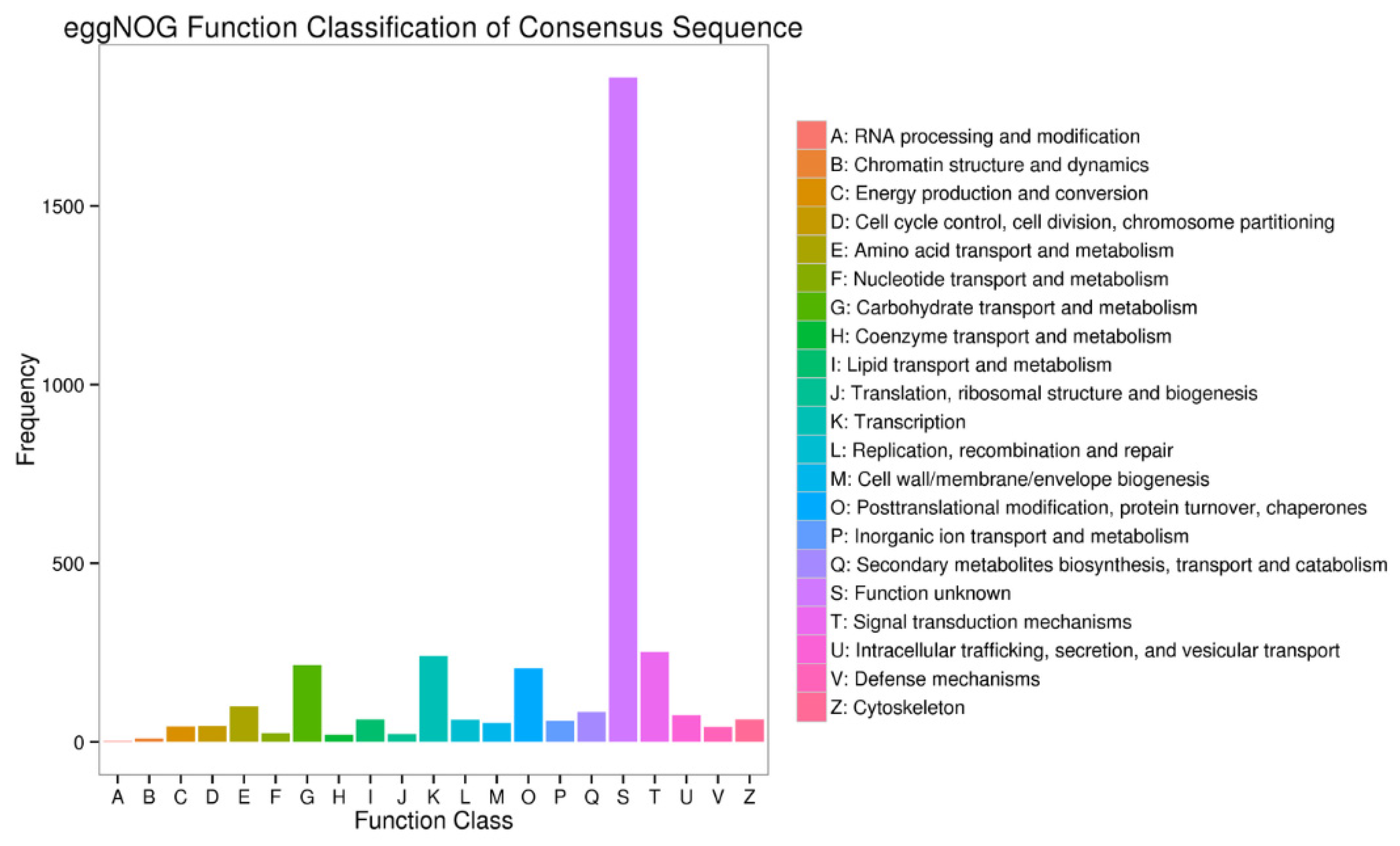
3.3. Functional Annotation of the Unigenes
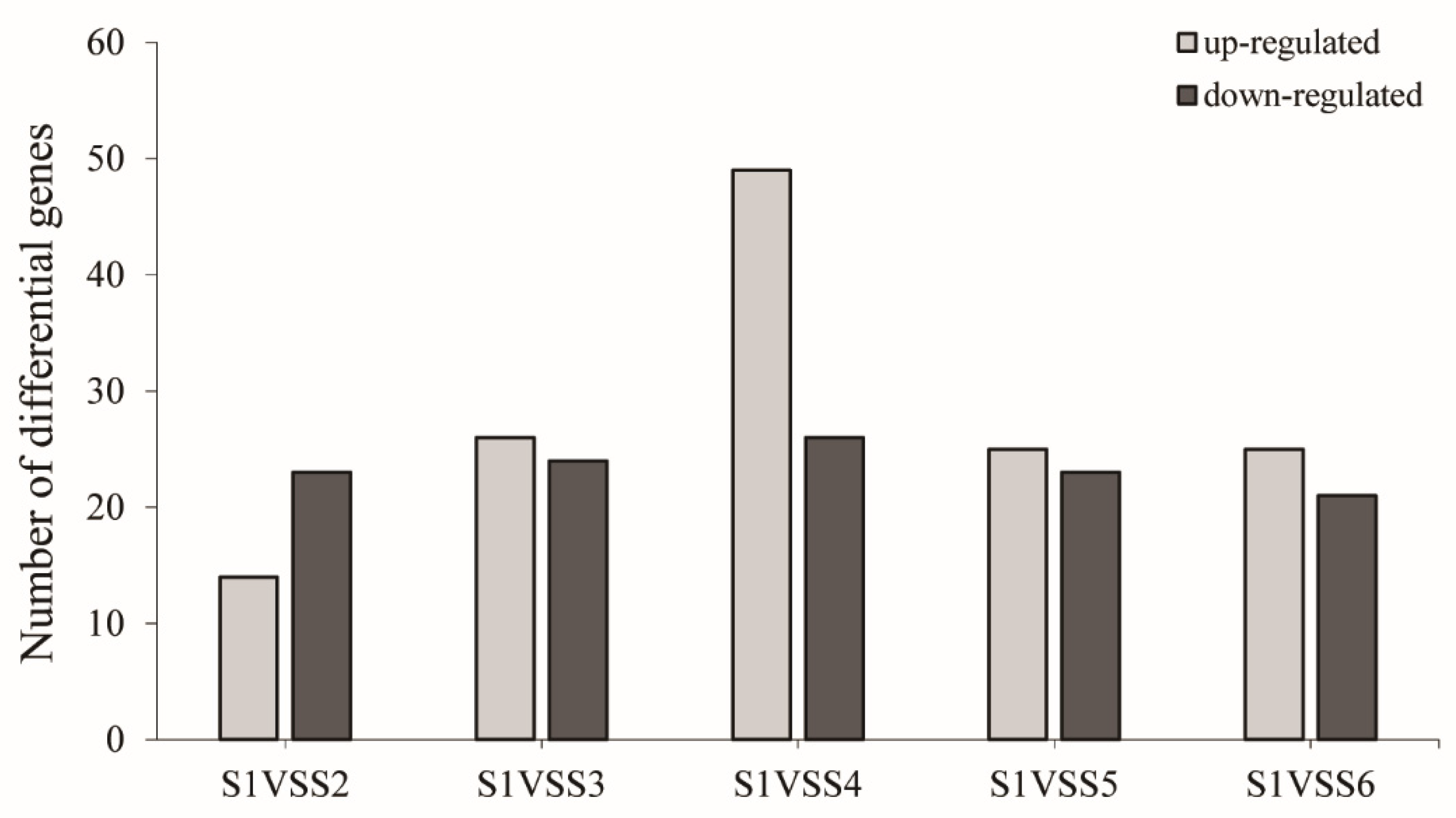
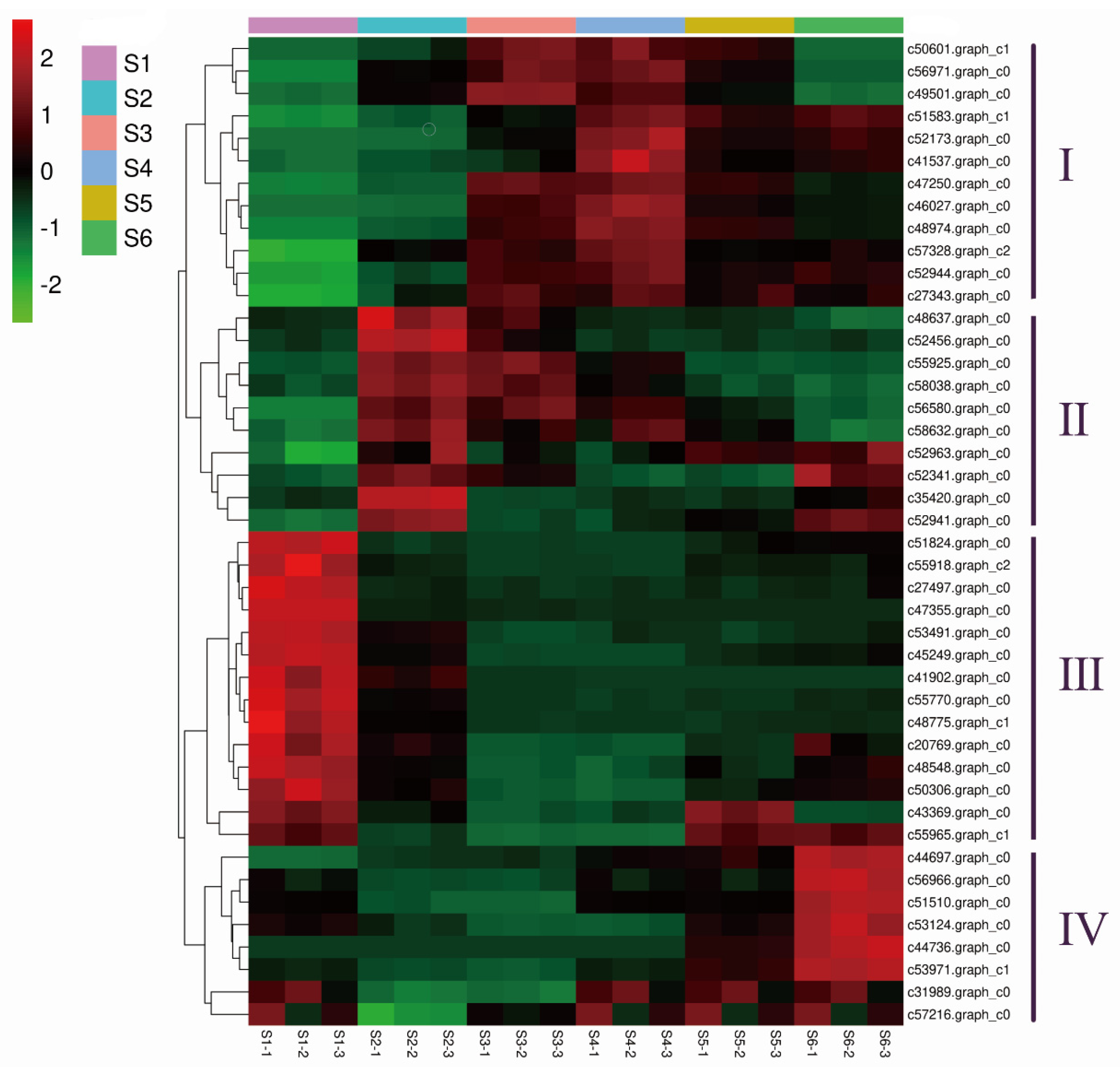
3.4. Identification of Differentially Expressed Genes (DEGs)
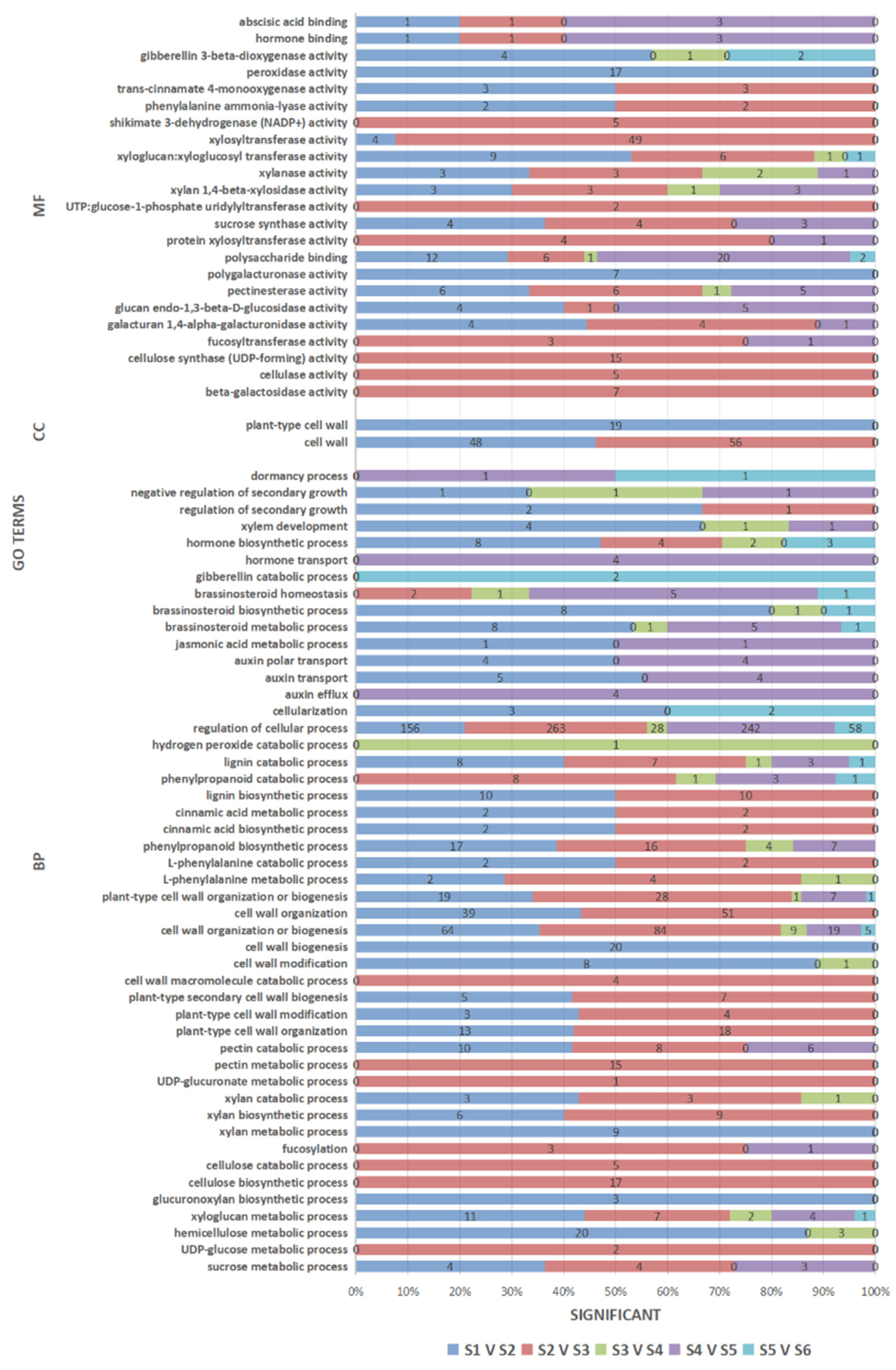
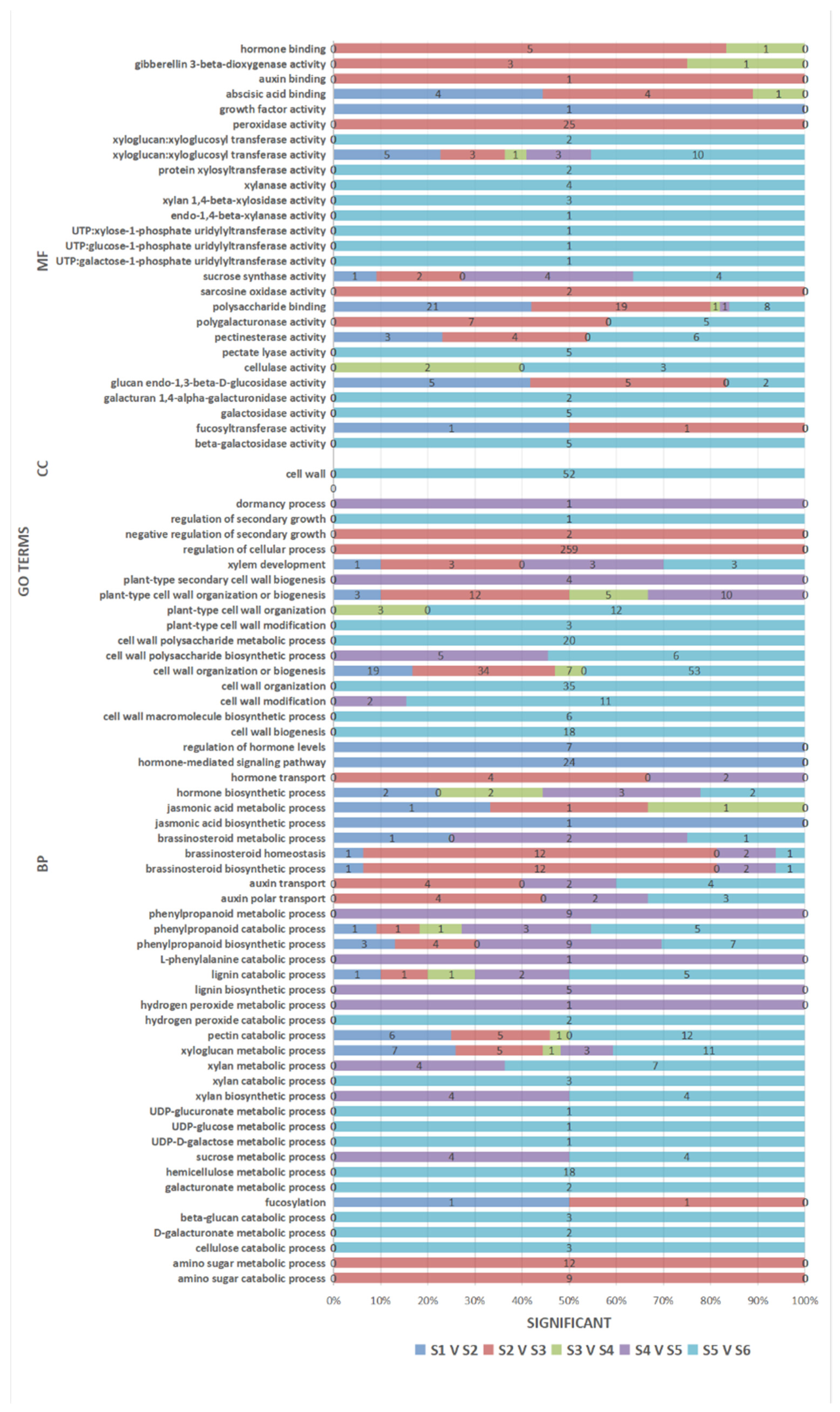
3.5. GO Enrichment Analysis of the DEGs
3.6. Pathway Enrichment Analysis of the DEGs
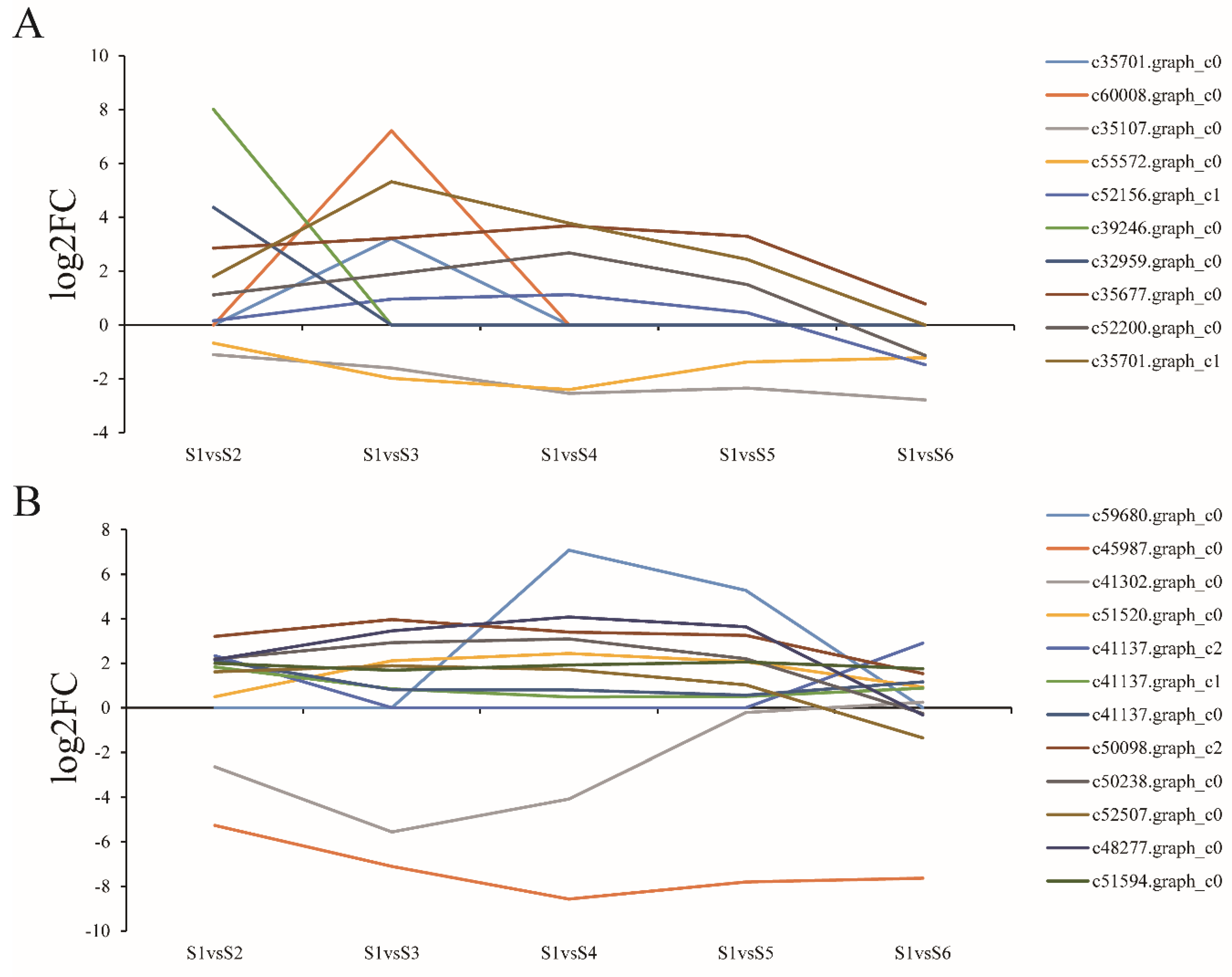
3.7. Expression of Genes Related to Cell Expansion and Cell Wall Biosynthesis
3.8. Expression of Genes Related to Polysaccharide Biosynthesis and Metabolism
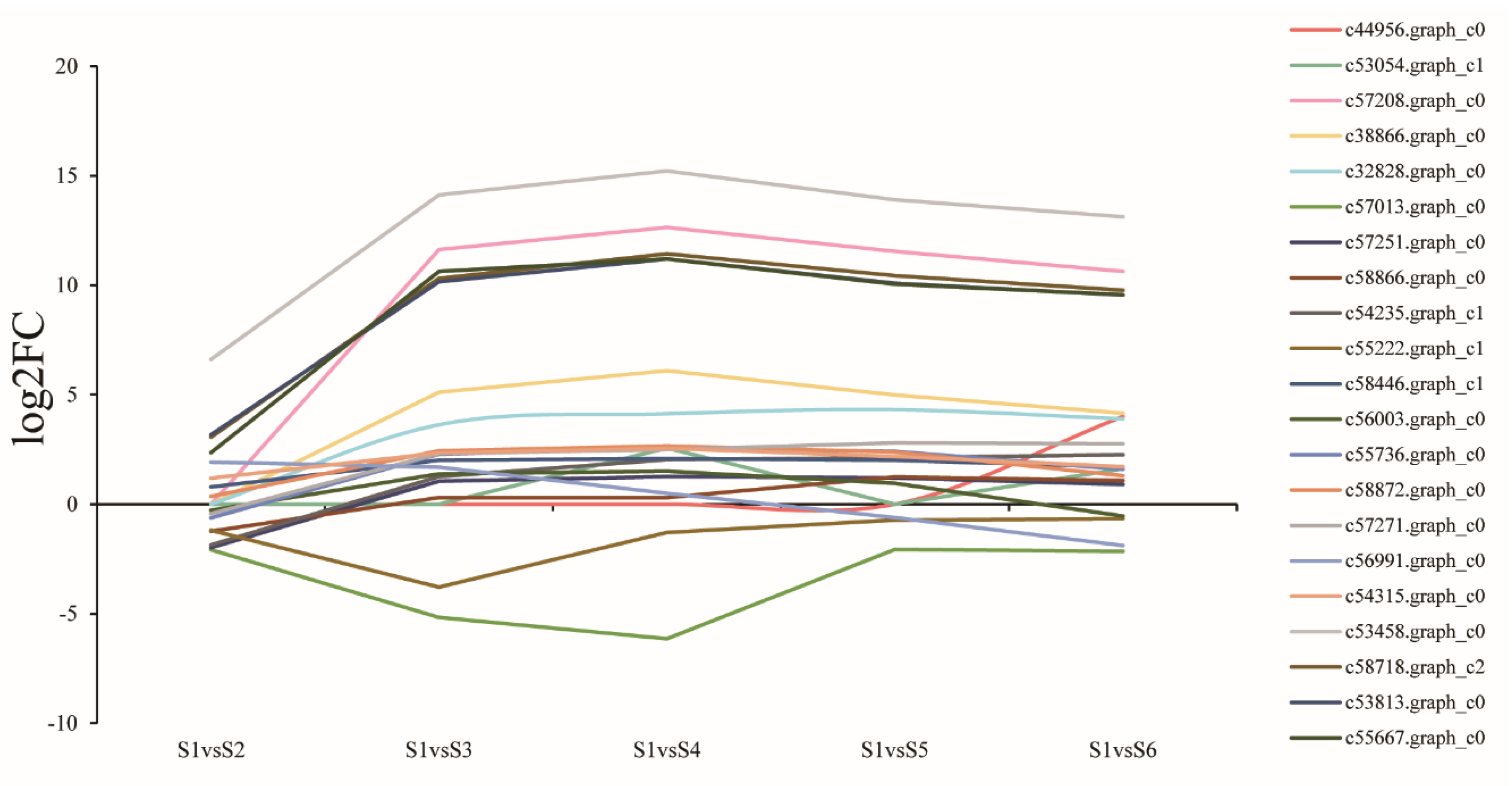
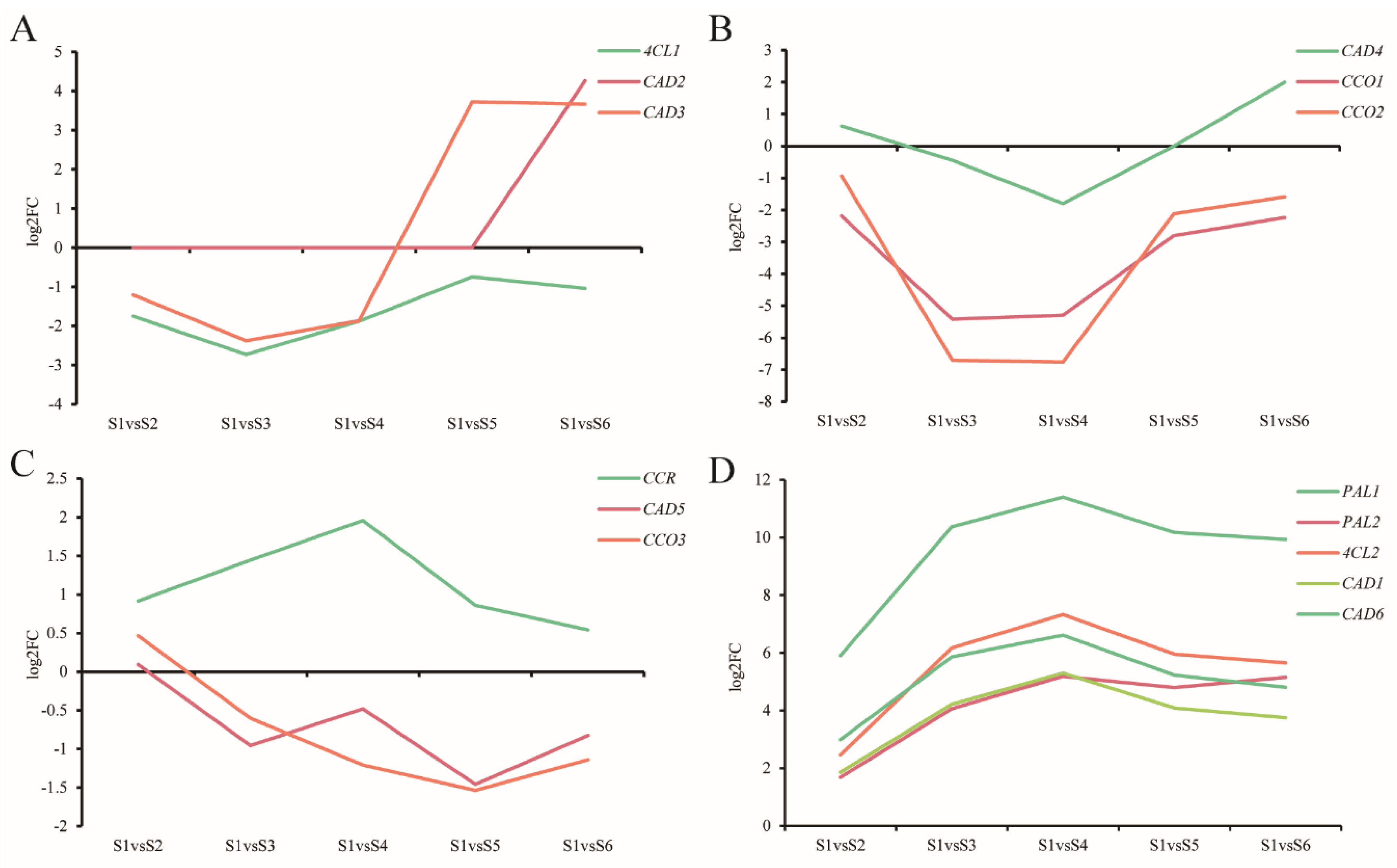
3.9. Expression of Genes Related to Lignin
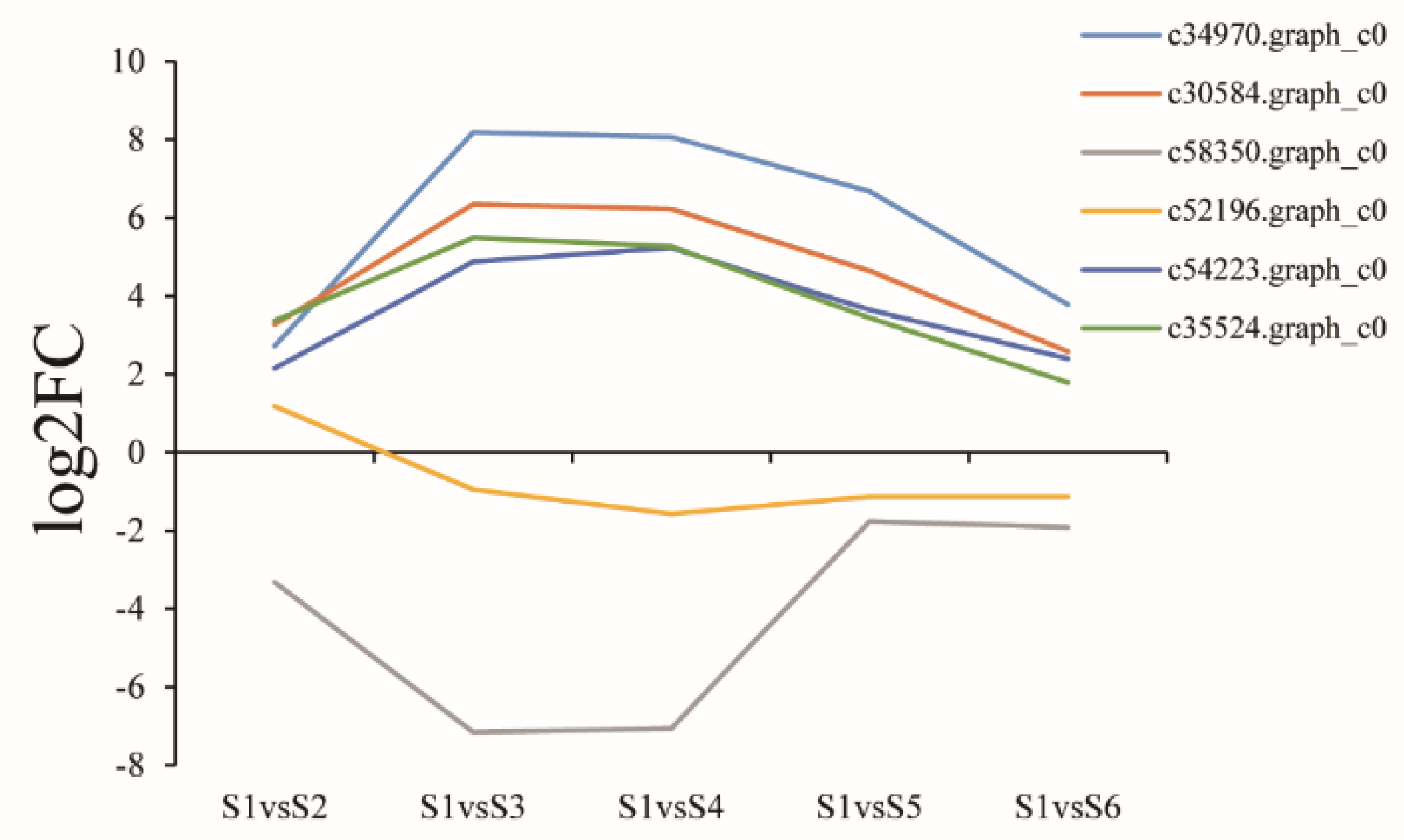
3.10. Identification of Phytohormone-Related Genes in the Xylem during the Seasonal Change
3.11. Identification of Transcription Factor (TF) Genes during the Seasonal Change
4. Discussion
4.1. Illumina Sequencing and Functional Annotation Can Provide Valuable Transcriptomic Resources for the Analysis of Seasonal Gene Expression
4.2. Gene Expression in Response to Seasonal Changes
4.3. Genes for Polysaccharide Biosynthesis and Metabolism Were Helpful for Xylem Development during Seasonal Change
4.4. Expression of Genes Related to Lignin Synthesis Varied in the Different Stages of Xylem Development
4.5. Phytohormone Signaling during the Seasonal Change
4.6. Transcription Factors Involved in Xylem Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Wang, C.; Liu, H.; Tang, R.; Zhang, H. An efficient Agrobacterium-mediated transformation and regeneration system for leaf explants of two elite aspen hybrid clones Populus alba × P. berolinensis and Populus davidiana × P. bolleana. Plant Cell Rep. 2011, 30, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.H.; Jiang, J.; Lin, S.J.; Wang, B.C.; Wang, Y.C.; Liu, G.F.; Yang, C.P. A ThCAP gene from Tamarix hispida confers cold tolerance in transgenic Populus (P. davidiana × P. bolleana). Biotechnol. Lett. 2009, 31, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Bollhöner, B.; Prestele, J.; Tuominen, H. Xylem cell death: Emerging understanding of regulation and function. J. Exp. Bot. 2012, 63, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Gong-Ke, Z.; Ruiqin, Z.; Himmelsbach, D.S.; Mcphail, B.T.; Zheng-Hua, Y. Molecular Characterization of PoGT8D and PoGT43B, Two Secondary Wall-Associated Glycosyltransferases in Poplar. Plant Cell Physiol. 2007, 48, 689–699. [Google Scholar]
- Jacobs, W.P. The Role of Auxin in Differentiation of Xylem Around a Wound. Amer. J. Bot 1952, 39, 301–309. [Google Scholar] [CrossRef]
- Hertzberg, M.; Aspeborg, H.; Schrader, J.; Andersson, A.; Erlandsson, R.; Blomqvist, K.; Bhalerao, R.; Uhlén, M.; Teeri, T.T.; Lundeberg, J.; et al. A transcriptional roadmap to wood formation. Proc. Natl. Acad. Sci. USA 2001, 98, 14732–14737. [Google Scholar] [CrossRef] [Green Version]
- Sundell, D.; Street, N.R.; Kumar, M.; Mellerowicz, E.J.; Kucukoglu, M.; Johnsson, C.; Kumar, V.; Mannapperuma, C.; Delhomme, N.; Nilsson, O. AspWood: High-Spatial-Resolution Transcriptome Profiles Reveal Uncharacterized Modularity of Wood Formation in Populus tremula. Plant Cell 2017, 153, 02017. [Google Scholar] [CrossRef] [Green Version]
- Plomion, C.H.; Leprovost, G.; Stokes, A. Wood Formation in Trees. Plant Physiol. 2002, 127, 1513–1523. [Google Scholar] [CrossRef]
- Kawana, A.; Doi, M.; Motoyama, Y. The Seasonal Patterns of Xylem Formation Denoted by Pin Marking Method (II): The Morphological Study of Denoted Date and Healed Wounds after Pin Markings on Sugi (Cryptomeria japonica) Trunks. J. Jpn. For. Soc. 1974, 56, 16–19. [Google Scholar]
- Vander Mijnsbrugge, K.; Meyermans, H.; Van Montagu, M.; Bauw, G.; Boerjan, W. Wood formation in poplar: Identification, characterization, and seasonal variation of xylem proteins. Planta 2000, 210, 589–598. [Google Scholar] [CrossRef]
- Wu, H.; Xu, H.; Li, H.; Wei, D.; Lin, J.; Li, X. Seasonal development of cambial activity in relation to xylem formation in Chinese fir. J. Plant Physiol. 2016, 195, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sergio Rossi, H.M.D. Causes and correlations in cambium phenology: Towards an integrated framework of xylogenesis. J. Exp. Bot. 2012, 63, 2117–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nieminen, K.; Serra, J.A.A.; Helariutta, Y. The formation of wood and its control. Curr. Opin. Plant Biol. 2014, 17, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Baier, M.; Goldberg, R.; Catesson, A.-M.; Liberman, M.; Bouchemal, N.; Michon, V.; Du Penhoat, C.H. Pectin changes in samples containing poplar cambium and inner bark in relation to the seasonal cycle. Planta Berl. 1994, 193, 446–454. [Google Scholar] [CrossRef]
- Zhao, L.; Ding, Q.; Zeng, J.; Wang, F.R.; Zhang, J.; Fan, S.J.; He, X.Q. An Improved CTAB–Ammonium Acetate Method for Total RNA Isolation from Cotton. Phytochem. Anal. 2012, 23, 647–650. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Thissen, D.; Steinberg, L.; Kuang, D. Quick and Easy Implementation of the Benjamini-Hochberg Procedure for Controlling the False Positive Rate in Multiple Comparisons. J. Educ. Behav. Stat. 2002, 27, 77–83. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kanehisa, M. KEGG GLYCAN for Integrated Analysis of Pathways, Genes, and Structures; Springer: Tokyo, Japan, 2008. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein databases search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R.; Eddy, S. HMMER User’s Guide—Biological sequence analysis using profile hidden Markov models. Dep. Genet. 2003. [Google Scholar]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacob, A.R. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database^1. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Bateman, A. The Pfam protein family’s database. Nucleic Acids Res. 2009, 38, D211–D222. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277. [Google Scholar]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P. Principles of Long Noncoding RNA Evolution Derived from Direct Comparison of Transcriptomes in 17 Species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Borghi, L.; Gutzat, R.; Futterer, J.; Laizet, Y.; Hennig, L.; Gruissem, W. Arabidopsis RETINOBLASTOMA-RELATED Is Required for Stem Cell Maintenance, Cell Differentiation, and Lateral Organ Production. Plant. Cell 2010, 22, 1792–1811. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Torii, Y.; Morita, S.; Onodera, R.; Hara, Y.; Yokoyama, R.; Nishitani, K.; Satoh, S. Cloning, characterization, and expression of xyloglucan endotransglucosylase/hydrolase and expansin genes associated with petal growth and development during carnation flower opening. J. Exp. Bot. 2011, 62, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Fernie, A.R.; Persson, S. Transition of primary to secondary cell wall synthesis. Sci. Bull. 2016, 61, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Betancur, L.; Singh, B.; Rapp, R.A.; Wendel, J.F.; Marks, M.D.; Roberts, A.W.; Haigler, C.H. Phylogenetically Distinct Cellulose Synthase Genes Support Secondary Wall Thickening in Arabidopsis Shoot Trichomes and Cotton Fiber. J. Integr. Plant. Biol. 2010, 52, 205–220. [Google Scholar] [CrossRef]
- Merwe, M.J.V.D.; Osorio, S.; Araújo, W.L.; Balbo, I.; Nunes-Nesi, A.; Maximova, E.; Carrari, F.; Bunik, V.I.; Fernie, P.A.R. Tricarboxylic Acid Cycle Activity Regulates Tomato Root Growth via Effects on Secondary Cell Wall Production. Plant. Physiol. 2010, 153, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Tsuyama, T.; Kawai, R.; Shitan, N.; Matoh, T.; Sugiyama, J.; Yoshinaga, A.; Takabe, K.; Fujita, M.; Yazaki, K. Proton-Dependent Coniferin Transport, a Common Major Transport Event in Differentiating Xylem Tissue of Woody Plants. Plant. Physiol. 2013, 162, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Cahill, D.M.; Mccomb, J.A. A comparison of changes in phenylalanine ammonia-lyase activity, lignin and phenolic synthesis in the roots of Eucalyptus calophylla (field resistant) and E. marginata (susceptible) when infected with Phytophthora cinnamomi. Physiol. Mol. Plant. Pathol. 1992, 40, 315–332. [Google Scholar] [CrossRef]
- Costa, M.A.; Bedgar, D.L.; Moinuddin, S.G.A.; Kim, K.W.; Cardenas, C.L.; Cochrane, F.C.; Shockey, J.M.; Helms, G.L.; Amakura, Y.; Takahashi, H. Characterization in vitro and in vivo of the putative multigene 4-coumarate:CoA ligase network in Arabidopsis: Syringyl lignin and sinapate/sinapyl alcohol derivative formation. Phytochemistry 2005, 66, 2072–2091. [Google Scholar] [CrossRef] [PubMed]
- Piquemal, J.; Lapierre, C.; Myton, K.; O’Connell, A.; Boudet, A.M. Down-regulation of Cinnamoyl-CoA Reductase induces significant changes of lignin profiles in transgenic tobacco plants. Plant J. 2010, 13, 71–83. [Google Scholar] [CrossRef]
- Halpin, C.; Knight, M.E.; Foxon, G.A.; Campbell, M.M.; Schuch, W. Manipulation of lignin quality by downregulation of cinnamyl alcohol dehydrogenase. Plant. J. 2010, 6, 339–350. [Google Scholar] [CrossRef]
- Xue, Y.J.; Tao, L.; Yang, Z.M. Aluminum-induced cell wall peroxidase activity and lignin synthesis are differentially regulated by jasmonate and nitric oxide. J. Agric. Food Chem. 2008, 56, 9676. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Zhao, Y.; Xiang, Y.; Jiang, H.; Zhu, S.; Cheng, B. Downregulation of caffeoyl-CoA O-methyltransferase (CCoAOMT) by RNA interference leads to reduced lignin production in maize straw. Genet. Mol. Biol. 2013, 36, 540–546. [Google Scholar] [CrossRef] [Green Version]
- Negi, S.; Tak, H.; Ganapathi, T.R. In vitro xylem vessel elements formation from banana embryogenic cells and expression analysis of vessel development-related genes. J. Plant Biotechnol. Rep. 2015, 9, 47–54. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohtani, M.; Mitsuda, N.; Kubo, M.; Ohme-Takagi, M.; Fukuda, H.; Demura, T. VND-INTERACTING2, a NAC Domain Transcription Factor, Negatively Regulates Xylem Vessel Formation in Arabidopsis. Plant. Cell 2010, 22, 1249–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, X.; Liu, J.H. Deep sequencing-based characterization of transcriptome of trifoliate orange (Poncirus trifoliata (L.) Raf.) in response to cold stress. BMC Genom. 2015, 16, 555. [Google Scholar] [CrossRef] [Green Version]
- Changzheng, X.; Xiaokang, F.; Rui, L.; Li, G.; Lingyu, R.; Chaofeng, L.; Qiaoyan, T.; Bo, J.; Bangjun, W.; Keming, L. PtoMYB170 positively regulates lignin deposition during wood formation in poplar and confers drought tolerance in transgenic Arabidopsis. Narnia 2017, 37, 1713–1726. [Google Scholar]
- Zhong, R.; Yuan, Y.; Spiekerman, J.J.; Guley, J.T.; Egbosiuba, J.C.; Ye, Z.-H. Functional Characterization of NAC and MYB Transcription Factors Involved in Regulation of Biomass Production in Switchgrass (Panicum virgatum). PLoS ONE 2015, 10, e0134611. [Google Scholar] [CrossRef]
- Huang, J.G.; Ma, Q.; Rossi, S.; Biondi, F.; Deslauriers, A.; Fonti, P.; Liang, E.; Mäkinen, H.; Oberhuber, W.; Rathgeber, C.B.K.; et al. Photoperiod and temperature as dominant environmental drivers triggering secondary growth resumption in Northern Hemisphere conifers. Proc. Natl. Acad. Sci. USA 2020, 117, 20645–20652. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Paredez, A.; Carroll, A.; Palsdottir, H.; Doblin, M.; Poindexter, P.; Khitrov, N.; Auer, M.; Somerville, C.R. Genetic evidence for three unique components in primary cell-wall cellulose synthase complexes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 15566–15571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalivendra, S.; Huber, S.C.; Sachs, M.M.; Rhoads, D. Sucrose Synthase. Plant. Signal. Behav. 2007, 2, 28–29. [Google Scholar] [CrossRef] [Green Version]
- Abid, G.; Muhovski, Y.; Jacquemin, J.M.; Mingeot, D.; Sassi, K.; Toussaint, A.; Baudoin, J.P. Characterization and expression profile analysis of a sucrose synthase gene from common bean (Phaseolus vulgaris L.) during seed development. Mol. Biol. Rep. 2012, 39, 1133–1143. [Google Scholar] [CrossRef]
- Appenzeller, L.; Doblin, M.; Barreiro, R.; Wang, H.; Niu, X.; Kollipara, K.; Carrigan, L.; Tomes, D.; Chapman, M.; Dhugga, K.S. Cellulose synthesis in maize: Isolation and expression analysis of the cellulose synthase (CesA) gene family. Cellulose 2004, 11, 287–299. [Google Scholar] [CrossRef]
- Allario, T.; Tixier, A.; Awad, H.; Lemaire, C.; Brunel, N.; Badel, E.; Barigah, T.S.; Julien, J.L.; Peyret, P.; Mellerowicz, E.J.; et al. PtxtPME1 and homogalacturonans influence xylem hydraulic properties in poplar. Physiol. Plant. 2018, 163, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Canam, T.; Kang, K.Y.; Unda, F.; Mansfield, S.D. Sucrose phosphate synthase expression influences poplar phenology. Tree Physiol. 2009, 29, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureno, A.; Rencoret, J.; Chemetova, C.; Gominho, J.; Pereira, H. Lignin Composition and Structure Differs between Xylem, Phloem and Phellem in Quercus suber L. Front. Plant. Sci. 2016, 7, 1612. [Google Scholar]
- Farquharson, K.L. Good neighbor hypothesis of lignification holds for xylem cells, but not for interfascicular fibers. Plant. Cell 2013, 25, 3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, M.; Suzuki, S.; Umezawa, T.; Sato, Y. Progress of lignification mediated by intercellular transportation of monolignols during tracheary element differentiation of isolated Zinnia mesophyll cells. Plant. Cell Physiol. 2001, 42, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, N.; Sakakibara, N.; Umezawa, T.; Ito, Y.; Fukuda, H.; Sato, Y. Involvement of extracellular dilignols in lignification during tracheary element differentiation of isolated Zinnia mesophyll cells. Plant Cell Physiol. 2005, 46, 224–232. [Google Scholar] [CrossRef]
- Emiliani, G.; Traversi, M.L.; Anichini, M.; Giachi, G.; Giovannelli, A. Transcript Accumulation Dynamics of Phenylpropanoid Pathway Genes in the Maturing Xylem and Phloem of Picea abies during Latewood Formation. J. Integr. Plant. Biol. 2011, 53, 783–799. [Google Scholar] [CrossRef]
- Li, L.; Zhou, Y.; Cheng, X.; Sun, J.; Marita, J.M.; Ralph, J.; Chiang, V.L. Combinatorial modification of multiple lignin traits in trees through multigene cotransformation. Proc. Natl. Acad. Sci. USA 2003, 100, 4939–4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Vashisth, D.; Misra, A.; Akhtar, M.Q.; Jalil, S.U.; Shanker, K.; Gupta, M.M.; Rout, P.K.; Gupta, A.K.; Shasany, A.K. RNAi down-regulation of cinnamate-4-hydroxylase increases artemisinin biosynthesis in Artemisia annua. Sci. Rep. 2016, 6, 26458. [Google Scholar] [CrossRef] [Green Version]
- Milhinhos, A.; Miguel, C.L.M. Hormone interactions in xylem development: A matter of signals. Plant. Cell Rep. 2013, 32, 867–883. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, S.; Li, P.; Yin, Y.; Niu, Q.; Yan, J.; Huang, D. Plant buffering against the high-light stress-induced accumulation of CsGA2ox8 transcripts via alternative splicing to finely tune gibberellin levels and maintain hypocotyl elongation. Hortic. Res. 2021, 8, 2. [Google Scholar] [CrossRef]
- Huiyan, G.; Yucheng, W.; Huizi, L.; Ping, H.; Yuanyuan, J.; Chunrui, Z.; Yanmin, W.; Shan, G.; Chuanping, Y.; Chao, W. Exogenous GA3 Application Enhances Xylem Development and Induces the Expression of Secondary Wall Biosynthesis Related Genes in Betula platyphylla. Int. J. Mol. Sci. 2015, 16, 22960–22975. [Google Scholar]
- Yuan, Y.; Chong, W.; Yunjun, L.; Jian, Y.; Luqi, H.; Ji-Hong, L. The Scutellaria baicalensis R2R3-MYB Transcription Factors Modulates Flavonoid Biosynthesis by Regulating GA Metabolism in Transgenic Tobacco Plants. PLoS ONE 2013, 8, e77275. [Google Scholar] [CrossRef] [Green Version]
- Goicoechea, M.; Lacombe, E.; Legay, S.; Mihaljevic, S.; Grima-Pettenati, J. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant. J. 2010, 43, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.H. Transcriptional activation of secondary wall biosynthesis by rice and maize NAC and MYB transcription factors. Plant. Cell Physiol. 2011, 52, 1856. [Google Scholar]
- Nakano, Y.; Yamaguchi, M.; Endo, H.; Rejab, N.A.; Ohtani, M. NAC-MYB-based transcriptional regulation of secondary cell wall biosynthesis in land plants. Front. Plant. Sci. 2015, 6, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaumie, S.; Mzid, R.; Méchin, V.; Léon, C.; Hichri, I.; Destrac-Irvine, A.; Trossat-Magnin, C.; Delrot, S.; Lauvergeat, V. The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco. Plant. Mol. Biol. 2010, 72, 215. [Google Scholar] [CrossRef]
- Ko, J. Ectopic expression of MYB46 identifies transcriptional regulatory genes involved in secondary wall biosynthesis in Arabidopsis. Plant. J. Cell Mol. Biol. 2010, 60, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Camargo, E.L.O.; Carocha, V.; Cassan-Wang, H.; Grima-Pettenati, J. The Eucalyptus grandis R2R3-MYB transcription factor family: Evidence for woody growth-related evolution and function. New Phytol. 2014, 206, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Bonke, M.; Thitamadee, S.; Mhnen, A.P.; Hauser, M.T.; Helariutta, Y. APL regulates vascular tissue identity in Arabidopsis. Nature 2003, 426, 181–186. [Google Scholar] [CrossRef]
- Cominelli, E.; Sala, T.; Calvi, D.; Gusmaroli, G.; Tonelli, C. Over-expression of the Arabidopsis AtMYB41 gene alters cell expansion and leaf surface permeability. Plant. J. 2010, 53, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Ye, R.Z.H. The MYB46 Transcription Factor Is a Direct Target of SND1 and Regulates Secondary Wall Biosynthesis in Arabidopsis. Plant. Cell Online 2007, 19, 2776–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, M.; Nishikubo, N.; Xu, B.; Yamaguchi, M.; Mitsuda, N.; Goué, N.; Shi, F.; Ohme-Takagi, M.; Demura, T. A NAC domain protein family contributing to the regulation of wood formation in poplar. Plant. J. 2011, 67, 499–512. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Transcript | Unigene |
|---|---|---|
| 200–300 | 39,799 (13.06%) | 33,221 (39.21%) |
| 300–500 | 31,720 (10.41%) | 19,620 (23.16%) |
| 500–1000 | 46,697 (15.33%) | 14,289 (16.86%) |
| 1000–2000 | 83,525 (27.41%) | 8068 (9.52%) |
| 2000+ | 102,953 (33.79%) | 9535 (11.25%) |
| Total Number | 304,694 | 84,733 |
| Total Length | 497,649,345 | 68,129,242 |
| N50 Length | 2437 | 1764 |
| Mean Length | 1633.28 | 804.05 |
| Public Database | Number of Annotated Genes |
|---|---|
| COG_Annotation | 13,860 |
| GO_Annotation | 25,234 |
| KEGG_Annotation | 15,363 |
| KOG_Annotation | 19,885 |
| Pfam_Annotation | 23,089 |
| Swissprot_Annotation | 24,234 |
| eggNOG_Annotation | 37,441 |
| nr_Annotation | 40,513 |
| All_Annotated | 43,525 |
| DEG_Set | All_DEGs | Upregulated | Downregulated |
|---|---|---|---|
| S1 vs._S2 | 4273 | 2091 | 2182 |
| S1 vs._S3 | 8282 | 4014 | 4268 |
| S1 vs._S4 | 8587 | 4266 | 4321 |
| S1 vs._S5 | 6852 | 3833 | 3019 |
| S1 vs._S6 | 5875 | 3308 | 2567 |
| S2 vs. S3 | 6470 | 3224 | 3246 |
| S3 vs. S4 | 907 | 481 | 426 |
| S4 vs. S5 | 3564 | 2807 | 757 |
| S5 vs. S6 | 1726 | 560 | 1166 |
| Pathway | DEGs with Pathway Annotation | All Genes with Pathway Annotation | Q Value | Pathway ID | |
|---|---|---|---|---|---|
| S1 vs. S2 | Phenylpropanoid biosynthesis | 43 | 198 | 1.90 × 10−10 | ko00940 |
| Plant hormone signal transduction | 49 | 337 | 1.08 × 10−5 | ko04075 | |
| Starch and sucrose metabolism | 52 | 368 | 1.11 × 10−5 | ko00500 | |
| Plant–pathogen interaction | 42 | 307 | 4.63 × 10−4 | ko04626 | |
| S1 vs. S3 | Starch and sucrose metabolism | 95 | 368 | 2.00 × 10−5 | ko00500 |
| Endocytosis | 79 | 314 | 6.57 × 10−4 | ko04144 | |
| Amino sugar and nucleotide sugar metabolism | 66 | 252 | 9.63 × 10−4 | ko00520 | |
| Phagosome | 44 | 159 | 7.78 × 10−3 | ko04145 | |
| Plant hormone signal transduction | 77 | 337 | 2.86 × 10−2 | ko04075 | |
| S1 vs. S4 | Endocytosis | 84 | 314 | 3.70 × 10−4 | ko04144 |
| Starch and sucrose metabolism | 92 | 368 | 2.56 × 10−3 | ko00500 | |
| Phagosome | 47 | 159 | 4.01 × 10−3 | ko04145 | |
| Amino sugar and nucleotide sugar metabolism | 66 | 252 | 8.58 × 10−3 | ko00520 | |
| Plant hormone signal transduction | 81 | 337 | 3.22 × 10−2 | ko04075 | |
| S1 vs. S5 | Starch and sucrose metabolism | 85 | 368 | 1.39 × 10−5 | ko00500 |
| Phenylpropanoid biosynthesis | 49 | 198 | 9.89 × 10−4 | ko00940 | |
| S1 vs. S6 | Phenylpropanoid biosynthesis | 54 | 198 | 2.04 × 10−8 | ko00940 |
| Starch and sucrose metabolism | 70 | 368 | 4.32 × 10−4 | ko00500 | |
| Diterpenoid biosynthesis | 14 | 33 | 5.18 × 10−4 | ko00904 | |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 10 | 24 | 1.50 × 10−2 | ko00945 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; He, Y.; Yu, Y.; Zhou, M.; Zhao, L.; Xia, X.; Wang, C. Transcriptomic Analysis of Seasonal Gene Expression and Regulation during Xylem Development in “Shanxin” Hybrid Poplar (Populus davidiana × Populus bolleana). Forests 2021, 12, 451. https://0-doi-org.brum.beds.ac.uk/10.3390/f12040451
Zhao C, He Y, Yu Y, Zhou M, Zhao L, Xia X, Wang C. Transcriptomic Analysis of Seasonal Gene Expression and Regulation during Xylem Development in “Shanxin” Hybrid Poplar (Populus davidiana × Populus bolleana). Forests. 2021; 12(4):451. https://0-doi-org.brum.beds.ac.uk/10.3390/f12040451
Chicago/Turabian StyleZhao, Chao, Youchao He, Ying Yu, Meiqi Zhou, Leifei Zhao, Xiangyou Xia, and Chao Wang. 2021. "Transcriptomic Analysis of Seasonal Gene Expression and Regulation during Xylem Development in “Shanxin” Hybrid Poplar (Populus davidiana × Populus bolleana)" Forests 12, no. 4: 451. https://0-doi-org.brum.beds.ac.uk/10.3390/f12040451