The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Horses and Data Collection
2.2. Genotypes and Quality Control
2.3. Parameters Within Populations
2.3.1. Inbreeding Coefficient
2.3.2. Linkage Disequilibrium
2.3.3. Pairwise Identity-by-Descent
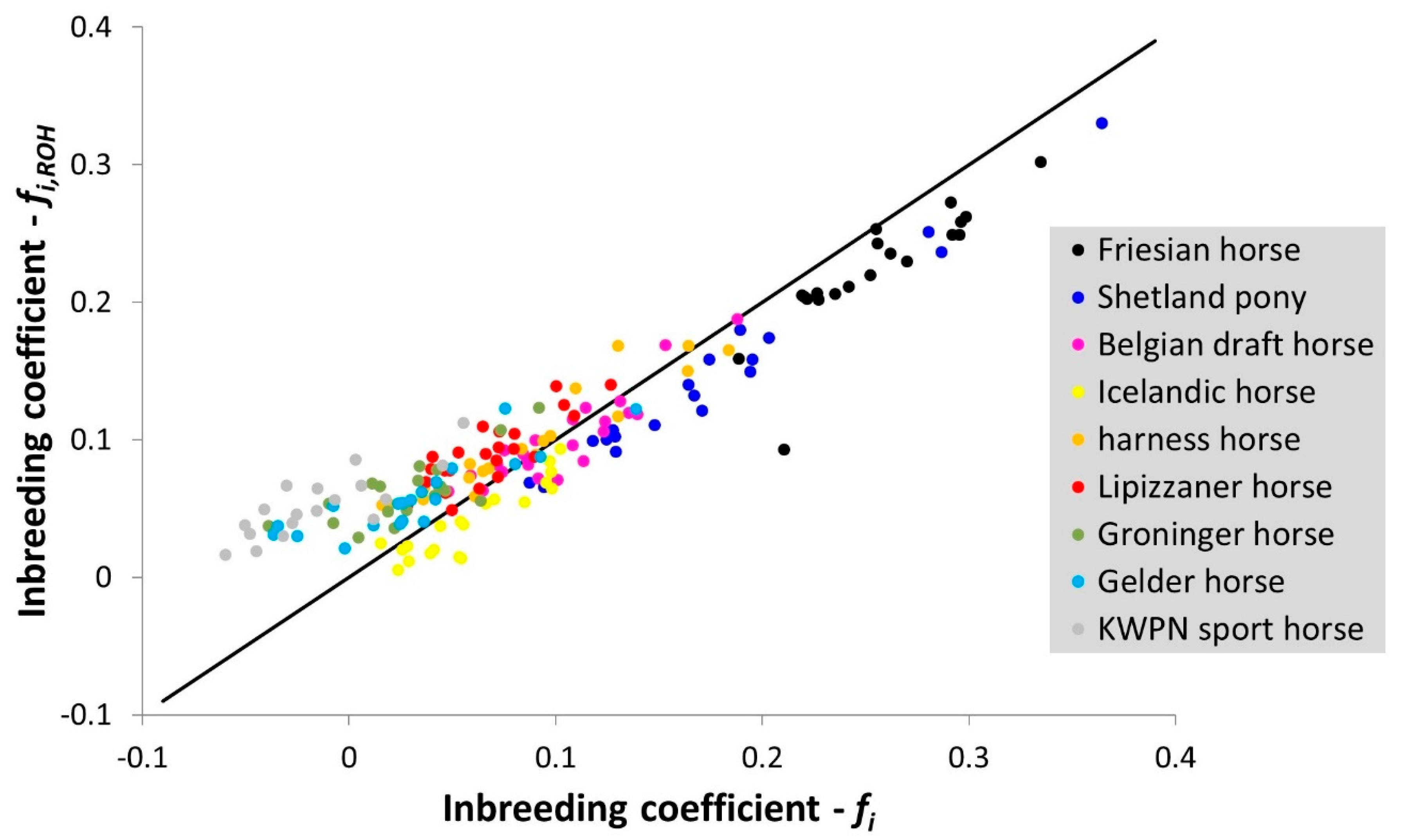
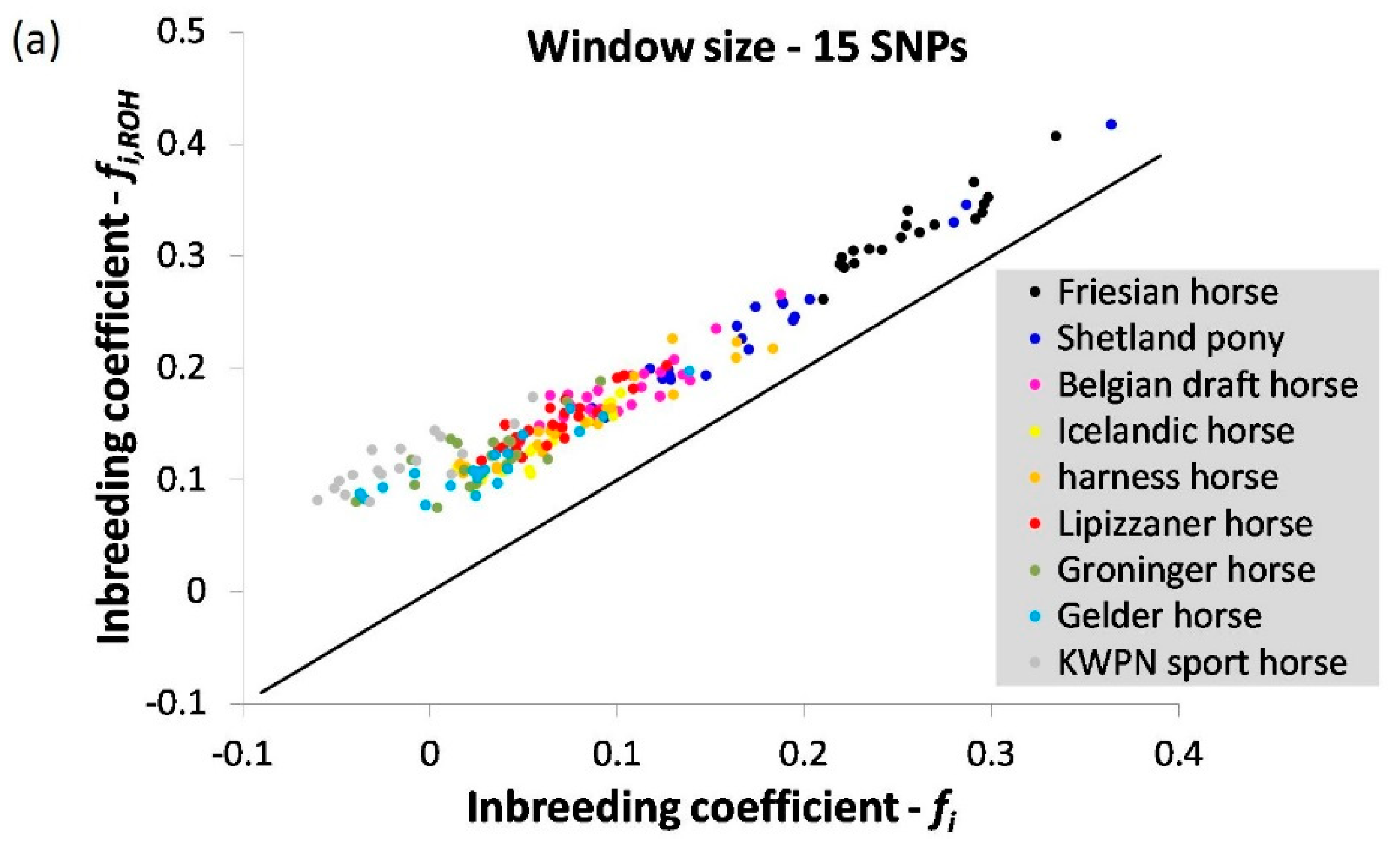
2.3.4. Runs of Homozygosity
2.4. Relationships Among Populations
2.4.1. Multidimensional Scaling
2.4.2. FST Approach
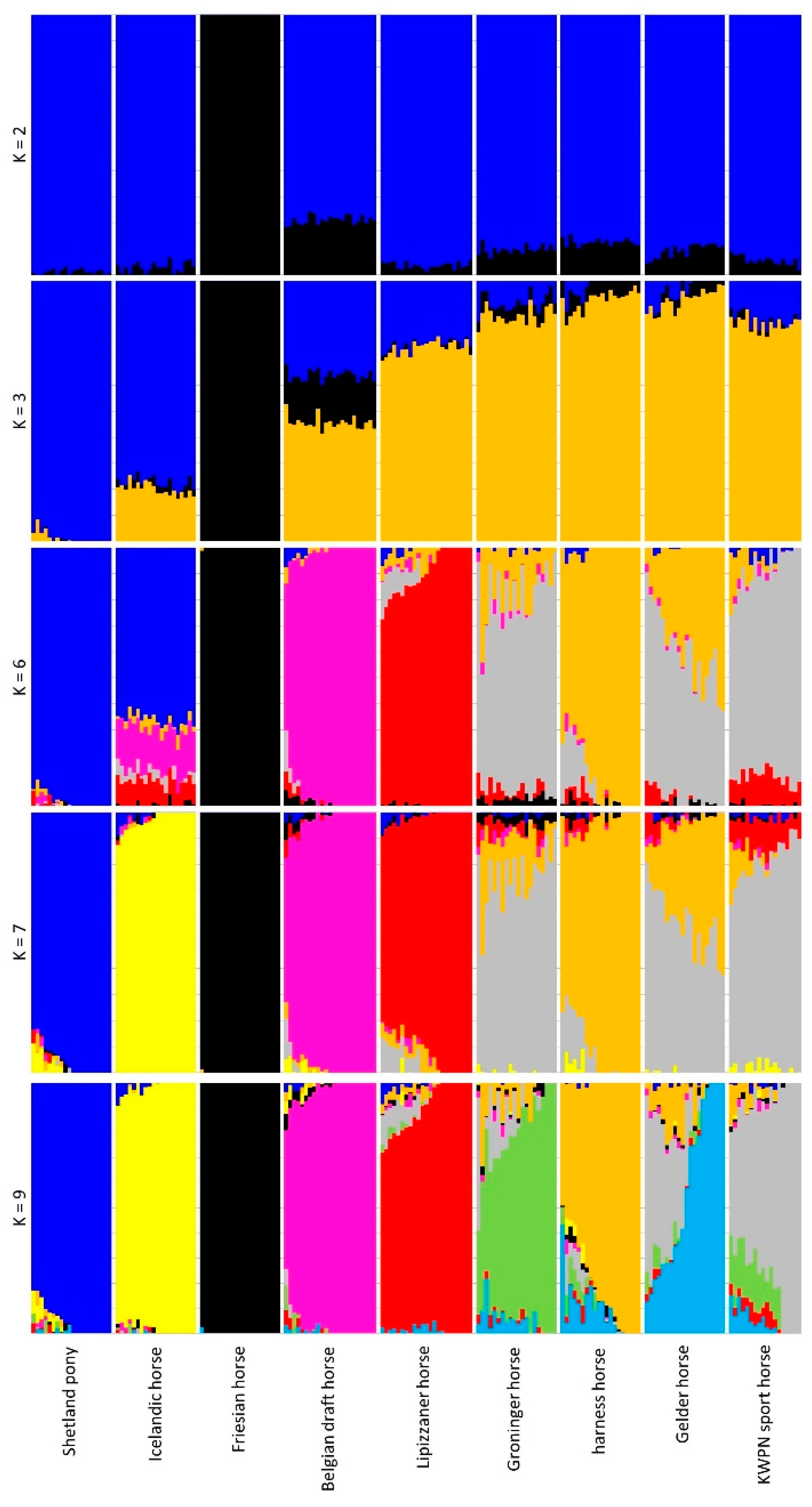
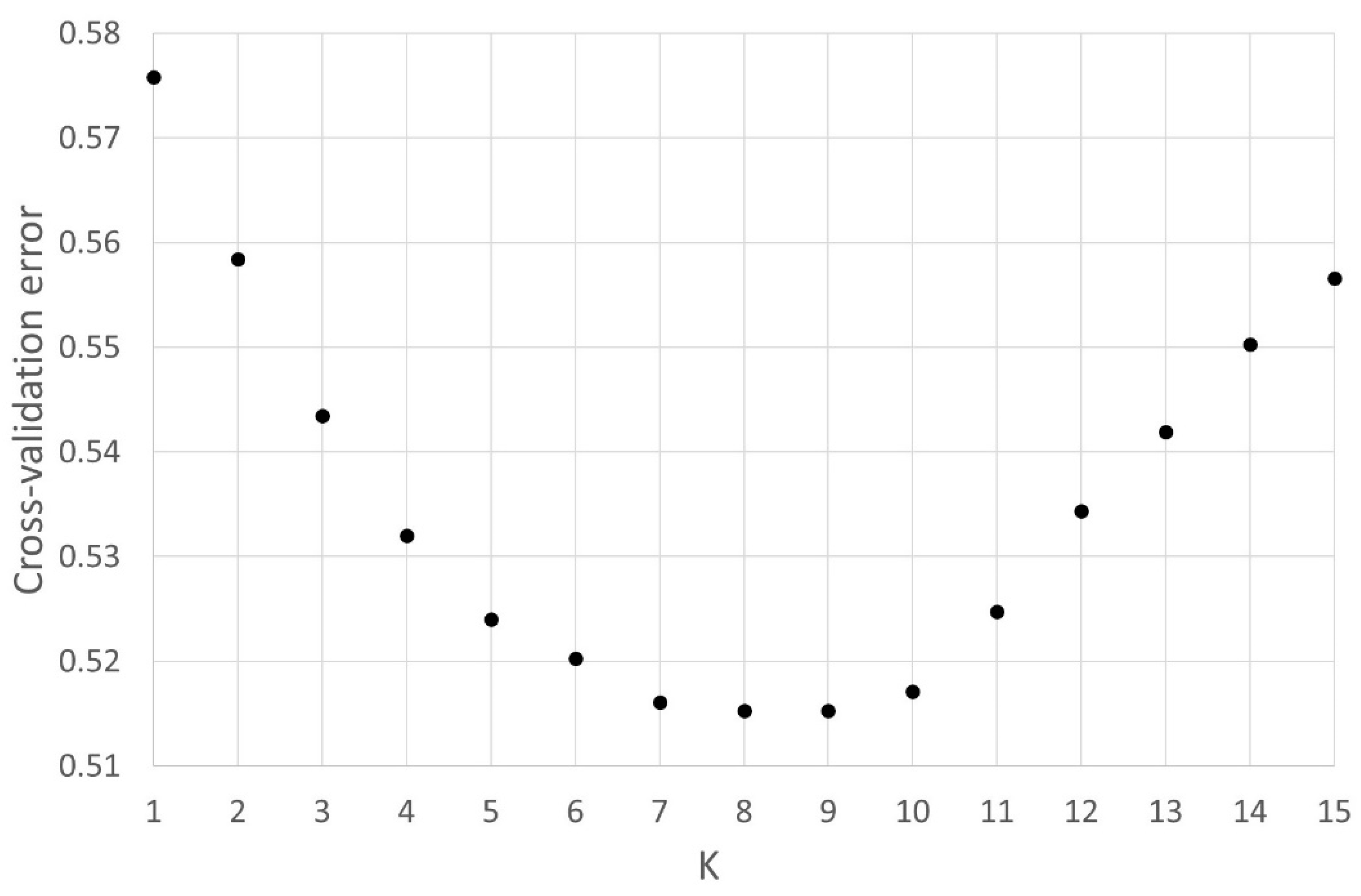
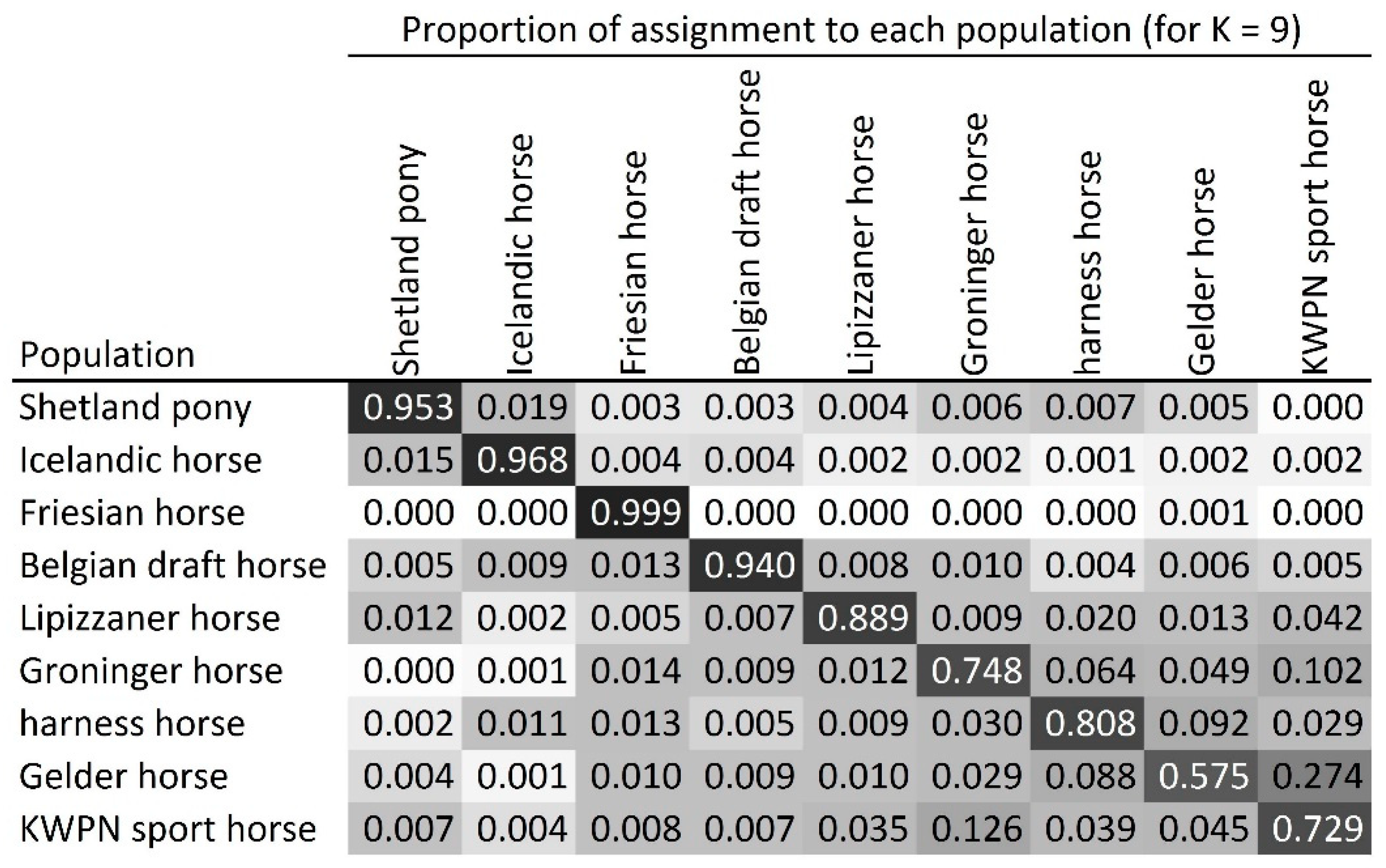
2.4.3. Admixture Analysis
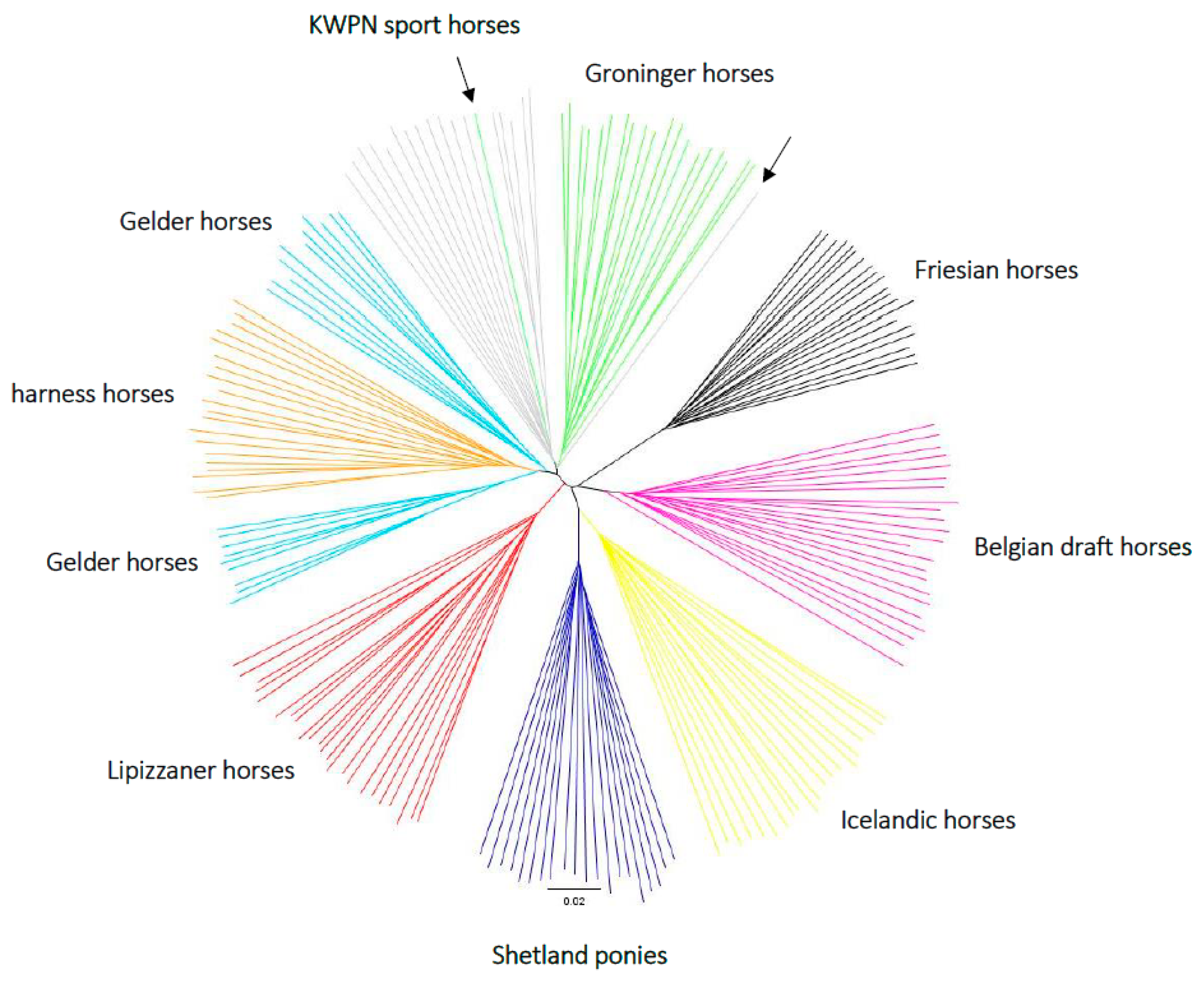
2.4.4. Phylogenetic Tree
3. Results
3.1. Parameters Within Populations
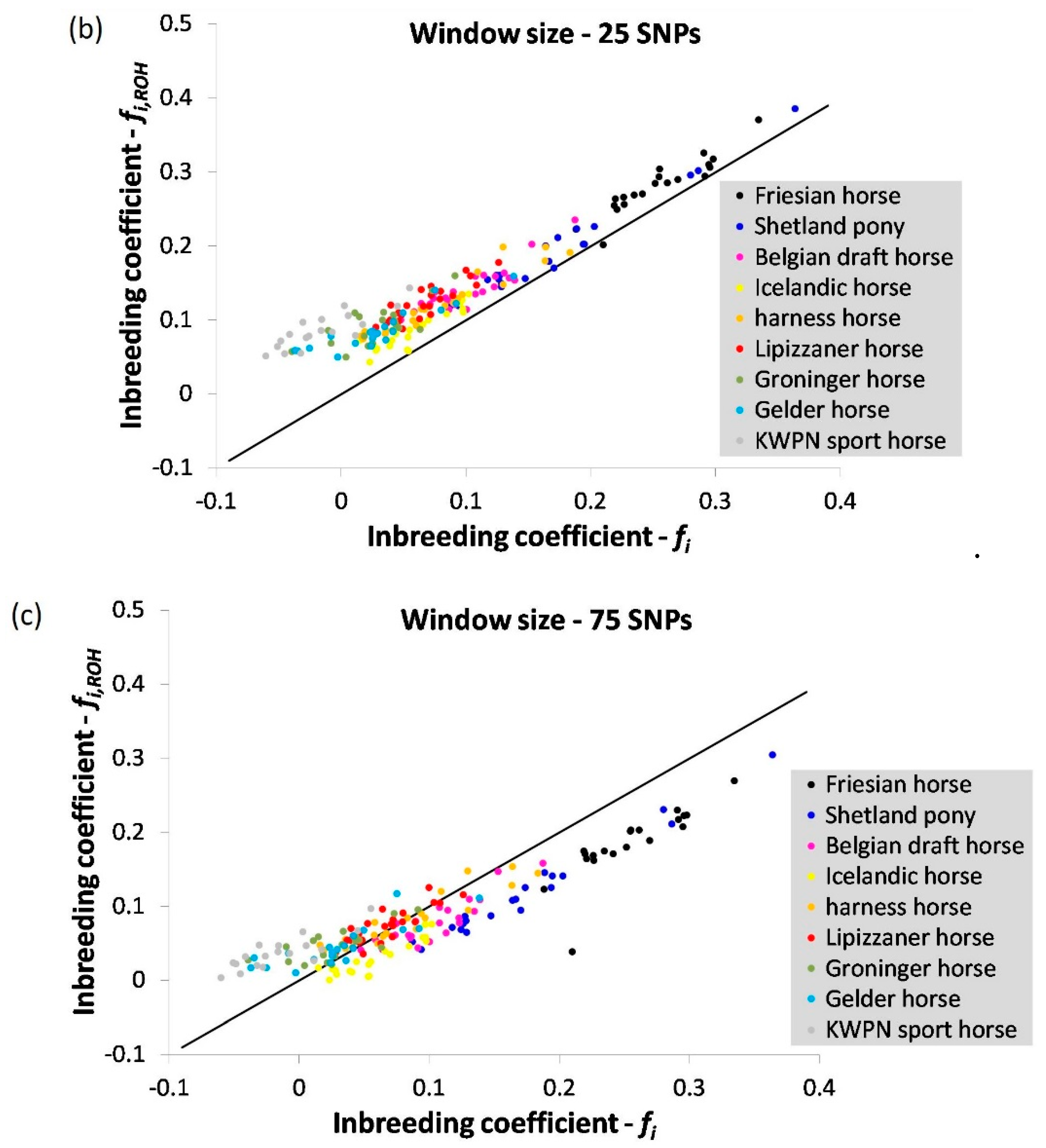
3.1.1. Inbreeding Coefficient
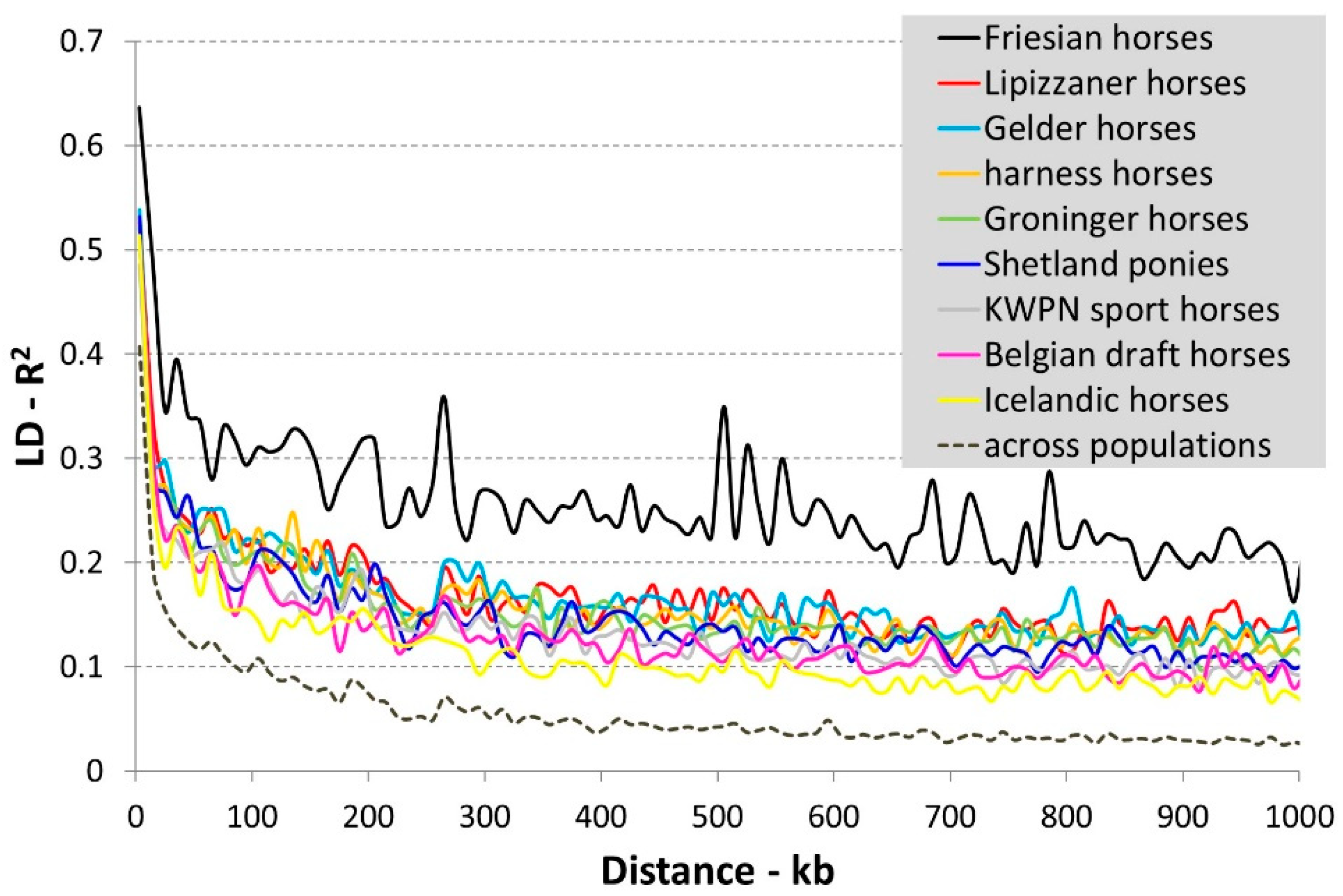
3.1.2. Linkage Disequilibrium
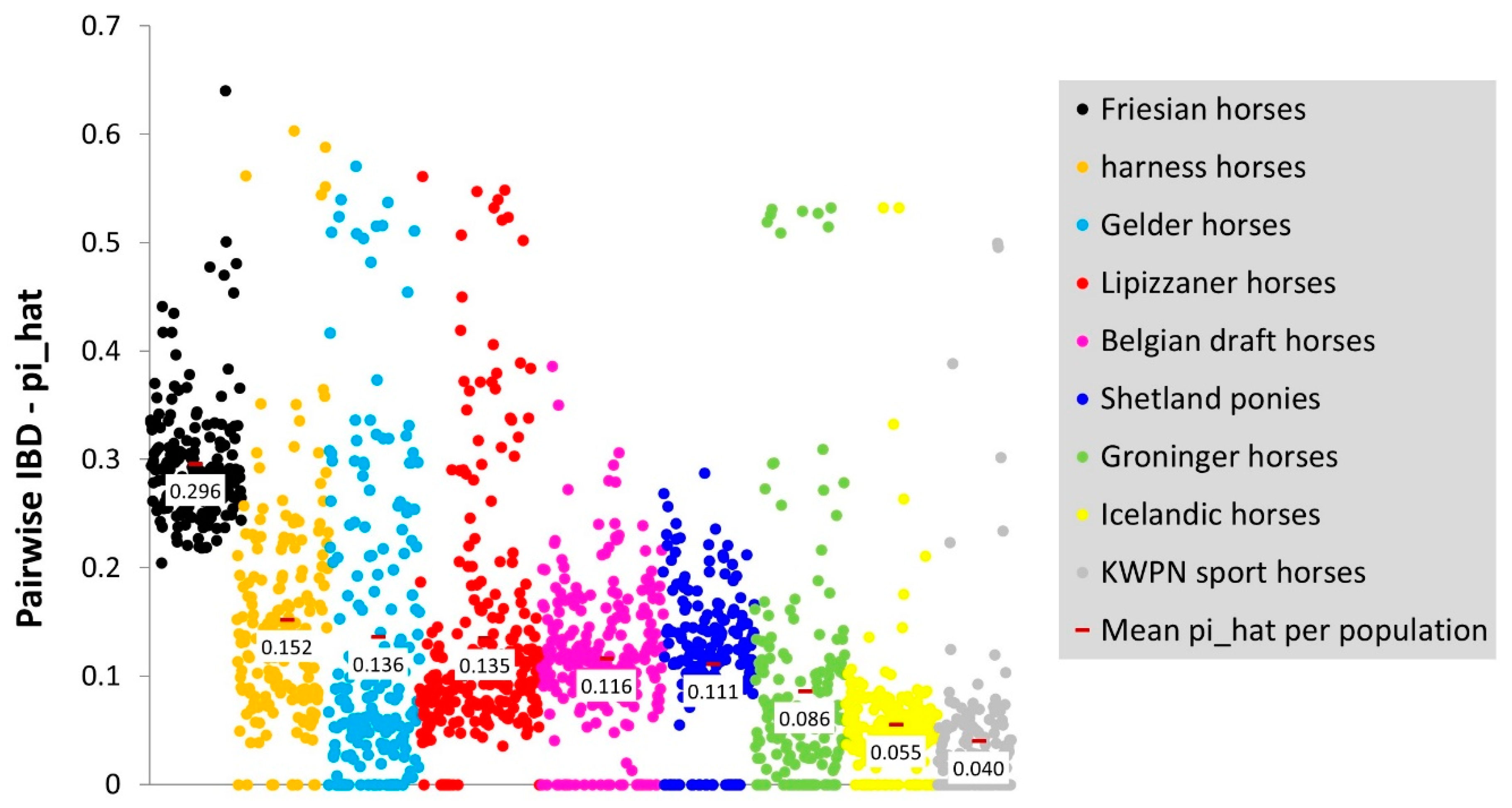
3.1.3. Pairwise IBD
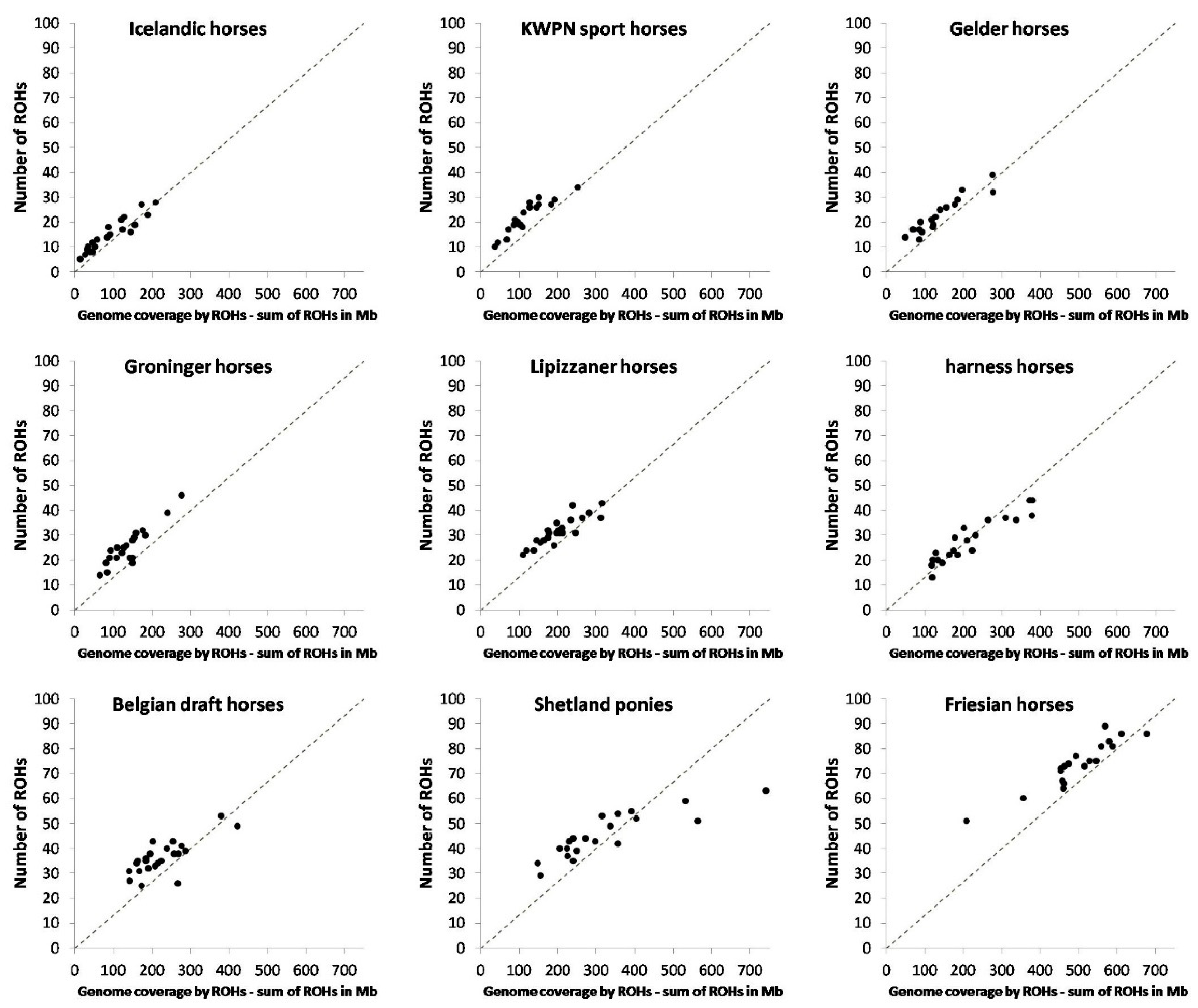
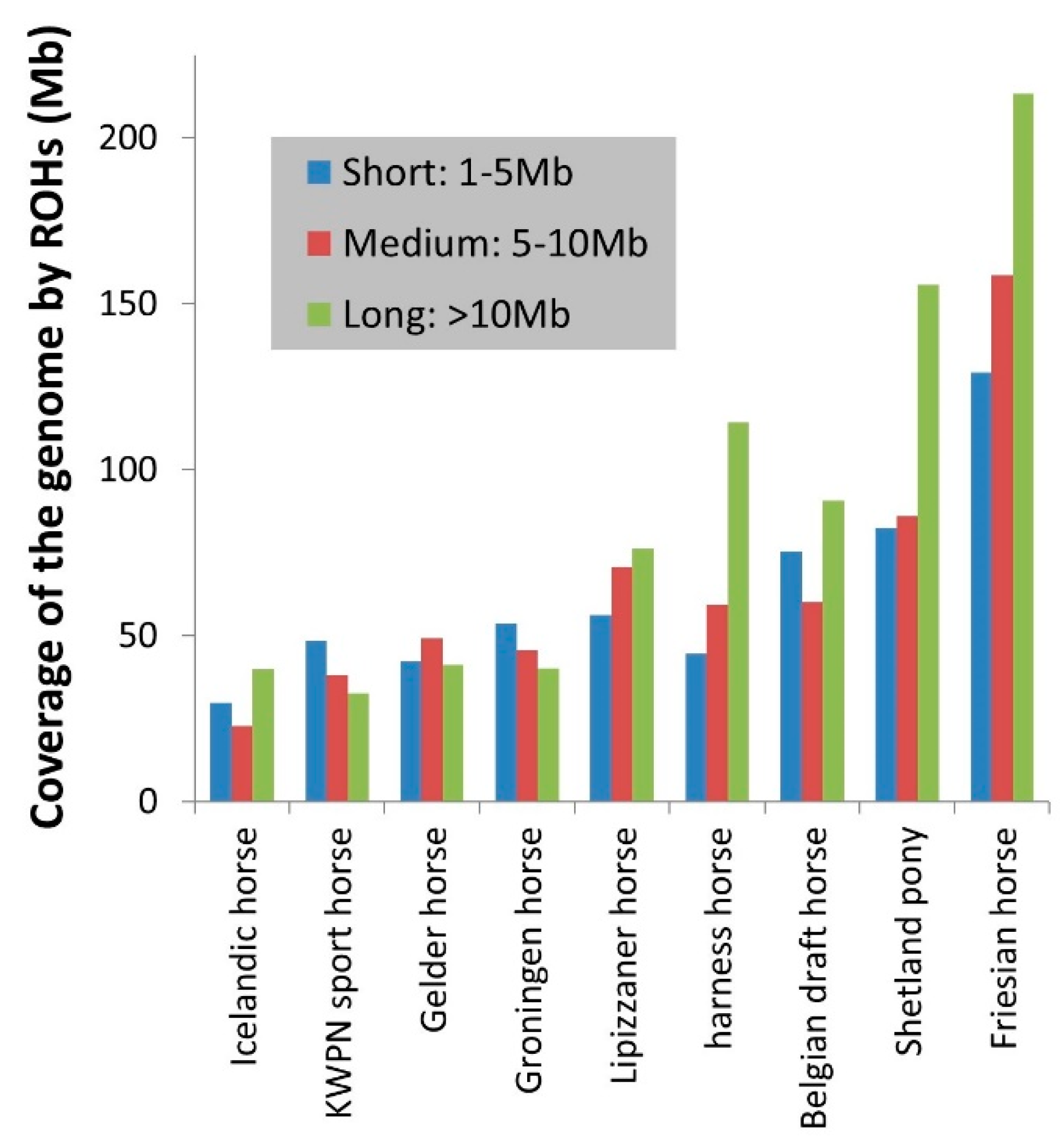
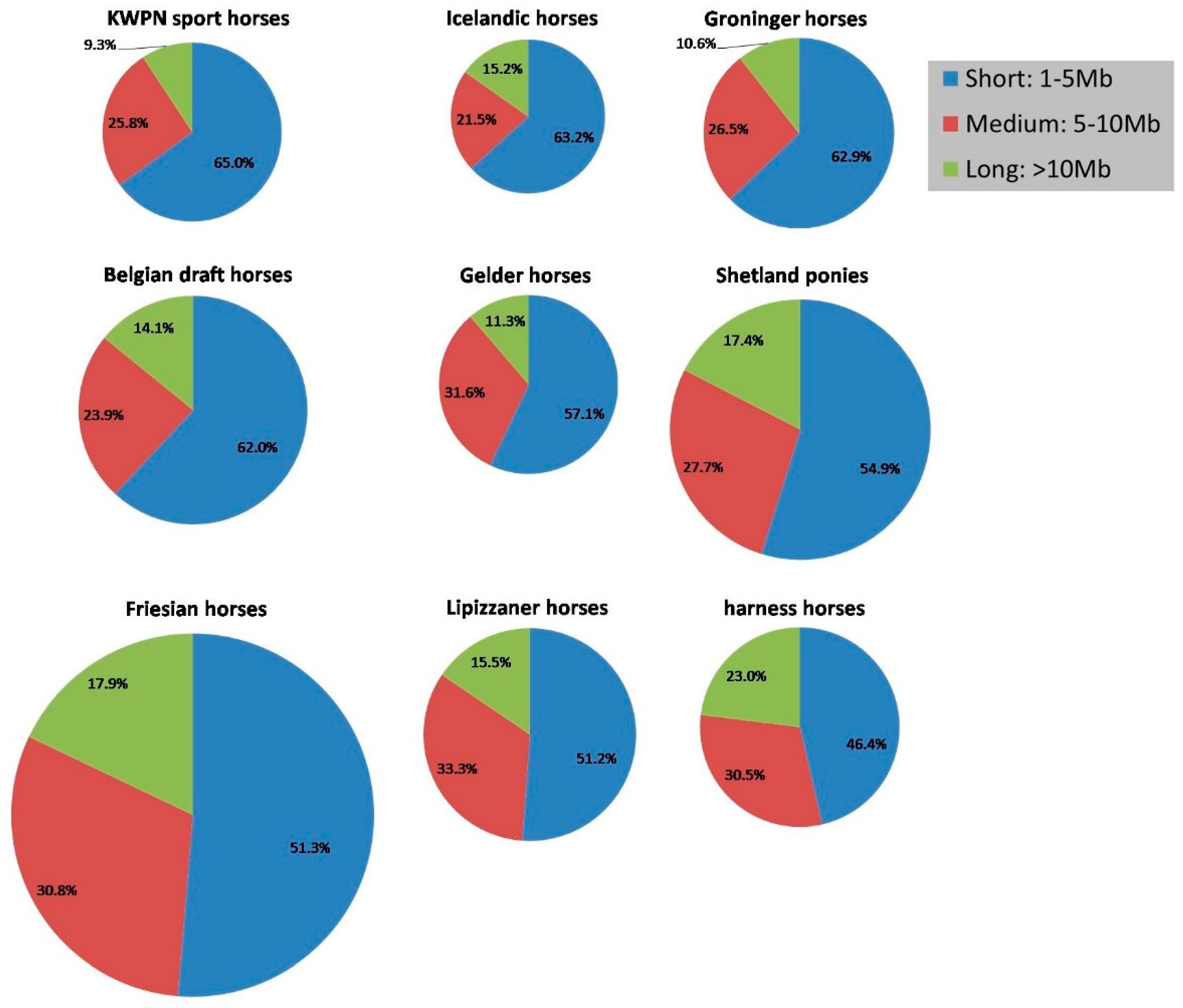
3.1.4. Runs of Homozygosity
3.2. Relationships Among Populations
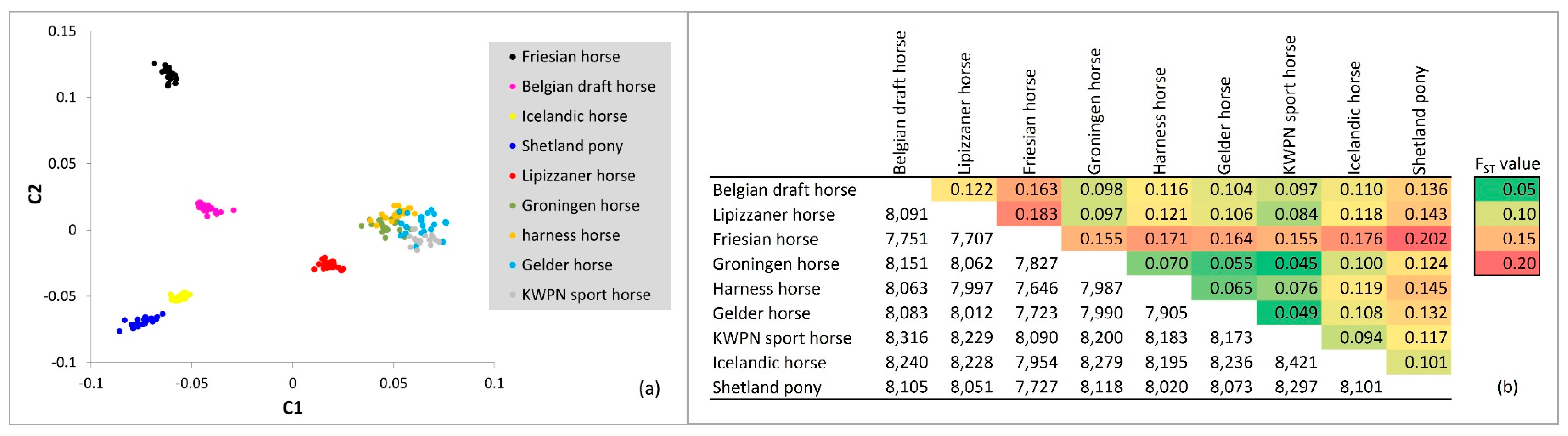
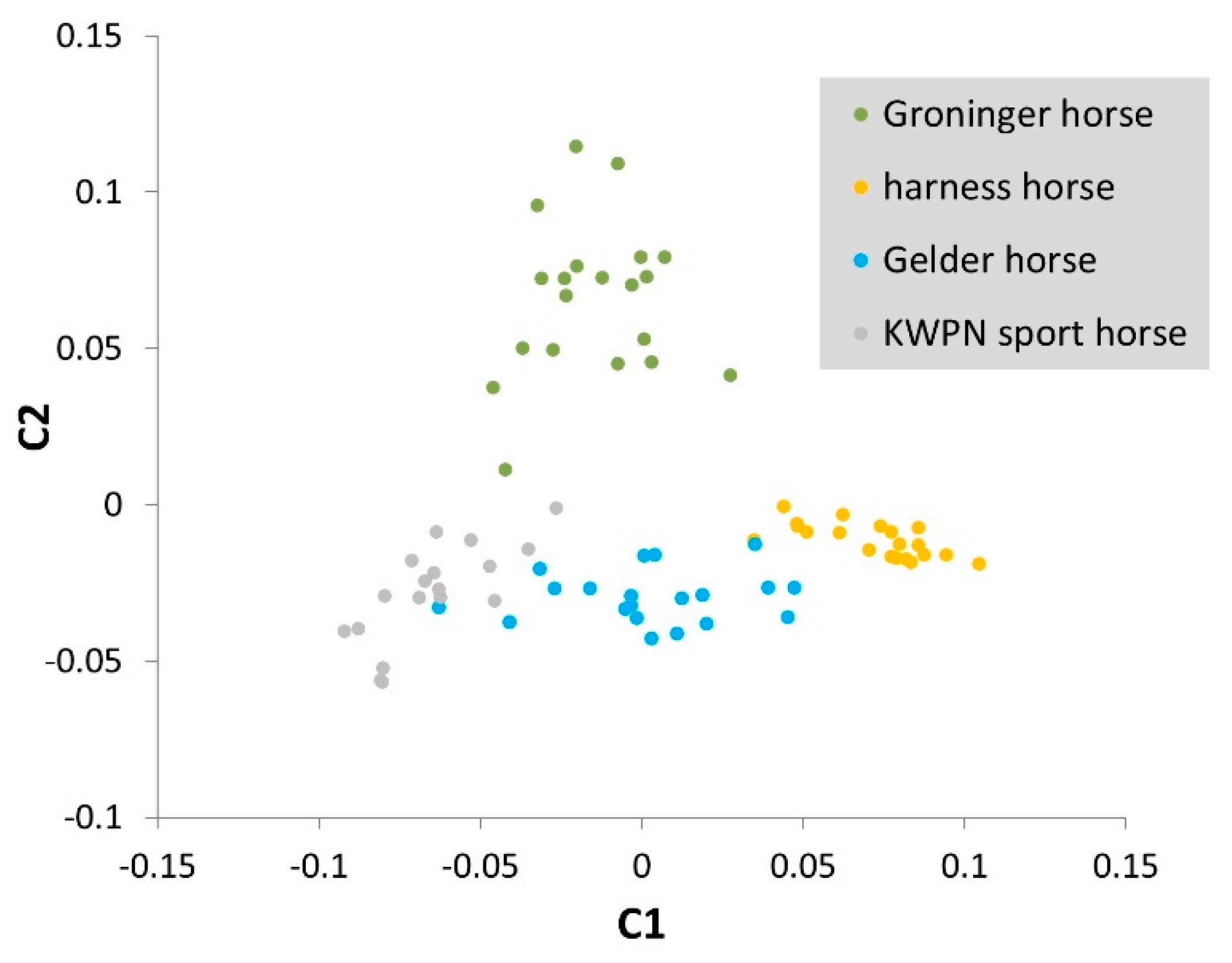
3.2.1. Multidimensional Scaling
3.2.2. FST Approach
3.2.3. Admixture Analysis
3.2.4. Phylogenetic Tree
4. Discussion
4.1. Population Specific Characteristics
4.1.1. Inbreeding
4.1.2. Runs of Homozygosity
4.1.3. Linkage Disequilibrium
4.2. Relationships Among Populations
4.3. Data Collection
5. Conclusions and Implications
Author Contributions
Funding
Conflicts of Interest
Appendix A



Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inbreeding Coefficient | ||||||||||||||||||||||||||||||
| Window Size 15 SNPs | Window Size 25 SNPs | Window Size 75 SNPs | ||||||||||||||||||||||||||||
| Population | Mean | SD | Min | Max | Mean | SD | Min | Max | Mean | SD | Min | Max | ||||||||||||||||||
| Belgian draft horse | 18.0 | 2.9 | 13.3 | 26.5 | 14.2 | 3.0 | 9.9 | 23.5 | 7.8 | 3.1 | 4.0 | 15.8 | ||||||||||||||||||
| Friesian horse | 31.9 | 3.5 | 25.9 | 40.7 | 28.1 | 3.7 | 20.1 | 37.0 | 18.4 | 4.7 | 3.8 | 26.9 | ||||||||||||||||||
| Gelder horse | 11.5 | 3.1 | 7.7 | 19.7 | 8.8 | 2.8 | 4.9 | 15.8 | 4.6 | 2.9 | 1.0 | 11.7 | ||||||||||||||||||
| Groninger horse | 12.0 | 2.7 | 7.5 | 18.8 | 9.2 | 2.6 | 4.9 | 15.9 | 4.8 | 2.1 | 1.9 | 9.6 | ||||||||||||||||||
| Harness horse | 15.5 | 4.0 | 10.6 | 22.6 | 12.6 | 4.1 | 7.4 | 19.9 | 8.2 | 3.7 | 4.0 | 15.3 | ||||||||||||||||||
| Icelandic horse | 12.9 | 2.7 | 9.1 | 17.8 | 8.5 | 2.7 | 4.3 | 13.5 | 3.0 | 2.4 | 0.0 | 7.6 | ||||||||||||||||||
| KWPN sport horse | 11.5 | 2.5 | 8.0 | 17.4 | 8.6 | 2.5 | 5.1 | 14.3 | 3.7 | 2.3 | 0.4 | 9.7 | ||||||||||||||||||
| Lipizzaner horse | 15.2 | 2.4 | 11.7 | 20.2 | 12.3 | 2.5 | 8.8 | 17.7 | 7.4 | 2.4 | 3.5 | 12.5 | ||||||||||||||||||
| Shetland pony | 23.5 | 6.5 | 15.6 | 41.7 | 19.5 | 6.7 | 11.7 | 38.5 | 11.8 | 6.5 | 4.2 | 30.4 | ||||||||||||||||||
| All 184 horses | 16.9 | 7.3 | 7.5 | 41.7 | 13.6 | 7.1 | 4.3 | 38.5 | 7.8 | 5.7 | 0.0 | 30.4 | ||||||||||||||||||
| Number of ROH | ||||||||||||||||||||||||||||||
| Window Size 15 SNPs | Window Size 25 SNPs | Window Size 75 SNPs | ||||||||||||||||||||||||||||
| Population | Total | Mean | SD | Min | Max | Total | Mean | SD | Min | Max | Total | Mean | SD | Min | Max | |||||||||||||||
| Belgian draft horse | 3401 | 147.9 | 12.0 | 130 | 169 | 2010 | 87.4 | 8.8 | 72 | 108 | 473 | 20.6 | 5.9 | 13 | 40 | |||||||||||||||
| Friesian horse | 3884 | 194.2 | 11.1 | 181 | 224 | 2833 | 141.7 | 11.3 | 124 | 165 | 957 | 48.9 | 10.0 | 17 | 65 | |||||||||||||||
| Gelder horse | 2071 | 103.6 | 14.0 | 81 | 143 | 1170 | 58.5 | 9.0 | 40 | 82 | 268 | 13.4 | 7.8 | 4 | 33 | |||||||||||||||
| Groninger horse | 2160 | 108.0 | 13.9 | 78 | 138 | 1261 | 63.1 | 12.4 | 38 | 92 | 298 | 14.9 | 5.6 | 7 | 28 | |||||||||||||||
| Harness horse | 2212 | 110.6 | 8.6 | 84 | 118 | 1290 | 64.5 | 10.7 | 43 | 77 | 370 | 18.5 | 6.8 | 9 | 32 | |||||||||||||||
| Icelandic horse | 2958 | 147.9 | 8.6 | 129 | 169 | 1506 | 75.3 | 8.8 | 57 | 94 | 142 | 7.1 | 4.6 | 0 | 16 | |||||||||||||||
| KWPN sport horse | 1979 | 109.9 | 11.3 | 83 | 130 | 1160 | 64.4 | 9.1 | 43 | 79 | 206 | 11.4 | 5.1 | 2 | 23 | |||||||||||||||
| Lipizzaner horse | 2725 | 118.5 | 8.1 | 105 | 134 | 1672 | 72.7 | 7.9 | 55 | 89 | 480 | 20.9 | 5.4 | 11 | 30 | |||||||||||||||
| Shetland pony | 3417 | 170.9 | 12.1 | 151 | 204 | 2184 | 109.2 | 11.6 | 94 | 136 | 551 | 27.6 | 8.1 | 16 | 45 | |||||||||||||||
| All 184 horses | 24,807 | 134.8 | 32.1 | 78 | 224 | 15,086 | 82.0 | 27.4 | 38 | 165 | 3745 | 20.4 | 13.0 | 0 | 65 | |||||||||||||||
| Length of ROH, Mb | ||||||||||||||||||||||||||||||
| Window Size 15 SNPs | Window Size 25 SNPs | Window Size 75 SNPs | ||||||||||||||||||||||||||||
| Population | Mean | SD | Min | Max | Mean | SD | Min | Max | Mean | SD | Min | Max | ||||||||||||||||||
| Belgian draft horse | 2.7 | 3.5 | 1.0 | 40.2 | 3.6 | 4.3 | 1.0 | 40.1 | 8.5 | 6.2 | 3.1 | 39.8 | ||||||||||||||||||
| Friesian horse | 3.7 | 4.3 | 1.0 | 47.7 | 4.5 | 4.7 | 1.0 | 47.6 | 8.6 | 5.7 | 3.0 | 46.1 | ||||||||||||||||||
| Gelder horse | 2.5 | 3.1 | 1.0 | 55.8 | 3.4 | 3.8 | 1.0 | 55.7 | 7.8 | 5.8 | 2.9 | 55.3 | ||||||||||||||||||
| Groninger horse | 2.5 | 2.7 | 1.0 | 33.2 | 3.3 | 3.2 | 1.0 | 33.2 | 7.2 | 4.3 | 3.1 | 33.1 | ||||||||||||||||||
| Harness horse | 3.1 | 4.5 | 1.0 | 52.9 | 4.4 | 5.4 | 1.0 | 52.8 | 10.0 | 7.3 | 3.1 | 52.7 | ||||||||||||||||||
| Icelandic horse | 2.0 | 2.5 | 1.0 | 56.8 | 2.5 | 3.3 | 1.0 | 56.6 | 9.4 | 7.3 | 3.3 | 56.3 | ||||||||||||||||||
| KWPN sport horse | 2.3 | 2.6 | 1.0 | 32.9 | 3.0 | 3.1 | 1.0 | 32.9 | 7.3 | 4.9 | 3.3 | 32.8 | ||||||||||||||||||
| Lipizzaner horse | 2.9 | 3.5 | 1.0 | 48.4 | 3.8 | 4.1 | 1.0 | 48.3 | 7.9 | 5.3 | 3.2 | 48.1 | ||||||||||||||||||
| Shetland pony | 3.1 | 4.8 | 1.0 | 92.0 | 4.0 | 5.7 | 1.0 | 91.8 | 9.6 | 8.9 | 3.0 | 91.3 | ||||||||||||||||||
| All 184 horses | 2.8 | 3.7 | 1.0 | 92.0 | 3.7 | 4.5 | 1.0 | 91.8 | 8.6 | 6.4 | 2.9 | 91.3 | ||||||||||||||||||
Appendix C

Appendix D

Appendix E

Appendix F

Appendix G

References
- McCue, M.E.; Bannasch, D.L.; Petersen, J.L.; Gurr, J.; Bailey, E.; Binns, M.M.; Distl, O.; Guérin, G.; Hasegawa, T.; Hill, E.W.; et al. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012, 8, e1002451. [Google Scholar] [CrossRef]
- Librado, P.; Fages, A.; Gaunitz, C.; Leonardi, M.; Wagner, S.; Khan, N.; Hanghøj, K.; Alquraishi, S.A.; Alfarhan, A.H.; Al-Rasheid, K.A.; et al. The evolutionary origin and genetic makeup of domestic horses. Genetics 2016, 204, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Cothran, E.G.; Luis, C. Genetic distance as a tool in the conservation of rare horse breeds. In Conservation Genetics of Endangered Horse Breeds, 1st ed.; Bodo, I., Alderson, L., Langlois, B., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2005; pp. 55–73. [Google Scholar]
- Hendricks, B.L. International Encyclopedia of Horse Breeds, 1st ed.; University of Oklahoma Press: Norman, OK, USA, 2007. [Google Scholar]
- The Shetland Pony Stud-Book Society—Breed History. Available online: http://www.shetlandponystudbooksociety.co.uk/about-the-breed (accessed on 18 July 2018).
- Bowling, A.T.; Ruvinsky, A. The Genetics of the Horse, 1st ed.; CABI Publishing: Wallingford, Oxon, UK, 2000. [Google Scholar]
- KWPN|Royal Dutch Sport Horse—History. Available online: https://www.kwpn.org/kwpn-horse/kwpn-studbook/history (accessed on 3 June 2019).
- Ducro, B.J. Relevance of Test Information in Horse Breeding. Ph.D. Thesis, Wageningen University & Research, Wageningen, The Netherlands, 2011. Available online: http://edepot.wur.nl/168395 (accessed on 5 June 2019).
- Schurink, A.; Arts, D.J.G.; Ducro, B.J. Genetic diversity in the Dutch Harness horse population using pedigree analysis. Livest. Sci. 2012, 143, 270–277. [Google Scholar] [CrossRef]
- Ducro, B.J.; Windig, J.; Hellinga, I.; Bovenhuis, H. Genetic diversity and measures to reduce inbreeding in Friesian horses. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- Van de Goor, L.H.P.; van Haeringen, W.A.; Lenstra, J.A. Population studies of 17 equine STR for forensic and phylogenetic analysis. Anim. Genet. 2011, 42, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Centre for Genetic Resources, the Netherlands—Species and Breed Information. Available online: https://www.wur.nl/upload_mm/1/2/8/84a41236-9591-485b-9d8e-0847ad501646_Status%20NL%20landbouwhuisdierrassen_apr2017.pdf (accessed on 5 June 2019).
- Peerlings, J.; van der Weerden, T.; van Hoof, W. Het Trekpaard; Roodbont|Agricultural Publishers: Zutphen, The Netherlands, 2007; Volume 1. [Google Scholar]
- European Farm Animal Biodiversity Information System (EFABIS)—Breeds by Species and Country. Available online: https://dadis-breed-4eff5.firebaseapp.com/?country=Netherlands&specie=Horse&breed=&callback=allbreeds (accessed on 5 June 2019).
- Purcell, S. PLINK. 2007. Available online: http://zzz.bwh.harvard.edu/plink/ (accessed on 24 June 2019).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Lopes, M.S.; Silva, F.F.; Harlizius, B.; Duijvesteijn, N.; Lopes, P.S.; Guimaraes, S.E.F.; Knol, E.F. Improved estimation of inbreeding and kinship in pigs using optimized SNP panels. BMC Genet. 2013, 14, 92. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.6; Department of Genomice Sciences, University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Wright, S. Evolution and the Genetics of Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 4th ed.; Sinauer Associates: Sunderland, UK, 2007; p. 283. [Google Scholar]
- Meuwissen, T.H.E. Operation of conservation schemes. In Utilisation and Conservation of Farm Animal Genetic Resources, 1st ed.; Oldenbroek, J.K., Ed.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2007; pp. 167–193. [Google Scholar]
- Petersen, J.L.; Mickelson, J.R.; Cothran, E.G.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; Brama, P.; et al. Genetic diversity in the modern horse illustrated from genome-wide SNP data. PLoS ONE 2013, 8, e54997. [Google Scholar] [CrossRef]
- Geurts, R.H.J.J. Genetische Analyse en Structuur van de Fokkerij van Het Friese Paard. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 1969. [Google Scholar]
- Osinga, A. Het Fokken van Het Friese Paard; Schaafsma & Brouwer Grafische bedrijven BV: Dokkum, The Netherlands, 2000. [Google Scholar]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Druml, T.; Neuditschko, M.; Grilz-Seger, G.; Horna, M.; Ricard, A.; Mesarič, M.; Cotman, M.; Pausch, H.; Brem, G. Population networks associated with runs of homozygosity reveal new insights into the breeding history of the Haflinger horse. J. Hered. 2018, 109, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Quinto-Cortés, C.D.; Woerner, A.E.; Watkins, J.C.; Hammer, M.F. Modelling SNP array ascertainment with Approximate Bayesian Computation for demographic inference. Sci. Rep. 2018, 8, 10209. [Google Scholar] [CrossRef]
- Bortoluzzi, C.; Crooijmans, R.P.M.A.; Bosse, M.; Hiemstra, S.J.; Groenen, M.A.M.; Megens, H.J. The effects of recent changes in breeding preferences on maintaining traditional Dutch chicken genomic diversity. Hereditary 2018, 121, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Consortium, B.H.P.; Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M.; et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Board Institute Genome Sequencing Platform; Board Institute Whole Genome Assembly Team; Lander, E.S.; Lindblad-Toh, K. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef]
- Wang, W.Y.S.; Barratt, B.J.; Clayton, D.G.; Todd, J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005, 6, 109–118. [Google Scholar] [CrossRef]
- Juras, R.; Cothran, E.G.; Klimas, R. Genetic analysis of three Lithuanian native horse breeds. Acta Agric. Scand. A 2003, 53, 180–185. [Google Scholar] [CrossRef]
- KWPN|Royal Dutch Sport Horse—Historie. Available online: https://www.kwpn.nl/kwpn-paard/het-stamboek/historie (accessed on 5 June 2019). (In Dutch).
- KWPN|Royal Dutch Sport Horse—Breeding. Available online: https://www.kwpn.org/kwpn-horse/selection--and-breedingprogram/breeding (accessed on 5 June 2019).
- Het Groninger Paard—History. Available online: https://zeldzamerassen.nl/het-groninger-paard/history/ (accessed on 5 June 2019).
- Het Groninger Paard. Available online: https://zeldzamerassen.nl/het-groninger-paard/het-groninger-paard/ (accessed on 5 June 2019).
- Pruett, C.L.; Winker, K. The effects of sample size on population genetic diversity estimates in song sparrows Melospiza melodia. J. Avian Biol. 2008, 39, 252–256. [Google Scholar] [CrossRef]
- Sánchez-Montes, G.; Ariño, A.H.; Vizmanos, J.L.; Wang, J.; Martínez-Solano, Í. Effects of sample size and full sibs on genetic diversity characterization: A case study of three syntopic Iberian pond-breeding amphibians. J. Hered. 2017, 108, 535–543. [Google Scholar] [CrossRef]
- Leegwater, P.A.; Vos-Loohuis, M.; Ducro, B.J.; Boegheim, I.J.; van Steenbeek, F.G.; Nijman, I.J.; Monroe, G.R.; Bastiaansen, J.W.M.; Dibbits, B.W.; van de Goor, L.H.; et al. Dwarfism with joint laxity in Friesian horses is associated with a splice site mutation in B4GALT7. BMC Genom. 2016, 17, 839. [Google Scholar] [CrossRef] [PubMed]
- Ducro, B.J.; Schurink, A.; Bastiaansen, J.W.M.; Boegheim, I.J.M.; van Steenbeek, F.G.; Vos-Loohuis, M.; Nijman, I.J.; Monroe, G.R.; Hellinga, I.; Dibbits, B.W.; et al. A nonsense mutation in B3GALNT2 is concordant with hydrocephalus in Friesian horses. BMC Genom. 2015, 16, 761. [Google Scholar] [CrossRef] [PubMed]
- Druml, T.; Curik, I.; Baumung, R.; Aberle, K.; Distl, O.; Sölkner, J. Individual-based assessment of population structure and admixture in Austrian, Croatian and German draught horses. Heredity 2007, 98, 114–122. [Google Scholar] [CrossRef]






| Population | N | Country of origin | Type | Breeding | Description |
|---|---|---|---|---|---|
| Belgian draft horse | 23 | Belgium | Coldblood | Closed | Heavy draft horse |
| Friesian horse | 20 | The Netherlands | Coldblood | Closed | Harness and riding horse |
| Gelder horse | 20 | The Netherlands | Warmblood | Open, gene-flow from warmblood horses | Light draft and riding horse |
| Groninger horse | 20 | The Netherlands | Warmblood | Open | Heavy draft and riding horse |
| Harness horse | 20 | The Netherlands | Warmblood | Open, gene-flow from Hackney and Saddlebred horses | Harness horse |
| Icelandic horse | 20 | Iceland | Pony | Closed | Gaited riding horse |
| KWPN sport horse | 18 | The Netherlands | Warmblood | Open, gene-flow from Thoroughbred and warmblood sport horses | Sport horse, jumping or dressage |
| Lipizzaner horse | 23 | Lipica, modern-day Slovenia | Warmblood | Closed, use of several sire and dam lines | Riding horse, Spanish riding school |
| Shetland pony | 20 | Shetland Isles, Scotland | Pony | Closed, four categories based on withers height | Harness and riding pony |
| Runs of Homozygosity (ROHs) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inbreeding Coefficient | Inbreeding Coefficient | Number of ROH | Length of ROH, Mb | ||||||||||||||
| Population | Mean | SD | Min | Max | Mean | SD | Min | Max | Total | Mean | SD | Min | Max | Mean | SD | Min | Max |
| Belgian draft horse | 10.3 | 3.3 | 4.8 | 18.8 | 10.1 | 3.1 | 6.3 | 18.8 | 836 | 36.3 | 6.8 | 25 | 53 | 6.2 | 5.5 | 1.6 | 39.9 |
| Friesian horse | 25.5 | 3.7 | 18.9 | 33.5 | 22.3 | 4.5 | 9.3 | 30.2 | 1485 | 74.3 | 9.5 | 51 | 89 | 6.7 | 5.4 | 1.7 | 47.5 |
| Gelder horse | 3.1 | 4.3 | -3.7 | 13.9 | 5.9 | 2.8 | 2.1 | 12.3 | 443 | 22.2 | 7.0 | 13 | 39 | 6.0 | 5.1 | 1.9 | 55.4 |
| Groninger horse | 2.9 | 3.0 | -3.9 | 9.2 | 6.2 | 2.3 | 2.9 | 12.3 | 509 | 25.5 | 7.7 | 14 | 46 | 5.5 | 4.0 | 1.7 | 33.1 |
| Harness horse | 8.4 | 5.0 | 1.6 | 18.4 | 9.7 | 4.1 | 5.2 | 16.9 | 560 | 28.0 | 8.9 | 13 | 44 | 7.8 | 6.8 | 1.7 | 52.8 |
| Icelandic horse | 5.9 | 2.9 | 1.5 | 10.2 | 4.1 | 2.7 | 0.6 | 9.4 | 302 | 15.1 | 6.7 | 5 | 28 | 6.1 | 6.0 | 1.7 | 56.4 |
| KWPN sport horse | -1.5 | 3.2 | -6.0 | 5.5 | 5.3 | 2.4 | 1.6 | 11.3 | 400 | 22.2 | 6.7 | 10 | 34 | 5.4 | 4.2 | 1.9 | 32.8 |
| Lipizzaner horse | 6.8 | 2.5 | 2.8 | 12.7 | 9.0 | 2.5 | 4.9 | 14.1 | 729 | 31.7 | 5.6 | 22 | 43 | 6.4 | 4.9 | 1.9 | 48.2 |
| Shetland pony | 17.4 | 6.9 | 8.7 | 36.4 | 14.4 | 6.6 | 6.6 | 33.1 | 906 | 45.3 | 8.9 | 29 | 63 | 7.2 | 7.7 | 1.9 | 91.6 |
| All 184 horses | 8.8 | 8.6 | -6.0 | 36.4 | 9.7 | 6.4 | 0.6 | 33.1 | 6170 | 33.5 | 18.1 | 5 | 89 | 6.5 | 5.8 | 1.6 | 91.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schurink, A.; Shrestha, M.; Eriksson, S.; Bosse, M.; Bovenhuis, H.; Back, W.; Johansson, A.M.; Ducro, B.J. The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands. Genes 2019, 10, 480. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10060480
Schurink A, Shrestha M, Eriksson S, Bosse M, Bovenhuis H, Back W, Johansson AM, Ducro BJ. The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands. Genes. 2019; 10(6):480. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10060480
Chicago/Turabian StyleSchurink, Anouk, Merina Shrestha, Susanne Eriksson, Mirte Bosse, Henk Bovenhuis, Willem Back, Anna M. Johansson, and Bart J. Ducro. 2019. "The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands" Genes 10, no. 6: 480. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10060480