De Novo Assembly and Comparative Transcriptome Profiling of Anser anser and Anser cygnoides Geese Species’ Embryonic Skin Feather Follicles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Sample Collection
2.3. RNA Isolation
2.4. Library Sequencing
2.5. De Novo Assembly and Functional Annotation
2.6. Differentially Expressed Genes (DEGs) Analysis
2.7. RNA-Seq Data Validation by Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
3. Results
3.1. Illumina cDNA Data Sequencing and De Novo Assembly
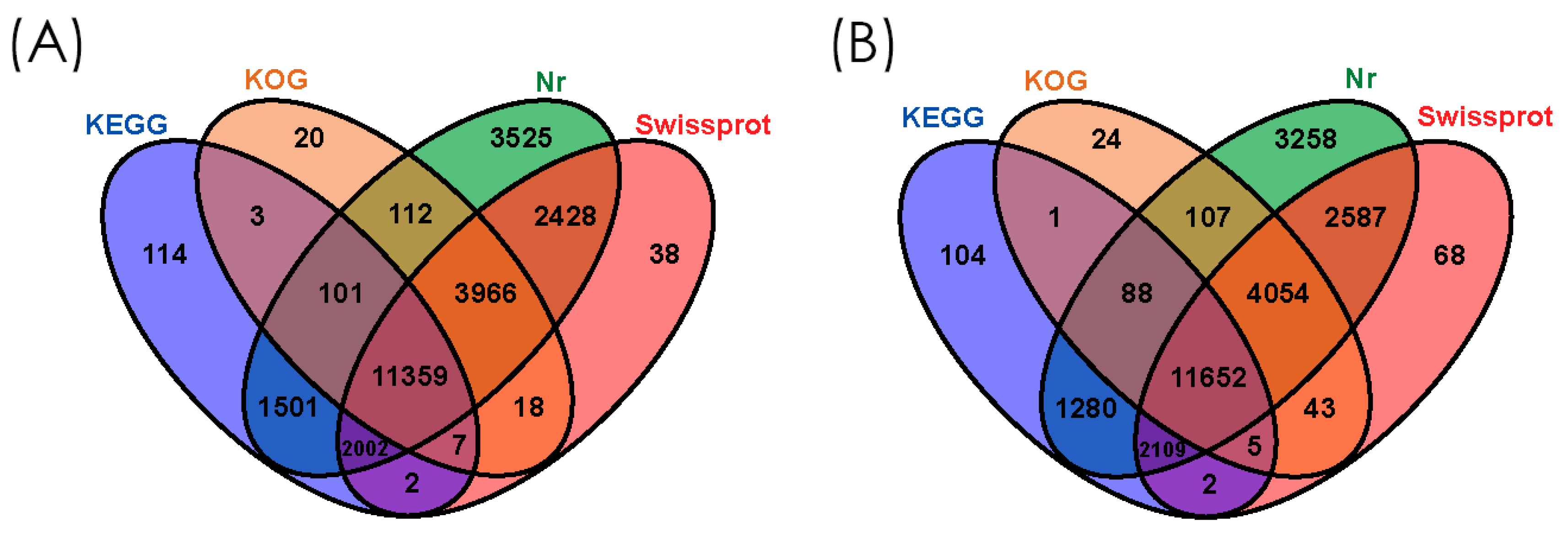
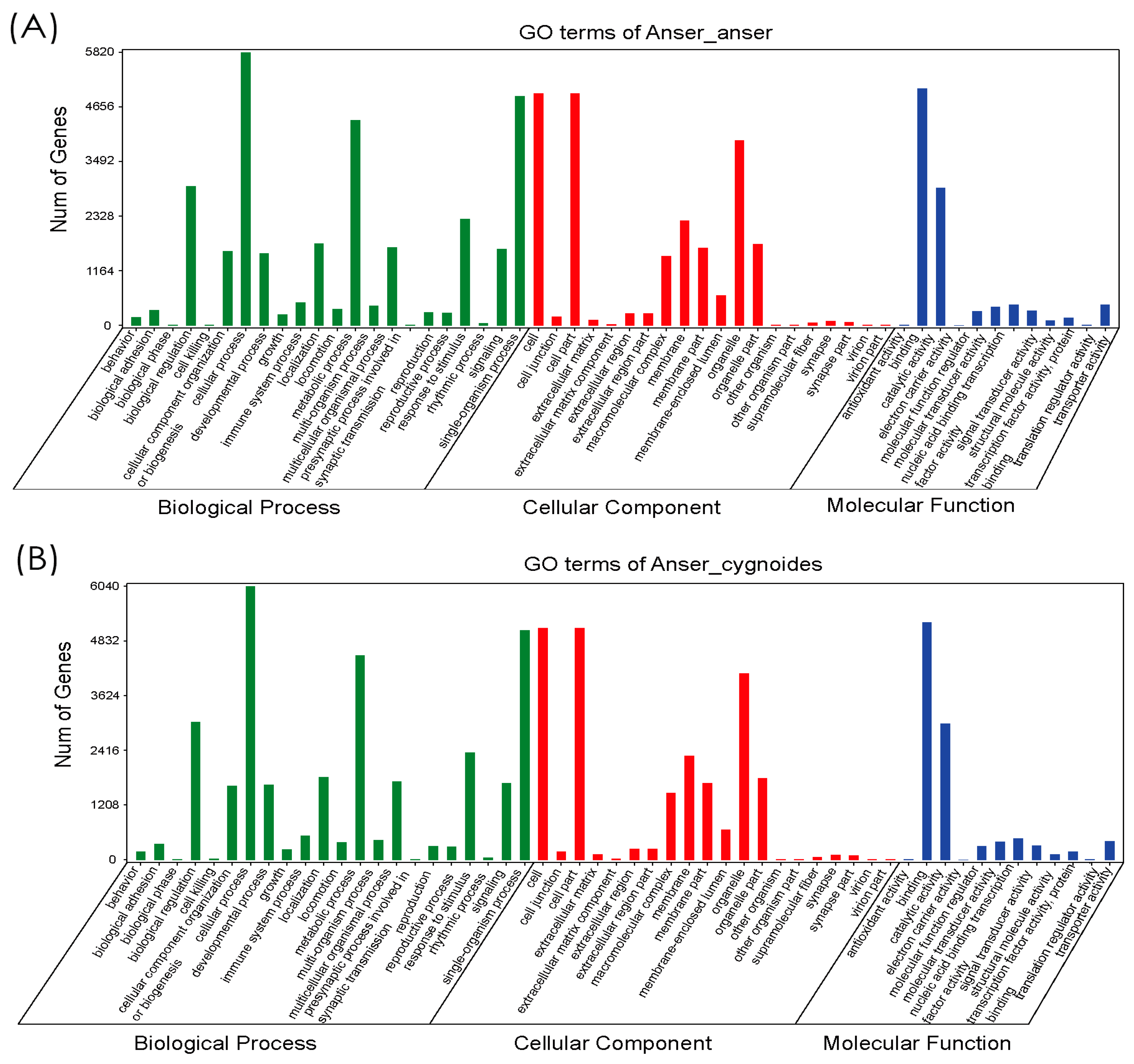
3.2. Functional Annotation of Gene Ontology (GO), Eukaryotic Orthologous Groups (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG)
3.3. Gene Homology Analysis
3.4. Differentially Expressed Genes (DEGs) Analysis
3.5. Validation of RNA-Seq by RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ding, N.; Han, Q.; Zhao, X.Z.; Li, Q.; Li, J.; Zhang, H.F.; Gao, G.L.; Luo, Y.; Xie, Y.H.; Su, J. Differential gene expression in pre-laying and laying period ovaries of Sichuan White geese (Anser cygnoides). Genet. Mol. Res. 2015, 14, 6773–6785. [Google Scholar] [CrossRef] [PubMed]
- Kozák, J. An overview of feathers formation, moults and down production in geese. Asian-Australas. J. Anim. Sci. 2011, 24, 881–887. [Google Scholar] [CrossRef]
- Yu, M.; Yue, Z.; Wu, P.; Wu, D.Y.; Mayer, J.A.; Medina, M.; Widelitz, R.B.; Jiang, T.X.; Chuong, C.M. The biology of feather follicles. Int. J. Dev. Biol. 2004, 48, 181–191. [Google Scholar] [CrossRef]
- Widelitz, R.B.; Veltmaat, J.M.; Mayer, J.A.; Foley, J.; Chuong, C.M. Mammary glands and feathers: Comparing two skin appendages which help define novel classes during vertebrate evolution. Semin. Cell Dev. Biol. 2007, 18, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.S.; Chen, C.K.; Fan, W.L.; Wu, P.; Wu, S.M.; Chen, J.J.; Lai, Y.T.; Mao, C.T.; Lu, M.Y.; Chen, D.R.; et al. Transcriptomic analyses of regenerating adult feathers in chicken. BMC Genom. 2015, 16, 756. [Google Scholar] [CrossRef]
- Widelitz, R.B.; Jiang, T.X.; Yu, M.; Shen, T.; Shen, J.Y.; Wu, P.; Yu, Z.; Chuong, C.M. Molecular biology of feather morphogenesis: A testable model for evo-devo research. J. Exp. Zool. B Mol. Dev. Evol. 2003, 298, 109–122. [Google Scholar] [CrossRef]
- Dhouailly, D.; Godefroit, P.; Martin, T.; Nonchev, S.; Caraguel, F.; Oftedal, O. Getting to the root of scales, feather and hair: As deep as odontodes? Exp. Dermatol. 2017, 28, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.F.; Wu, W.; Xu, H. Investigation of feather follicle development in embryonic geese. Poult. Sci. 2007, 86, 2000–2007. [Google Scholar] [CrossRef]
- Leeson, S.; Walsh, T. Feathering in commercial poultry II. Factors influencing feather growth and feather loss. World Poultry Sci. J. 2004, 60, 52–63. [Google Scholar] [CrossRef]
- Chen, X.; Bai, H.; Li, L.; Zhang, W.; Jiang, R.; Geng, Z. Follicle characteristics and follicle developmental related Wnt6 polymorphism in Chinese indigenous Wanxi-white goose. Mol. Biol. Rep. 2012, 39, 9843–9848. [Google Scholar] [CrossRef]
- Wu, W.; Xu, R.F.; Li, C.H.; Wu, C.X. Characterization of embryonic feather follicle development in the Chinese indigenous Jilin white goose. Asian-Australas. J. Anim. Sci. 2008, 21, 346–352. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, L.; Gong, G.; Zhang, W.; Zhu, B.; Hu, Y. De novo comparative transcriptome analysis of Acremonium chrysogenum: High-yield and wild-type strains of cephalosporin C producer. PLoS ONE 2014, 9, e104542. [Google Scholar] [CrossRef]
- Torre, S.; Tattini, M.; Brunetti, C.; Guidi, L.; Gori, A.; Marzano, C.; Landi, M.; Sebastiani, F. De novo assembly and comparative transcriptome analyses of red and green morphs of sweet basil grown in full sunlight. PLoS ONE 2016, 11, e0160370. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tomiyama, T.; Maruta, T.; Tomita, M.; Ishikawa, T.; Arakawa, K. De novo assembly and comparative transcriptome analysis of Euglena gracilis in response to anaerobic conditions. BMC Genom. 2016, 17, 182. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Zhang, L.; Yin, Y.; Wu, J.; Yu, L.; Zhou, Y.; Li, M. Sequencing, de novo assembly and comparative analysis of Raphanus sativus transcriptome. Front. Plant Sci. 2015, 6, 198. [Google Scholar] [CrossRef]
- Li, H.F.; Zhu, W.Q.; Chen, K.W.; H, Y.; Xu, W.J.; Song, W. Two maternal origins of Chinese domestic goose. Poult Sci. 2011, 90, 2705–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Sello, C.T.; Sun, Y.; Zhou, Y.; Lu, H.; Sui, Y.; Hu, J.; Xu, C.; Sun, Y.; Liu, J.; et al. De novo transcriptome sequencing analysis of goose (anser anser) embryonic skin and the identification of genes related to feather follicle morphogenesis at three stages of development. Int. J. Mol. Sci. 2018, 19, 3170. [Google Scholar] [CrossRef] [PubMed]
- Pernthaler, A.; Amann, R. Simultaneous fluorescence in situ hybridization of mRNA and rRNA in environmental bacteria. Appl. Environ. Microbiol. 2004, 70, 5426–5433. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef]
- Gluckman, T.L.; Mundy, N.I. The differential expression of MC1R regulators in dorsal and ventral quail plumages during embryogenesis: Implications for plumage pattern formation. PloS ONE 2017, 12, e0174714. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.S.; Li, W.H. Genetic and molecular basis of feather diversity in birds. Genome Biol. Evol. 2018, 10, 2572–2586. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Foley, J.; Jiang, T.X.; Yeh, C.Y.; Wu, P.; Foley, A.; Yen, C.M.; Huang, Y.C.; Cheng, H.; Chen, C.F.; et al. Topology of feather melanocyte progenitor niche allows complex pigment patterns to emerge. Science 2013, 340, 1442–1445. [Google Scholar] [CrossRef] [Green Version]
- Haupaix, N.; Curantz, C.; Bailleul, R.; Beck, S.; Robic, A.; Manceau, M. The periodic coloration in birds forms through a prepattern of somite origin. Science 2018, 361, eaar4777. [Google Scholar] [CrossRef]
- Ainsworth, S.J.; Stanley, R.L.; Evans, D.J.R. Developmental stages of the Japanese quail. J. Anat. 2010, 216, 3–15. [Google Scholar] [CrossRef]
- Chen, X.; Ge, K.; Wang, M.; Zhang, C.; Geng, Z. Integrative analysis of the Pekin duck (Anas anas) MicroRNAome during feather follicle development. BMC Dev. Biol. 2017, 17, 12. [Google Scholar] [CrossRef]
- Watt, F.M.; Fujiwara, H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb. Perspect. Biol. 2011, 3, a005124. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.W.; Huang, N.A.; Liu, I.H.; Wang, Y.H.; Wu, P.; Tseng, Y.T.; Hughes, M.W.; Jiang, T.X.; Tsai, M.H.; Chen, C.Y.; et al. Emergence of differentially regulated pathways associated with the development of regional specificity in chicken skin. BMC Genom. 2015, 16, 22. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, T.; Yuan, J.; Guo, X.; Liu, D. Transcriptome sequencing reveals differences between primary and secondary hair follicle-derived dermal papilla cells of the Cashmere goat (Capra hircus). PLoS ONE 2013, 8, e76282. [Google Scholar] [CrossRef]
- Michalik, L.; Wahli, W. Peroxisome proliferator-activated receptors (PPARs) in skin health, repair and disease. Biochim. Biophys. Acta 2007, 1771, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Van Zeeland, Y.R.; Schoemaker, N.J. Plumage disorders in psittacine birds-part 1: Feather abnormalities. EJCAP 2014, 24, 34–47. [Google Scholar]
- McGraw, K.J. An update on the honesty of melanin-based color signals in birds. Pigment Cell Melanoma Res. 2008, 21, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Pannkuk, E.L.; Mcguire, L.P.; Gilmore, D.F.; Savary, B.J.; Risch, T.S. Glycerophospholipid analysis of eastern red bat (Lasiurus borealis) hair by electrospray ionization tandem mass spectrometry. J. Chem. Ecol. 2014, 40, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Johnston, A.; Stoll, S.W.; Riblett, M.B.; Xing, X.; Kochkodan, J.J.; Ding, J.; Nair, R.P.; Aphale, A.; Voorhees, J.J.; et al. Evidence for altered Wnt signaling in psoriatic skin. J. Investig. Dermatol. 2010, 130, 1849–1859. [Google Scholar] [CrossRef]
- Chodankar, R.; Chang, C.H.; Yue, Z.; Jiang, T.X.; Suksaweang, S.; Burrus, L.; Chuong, C.M.; Widelitz, R. Shift of localized growth zones contributes to skin appendage morphogenesis: Role of the Wnt/β-catenin pathway. J. Investig. Dermatol. 2003, 120, 20–26. [Google Scholar] [CrossRef]
- Truzzi, F.; Marconi, A.; Pincelli, C. Neurotrophins in healthy and diseased skin. Dermatoendocrinol 2011, 3, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Thomson, T.M.; Rettig, W.J.; Chesa, P.G.; Green, S.H.; Mena, A.C.; Old, L.J. Expression of human nerve growth factor receptor on cells derived from all three germ layers. Exp. Cell Res. 1988, 174, 533–539. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Maddaluno, L.; Urwyler, C.; Werner, S. Fibroblast growth factors: Key players in regeneration and tissue repair. Development 2017, 144, 4047–4060. [Google Scholar] [CrossRef]
- Chen, G.J.; Forough, R. Fibroblast growth factors, fibroblast growth factor receptors, diseases, and drugs. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 211–224. [Google Scholar] [CrossRef]
- Botchkarev, V.A.; Sharov, A.A. BMP signaling in the control of skin development and hair follicle growth. Differentiation 2004, 72, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A. Bone morphogenetic proteins and their antagonists in skin and hair follicle biology. J. Investig. Dermatol. 2003, 120, 36–47. [Google Scholar] [CrossRef]
- Michon, F.; Forest, L.; Collomb, E.; Demongeot, J.; Dhouailly, D. BMP2 and BMP7 play antagonistic roles in feather induction. Development 2008, 135, 2797–2805. [Google Scholar] [CrossRef]
- Jahan, S.; Sun, J.M.; He, S.; Davie, J.R. Transcription-dependent association of HDAC2 with active chromatin. J. Cell. Physiol. 2018, 233, 1650–1657. [Google Scholar] [CrossRef]
- Hughes, M.W.; Jiang, T.X.; Lin, S.J.; Leung, Y.; Kobielak, K.; Widelitz, R.B.; Chuong, C.M. Disrupted ectodermal organ morphogenesis in mice with a conditional histone deacetylase 1, 2 deletion in the epidermis. J. Investig. Dermatol. 2014, 134, 24–32. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2010, 19, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Sharp, L.L.; Jameson, J.M.; Cauvi, G.; Havran, W.L. Dendritic epidermal T cells regulate skin homeostasis through local production of insulin-like growth factor 1. Nat. Immunol. 2005, 6, 73–79. [Google Scholar] [CrossRef]
- Vincent, A.M.; Feldman, E.L. Control of cell survival by IGF signaling pathways. Growth Horm. IGF Res. 2002, 12, 193–197. [Google Scholar] [CrossRef]
- Stentz, F.B.; Kitabchi, A.E. De novo emergence of growth factor receptors in activated human CD4+ and CD8+ T lymphocytes. Metabolism 2004, 53, 117–122. [Google Scholar] [CrossRef]
- Detmar, M. The role of VEGF and thrombospondins in skin angiogenesis. J. Dermatol. Sci. 2000, 24, S78–S84. [Google Scholar] [CrossRef]
- Jeltsch, M.; Kaipainen, A.; Joukov, V.; Meng, X.; Lakso, M.; Rauvala, H.; Swartz, M.; Fukumura, D.; Jain, R.K.; Alitalo, K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 1997, 276, 1423–1425. [Google Scholar] [CrossRef]
- Cegielski, M.; Izykowska, I.; Chmielewska, M.; Dziewiszek, W.; Bochnia, M.; Calkosinski, I.; Dziegiel, P. Characteristics of MIC-1 antlerogenic stem cells and their effect on hair growth in rabbits. In Vivo 2013, 27, 97–106. [Google Scholar] [CrossRef]
- Tomita, Y.; Akiyama, M.; Shimizu, H. PDGF isoforms induce and maintain anagen phase of murine hair follicles. J. Dermatol. Sci. 2006, 43, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Kamp, H.; Geilen, C.; Sommer, C.; Blume-Peytavi, U. Regulation of PDGF and PDGF receptor in cultured dermal papilla cells and follicular keratinocytes of the human hair follicle. Exp. Dermatol. 2003, 12, 662–672. [Google Scholar] [CrossRef]
- Bigot, N.; Beauchef, G.; Hervieu, M.; Oddos, T.; Demoor, M.; Boumediene, K.; Galéra, P. NF-κB accumulation associated with COL1A1 transactivators defects during chronological aging represses type I collagen expression through a–112/–61-bp region of the COL1A1 promoter in human skin fibroblasts. J. Investig. Dermatol. 2012, 32, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Peng, Z.; Xiao, S.; Geng, S.; Yuan, J.; Li, Z. RNAi-mediated inhibition of COL1A1 and COL3A1 in human skin fibroblasts. Exp. Dermatol. 2007, 16, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, Y.; Zhang, J.; Zhong, J.; Yang, R. COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Rep. 2018, 17, 5037–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, P.H.; Pyott, S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012, 46, 475–497. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anser anser | Anser cygnoides | |
|---|---|---|
| Total number of raw reads | 170,246,914 | 147,754,320 |
| Total number of clean reads | 165,564,278 | 144,595,262 |
| Total length of clean reads (bp) | 24,315,965,489 | 21,274,765,140 |
| Q20 percentage after filter (%) | 98.31 | 98.46 |
| Total number of unigenes | 77,134 | 66,041 |
| GC percentage of unigenes (%) | 49.3170 | 49.8801 |
| N50 length of unigenes (bp) | 2318 | 2206 |
| Max length of unigenes (bp) | 24,412 | 22,078 |
| Min length of unigenes (bp) | 201 | 201 |
| Pathways | DEGs in Anser anser (2200) | All genes in Anser anser (6850) | DEGs in Anser cygnoides (2100) | All genes in Anser cygnoides (6777) | Pathway ID |
|---|---|---|---|---|---|
| Cytokine-cytokine receptor interaction | 71 (3.23%) | 230 (3.36%) | 49 (2.33%) | 210 (3.1%) | ko04060 |
| Adherens junction | 26 (1.18%) | 98 (1.43%) | 48 (2.29%) | 120 (1.77%) | ko04520 |
| Cell cycle | 32 (1.45%) | 170 (2.48%) | 55 (2.62%) | 190 (2.8%) | ko04110 |
| TGF-beta signaling pathway | 24 (1.09%) | 108 (1.58%) | 17 (0.81%) | 95 (1.4%) | ko04350 |
| Wnt signaling pathway | 52 (2.36%) | 182 (2.66%) | 50 (2.38%) | 192 (2.83%) | ko04310 |
| Endocytosis | 125 (5.68%) | 388 (5.66%) | 120 (5.71%) | 376 (5.55%) | ko04144 |
| Metabolic pathways | 569 (25.86%) | 1688 (24.64%) | 513 (24.43%) | 1623 (23.95%) | ko01100 |
| MAPK signaling pathway | 129 (5.86%) | 372 (5.43%) | 149 (7.1%) | 393 (5.8%) | ko04010 |
| Hedgehog signaling pathway | 16 (0.73%) | 61 (0.89%) | 18 (0.86%) | 61 (0.9%) | ko04340 |
| Symbol | Anser anser | Anser cygnoides | KEGG Pathway | GO Function | ||
|---|---|---|---|---|---|---|
| Gene ID | RPKM | Gene ID | RPKM | |||
| Wnt6 | Unigene0008642 | 5.0506 | Unigene0034364 | 3.4138 | ko04310//Wnt signaling pathway | GO:0050789//regulation of biological process |
| NGFR | Unigene0002367 | 24.2656 | Unigene0045291 | 12.6814 | ko04060//Cytokine-cytokine receptor interaction | GO:0001942//hair follicle development |
| FGF16 | Unigene0016221 | 2.0958 | Unigene0019329 | 0.8415 | ko04010//MAPK signaling pathway | GO:0032502//developmental process |
| BMP7 | Unigene0043457 | 1.8824 | Unigene0010378 | 0.6135 | ko04060//Cytokine-cytokine receptor interaction | GO:0060429//epithelium development |
| HDAC2 | Unigene0039479 | 17.7176 | Unigene0022448 | 0.0936 | ko04110//Cell cycle | GO:0043588//skin development |
| IGF1R | Unigene0054392 | 2.1838 | Unigene0005210 | 1.695 | ko04144//Endocytosis | GO:0002009//morphogenesis of an epithelium |
| VEGFC | Unigene0019611 | 3.3092 | Unigene0022184 | 2.561 | ko04510//Focal adhesion | GO:0065007//biological regulation |
| PDGFB | Unigene0053351 | 8.9492 | Unigene0044387 | 5.1886 | ko04010//MAPK signaling pathway | GO:0060429//epithelium development |
| KCNMA1 | Unigene0054701 | 0.4124 | Unigene0042712 | 0.6262 | ko04270//Vasc-ular smooth muscle contraction | GO:0008544//epidermis development |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sello, C.T.; Liu, C.; Sun, Y.; Msuthwana, P.; Hu, J.; Sui, Y.; Chen, S.; Zhou, Y.; Lu, H.; Xu, C.; et al. De Novo Assembly and Comparative Transcriptome Profiling of Anser anser and Anser cygnoides Geese Species’ Embryonic Skin Feather Follicles. Genes 2019, 10, 351. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10050351
Sello CT, Liu C, Sun Y, Msuthwana P, Hu J, Sui Y, Chen S, Zhou Y, Lu H, Xu C, et al. De Novo Assembly and Comparative Transcriptome Profiling of Anser anser and Anser cygnoides Geese Species’ Embryonic Skin Feather Follicles. Genes. 2019; 10(5):351. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10050351
Chicago/Turabian StyleSello, Cornelius Tlotliso, Chang Liu, Yongfeng Sun, Petunia Msuthwana, Jingtao Hu, Yujian Sui, Shaokang Chen, Yuxuan Zhou, Hongtao Lu, Chenguang Xu, and et al. 2019. "De Novo Assembly and Comparative Transcriptome Profiling of Anser anser and Anser cygnoides Geese Species’ Embryonic Skin Feather Follicles" Genes 10, no. 5: 351. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10050351