
Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A Case Series Study of 155 Patients

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Genomic DNA Preparation
2.2. Libraries Establishment
2.3. Sequencing and Data Analysis
2.4. Gene Ranking
2.5. Statistical Analysis
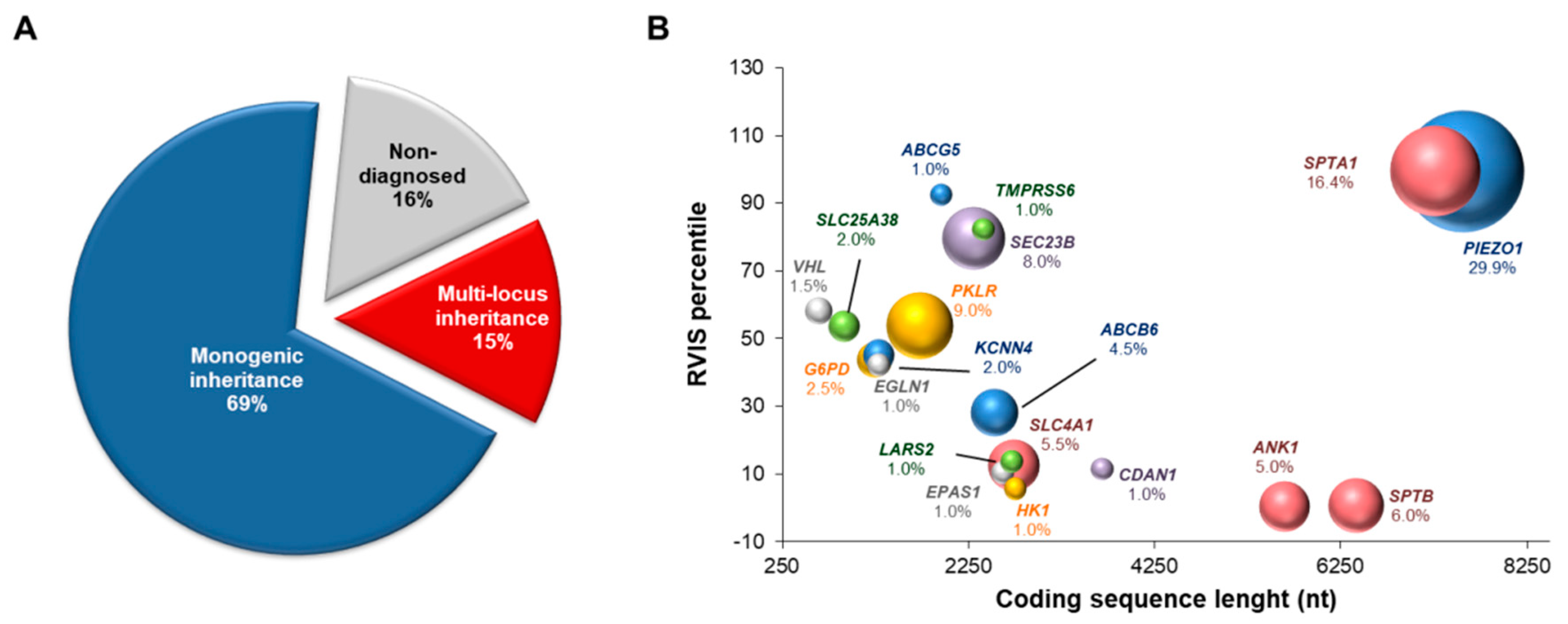
3. Results
3.1. NGS-Based Genetic Testing for Identification of Multiple Disease-Causing Genotypes
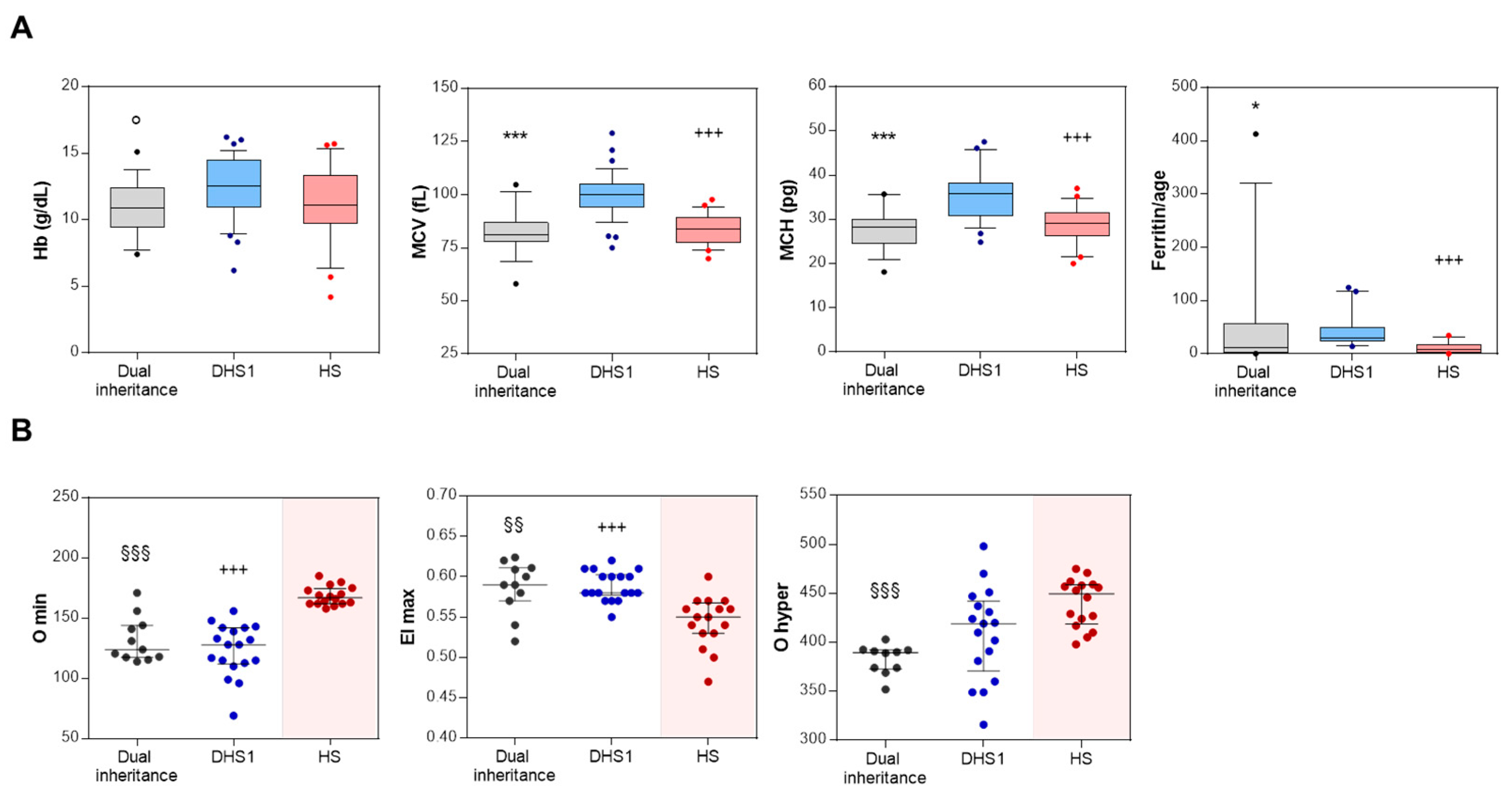
3.2. Blood Count, Hemolytic Markers, and Iron Balance of Dual Inheritance Patients
3.3. Hydration and Deformability Status of Dual Inheritance Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Advances in understanding the pathogenesis of red cell membrane disorders. Br. J. Haematol. 2019, 187, 13–24. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. Hereditary stomatocytosis: An underdiagnosed condition. Am. J. Hematol. 2018, 93, 107–121. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. New insights on hereditary erythrocyte membrane defects. Haematologica 2016, 101, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Bianchi, P.; Fermo, E.; Lezon-Geyda, K.; van Beers, E.J.; Morton, H.D.; Barcellini, W.; Glader, B.; Chonat, S.; Ravindranath, Y.; Newburger, P.E.; et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am. J. Hematol. 2020, 95, 472–482. [Google Scholar] [CrossRef]
- Roy, N.B.A.; Babbs, C. The pathogenesis, diagnosis and management of congenital dyserythropoietic anaemia type I. Br. J. Haematol. 2019, 185, 436–449. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.; Marra, R.; Rosato, B.E.; Iolascon, A.; Andolfo, I. Genetics and Genomics Approaches for Diagnosis and Research Into Hereditary Anemias. Front. Physiol. 2020, 11, 613559. [Google Scholar] [CrossRef]
- Posey, J.E. Genome sequencing and implications for rare disorders. Orphanet J. Rare Dis. 2019, 14, 153. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.; Langella, C.; Esposito, M.R.; Gambale, A.; Vitiello, F.; Vallefuoco, F.; Ek, T.; Yang, E.; Iolascon, A. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol. Dis. 2013, 51, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andolfo, I.; Martone, S.; Ribersani, M.; Bianchi, S.; Manna, F.; Genesio, R.; Gambale, A.; Pignataro, P.; Testi, A.M.; Iolascon, A.; et al. Apparent recessive inheritance of sideroblastic anemia type 2 due to uniparental isodisomy at the SLC25A38 locus. Haematologica 2020, 105, 2883–2886. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Manna, F.; Gambale, A.; Marra, R.; Rosato, B.E.; Caforio, P.; Pinto, V.; Pignataro, P.; Radhakrishnan, K.; et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol. 2018, 93, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Rosato, B.E.; Manna, F.; Gambale, A.; Brugnara, C.; Iolascon, A. Genotype-phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am. J. Hematol. 2018, 93, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Huisjes, R.; Makhro, A.; Llaudet-Planas, E.; Hertz, L.; Petkova-Kirova, P.; Verhagen, L.P.; Pignatelli, S.; Rab, M.A.; Schiffelers, R.M.; Seiler, E.; et al. Density, heterogeneity and deformability of red cells as markers of clinical severity in hereditary spherocytosis. Haematologica 2020, 105, 338–347. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef]
- Barreda Fierro, R.; Herrera Mora, P.; Zenteno, J.C.; Villarroel Cortes, C.E. Clinical and molecular evidence of possible digenic inheritance for MFN2/GDAP1 genes in Charcot-Marie-Tooth disease. Neuromuscul. Disord. 2020, 30, 986–990. [Google Scholar] [CrossRef]
- Durkie, M.; Chong, J.; Valluru, M.K.; Harris, P.C.; Ong, A.C.M. Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet. Med. 2021, 23, 689–697. [Google Scholar] [CrossRef]
- Neocleous, V.; Fanis, P.; Toumba, M.; Tanteles, G.A.; Schiza, M.; Cinarli, F.; Nicolaides, N.C.; Oulas, A.; Spyrou, G.M.; Mantzoros, C.S.; et al. GnRH Deficient Patients with Congenital Hypogonadotropic Hypogonadism: Novel Genetic Findings in ANOS1, RNF216, WDR11, FGFR1, CHD7, and POLR3A Genes in a Case Series and Review of the Literature. Front. Endocrinol. 2020, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Al-Riyami, A.Z.; Iolascon, A.; Al-Zadjali, S.; Andolfo, I.; Al-Mammari, S.; Manna, F.; Al Rawas, A.; King, M.J.; Russo, R. Targeted next generation sequencing identifies a novel β-spectrin gene mutation A2059P in two Omani children with hereditary pyropoikilocytosis. Am. J. Hematol. 2017, 92, E607–E609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Shyr, C.; Ross, C.J.; Horvath, G.A.; Salvarinova, R.; Ye, X.C.; Zhang, L.H.; Bhavsar, A.P.; Lee, J.J.; Drögemöller, B.I.; et al. Exome Sequencing and the Management of Neurometabolic Disorders. N. Engl. J. Med. 2016, 374, 2246–2255. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.; Posey, J.E.; Akdemir, Z.C.; Pehlivan, D.; Harel, T.; Jhangiani, S.N.; Bayram, Y.; Song, X.; Bahrambeigi, V.; Yuregir, O.O.; et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 2018, 20, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.G.; Murray, M.L.; Bailey, S.; Leistritz-Kessler, D.; Schwarze, U.; Byers, P.H. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet. Med. 2016, 18, 20–24. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Gene | HGVS Nomenclature | ACMG Rules † | Method | Class | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA-level | Protein-level | PVS1 | PS1 | PS2 | PS3 | PS4 | PS5 | PM1 | PM2 | PM3 | PM4 | PM5 | PM6 | PM7 | PP1 | PP2 | PP3 | PP4 | PP5 | PP6 | BA1 | BS1 | BS2 | BS3 | BS4 | BS5 | BP1 | BP2 | BP3 | BP4 | BP5 | BP6 | BP7 | BP8 | |||
| ABCB6 | c.1361T>C | p.Val454Ala | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| ABCB6 | c.1402G>T | p.Ala468Ser | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| ABCB6 | c.1474G>A | p.Ala492Thr | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| ABCB6 | c.1691T>C | p.Met564Thr | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| ABCB6 | c.1762G>A | p.Gly588Ser | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| ABCB6 | c.2215C>T | p.Arg739Cys | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| ANK1 | c.613-1G>C | - | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| ANK1 | c.1540G>T | p.Gly514Cys | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| G6PD | c.1360C>T | p.Arg454Cys | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| KCNN4 | c.983A>G | p.His328Arg | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| KCNN4 | c.1018C>A | p.His340Asn | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| LARS2 | c.457A>C | p.Asn153His | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.608T>C | p.Leu203Pro | Automated | LB | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.1001C>T | p.Ala334Val | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.1447G>A | p.Val483Met | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.1813A>G | p.Met605Val | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.3935C>T | p.Ala1312Val | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.4481A>C | p.Glu1494Ala | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.5195C>T | p.Thr1732Met | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.5835C>G | p.Phe1945Leu | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.5981C>G | p.Ser1994Cys | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.6205G>A | p.Val2069Met | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.6796G>A | p.Val2266Ile | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | P | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.7180G>A | p.Gly2394Ser | Automated | LB | |||||||||||||||||||||||||||||||||
| Adjusted | P | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.7219G>C | p.Glu2407Gln | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | P | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.7367G>A | p.Arg2456His | Automated | LP | |||||||||||||||||||||||||||||||||
| Adjusted | P | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.7529C>T | p.Pro2510Leu | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PIEZO1 | c.7558A>G | p.Lys2520Glu | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| PKLR | c.1675C>T | p.Arg559* | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| SEC23B | c.1233+4C>T | - | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| SLC4A1 | c.1462G>A | p.Val488Met | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| SLC4A1 | c.2608C>T | p.Arg870Trp | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| SLC4A1 | c.2621T>C | p.Leu874Pro | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.460_462dupTTG | p.Leu155dup | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | P | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.1958A>G | p.Tyr653Cys | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.2173C>T | p.Arg725* | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.2464+1G>A | - | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.4708G>A | p.Ala1570Thr | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.5029G>A | p.Gly1677Arg | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| SPTA1 | c.5183G>A | p.Trp1728* | Automated | P | |||||||||||||||||||||||||||||||||
| - | - | ||||||||||||||||||||||||||||||||||||
| SPTB | c.40C>T | p.Pro14Ser | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| SPTB | c.871G>A | p.Gly291Ser | Automated | V | |||||||||||||||||||||||||||||||||
| Adjusted | LP | ||||||||||||||||||||||||||||||||||||
| SPTB | c.1606G>A | p.Asp536Asn | Automated | B | |||||||||||||||||||||||||||||||||
| Adjusted | V | ||||||||||||||||||||||||||||||||||||
| Patient ID | Disease | Gene | HGVS Nomenclature | Zygosity | RefSeq ID | AF | HGMD ID | |
|---|---|---|---|---|---|---|---|---|
| cDNA-Level | Protein-Level | gnomAD § | ||||||
| P1 | HS/FP | ABCB6 | NM_005689:c.C2215T | p.Arg739Cys | Het | rs141840760 | 0.0004 | - |
| SPTA1 | NM_003126:c.5183G>A | p.Trp1728* | Comp het | - | - | - | ||
| SPTA1 | NM_003126:c.6531-12C>T | - | Comp het | rs28525570 | 0.23 | CS995155 | ||
| P2 | HS/CDAII | SEC23B | NM_006363:c.1233+4C>T | - | Hom | rs201883785 | - | - |
| SPTA1 | NM_003126:c.4708G>A | p.Ala1570Thr | Hom | rs778626016 | - | - | ||
| SPTA1 | NM_003126:c.6531-12C>T | - | Hom | rs28525570 | 0.23 | CS995155 | ||
| P3 | DHS1/HS | SPTA1 | NM_003126:c.6531-12C>T | - | Comp het | rs28525570 | 0.23 | CS995155 |
| SPTA1 | NM_003126:c.5029G>A | p.Gly1677Arg | Comp het | rs771033064 | 0 | CM187374 | ||
| PIEZO1 | NM_001142864:c.7558A>G | p.Lys2520Glu | Het | rs570744198 | 0.001 | CM187408 | ||
| P4 | DHS1/HE | SPTB | NM_001355437:c.871G>A | p.Gly291Ser | Het | rs143599352 | 0.0002 | - |
| PIEZO1 | NM_001142864:c.7219G>C | p.Glu2407Gln | Het | rs200291894 | 0.0001 | CM1922287 | ||
| P5 | DHS1/HE | SPTB | NM_001355437:c.40C>T | p.Pro14Ser | Het | rs147059670 | 0.0001 | - |
| PIEZO1 | NM_001142864:c.7180G>A | p.Gly2394Ser | Het | rs201950081 | 0.0001 | CM187364 | ||
| P6 | DHS1/HS | PIEZO1 | NM_001142864:c.608T>C | p.Leu203Pro | Het | rs977249154 | 0 | - |
| SPTA1 | NM_003126:c.6531-12C>T | - | Het | rs28525570 | 0.23 | CS995155 | ||
| SPTA1 | NM_003126:c.1958A>G | p.Tyr653Cys | Het | rs148912436 | 0.008 | CM187425 | ||
| P7 | DHS1/HS | PIEZO1 | NM_001142864:c.4481A>C | p.Glu1494Ala | Het | - | - | - |
| SPTA1 | NM_003126:c.2464+1G>A | - | Hom | rs774632615 | 0 | - | ||
| P8 | DHS2/FP/HLASA | ABCB6 | NM_005689:c.1402G>T | p.Ala468Ser | Het | rs777270402 | 0 | - |
| LARS2 | NM_015340:c.457A>C | p.Asn153His | Hom | rs786205560 | - | CM1615275 | ||
| KCNN4 | NM_002250:c.1018C>A | p.His340Asn | Het | rs76935412 | 0.002 | - | ||
| P9 | HS/FP | ABCB6 | NM_005689:c.1762G>A | p.Gly588Ser | Het | rs145526996 | 0.004 | CM128905 |
| SLC4A1 | NM_000342:c.2621T>C | p.Leu874Pro | Het | - | - | - | ||
| P10 | PKD/DHS1 | PKLR | NM_000298:c.1675C>T | p.Arg559* | Hom | rs532230312 | 0 | CM981585 |
| PIEZO1 | NM_001142864:c.6796G>A | p.Val2266Ile | Het | rs546338962 | 0 | CM187363 | ||
| P11 | DHS1/HE | PIEZO1 | NM_001142864:c.5195C>T | p.Thr1732Met | Het | rs139051768 | 0.011 | - |
| SPTB | NM_001024858:c.1606G>A | p.Asp536Asn | Het | rs145675502 | 0.001 | CM187385 | ||
| P12 | DHS1/FP | PIEZO1 | NM_001142864:c.7367G>A | p.Arg2456His | Het | rs587776988 | - | CM127746 |
| ABCB6 | NM_005689:c.1474G>A | p.Ala492Thr | Het | rs147445258 | 0.007 | CM169662 | ||
| P13 | DHS1/FP | ABCB6 | NM_005689:c.1361T>C | p.Val454Ala | Het | rs61733629 | 0.006 | CM169864 |
| PIEZO1 | NM_001142864:c.5981C>G | p.Ser1994Cys | Het | - | - | - | ||
| P14 | DHS1/HS | SLC4A1 | NM_000342:c.1462G>A | p.Val488Met | Het | rs28931584 | 0 | CM971385 |
| PIEZO1 | NM_001142864:c.5195C>T | p.Thr1732Met | Het | rs139051768 | 0.011 | CM200163 | ||
| P15 | DHS1/HS | SLC4A1 | NM_000342:c.2608C>T | p.Arg870Trp | Het | rs28931585 | - | CM951173 |
| PIEZO1 | NM_001142864:c.1001C>T | p.Ala334Val | Het | rs574402639 | 0.0003 | - | ||
| P16 | DHS2/HS | KCNN4 | NM_002250: c.1018C>A | p.His340Asn | Het | rs76935412 | 0.002 | - |
| SPTA1 | NM_003126:c.460_462dupTTG | p.Leu155dup | Het | rs757679761 | 0 | CI890173 | ||
| SPTA1 | NM_003126:c.6531-12C>T | - | Het | rs28525570 | 0.23 | CS995155 | ||
| P17 | DHS1/DHS2 | PIEZO1 | NM_001142864:c.1813A>G | p.Met605Val | Het | rs1490094586 | - | CM1911810 |
| KCNN4 | NM_002250:c.983A>G | p.His328Arg | Het | rs780167756 | - | - | ||
| P18 | DHS1/HS | ANK1 | NM_020476:c.613-1G>C | - | Het | - | - | - |
| PIEZO1 | NM_001142864:c.3935C>T | p.Ala1312Val | Het | rs34246477 | 0.0014 | - | ||
| P19 | DHS1/HS | PIEZO1 | NM_001142864:c.3935C>T | p.Ala1312Val | Het | rs34246477 | 0.001 | - |
| ANK1 | NM_020476:c.613-1G>C | - | Het | - | - | - | ||
| P20 | DHS1/HS | PIEZO1 | NM_001355436:c.7529C>T | p.Pro2510Leu | Het | rs61745086 | 0.007 | CM1812923 |
| ANK1 | NM_000037:c.1540G>T | p.Gly514Cys | Het | rs199975878 | 0 | - | ||
| P21 | DHS1/HS | PIEZO1 | NM_001142864:c.1447G>A | p.Val483Met | Het | rs747301309 | 0 | - |
| SPTA1 | NM_003126:c.2173C>T | p.Arg725* | Het | - | - | - | ||
| SPTA1 | NM_003126:c.2909C>A | p.Ala970Asp | Het | rs35948326 | 0.03 | CM930690 | ||
| P22 | DHS1/FP | PIEZO1 | NM_001142864:c.5835C>G | p.Phe1945Leu | Het | rs776602133 | 0 | - |
| ABCB6 | NM_005689:c.1691T>C | p.Met564Thr | Het | rs1233572695 | - | - | ||
| P23 | DHS1/G6PD | PIEZO1 | NM_001142864:c.6205G>A | p.Val2069Met | Het | rs199752762 | 0.001 | - |
| G6PD | NM_001042351:c.1360C>T | p.Arg454Cys | Hem | rs398123546 | 0.0001 | CM920290 | ||
| Analysis | Unit | DHS1 Cases | HS Cases | Dual Inheritance | Reference Range | P § | P1 | P2 | P3 |
|---|---|---|---|---|---|---|---|---|---|
| (n = 37) | (n = 21) | (n = 16) | |||||||
| Age | years | 20.5 ± 3.2 (20.0; 28) | 27.8 ± 4.0 (25.0; 20) | 23.9 ± 5.1 (18.5; 16) | - | 0.32 | - | - | - |
| Gender | female/male | 17 (45.9)/20 (54.1) | 16 (76.2)/5 (23.8) | 5 (29.4)/11 (70.6) | - | 0.02 | 0.32 | <0.01 | 0.03 |
| Hematological data | |||||||||
| RBCs | × 106/µL | 3.1 ± 0.1 (3.0; 36) | 3.9 ± 0.2 (4.3; 19) | 4.0 ± 0.2 (3.9; 15) | 4.0–5.2 | <0.001 | 0.004 | 1.00 | 0.004 |
| Hb | g/dL | 12.5 ± 0.4 (12.7; 36) | 11.2 ± 0.6 (11.1; 21) | 10.9 ± 0.5 (10.8; 16) | 11.5–15.5 | 0.06 | - | - | - |
| Ht | % | 36.3 ± 1.7 (37.3; 36) | 33.1 ± 1.6 (33.1; 19) | 31.8 ± 1.6 (30.5; 14) | 35–45 | 0.04 | 0.11 | 1.00 | 0.14 |
| MCV | fL | 100.5 ± 1.7 (100.0; 36) | 83.2 ± 1.6 (83.8; 21) | 82.5 ± 2. 5 (80.9; 16) | 77–95 | <0.001 | <0.001 | 1.00 | <0.001 |
| MCH | pg | 35.9 ± 1.1 (36.0;27) | 28.8 ± 0.9 (29.0; 21) | 27.6 ± 1.1 (27.4; 16) | 25–33 | <0.001 | <0.001 | 1.00 | <0.001 |
| MCHC | g/dL | 35.5 ± 0.7 (34.3; 36) | 34.2 ± 0.9 (34.5; 19) | 34.1 ± 0.5 (34.1; 16) | 32–36 | 0.81 | - | - | - |
| PLTs | × 103/µL | 414.2 ± 55.3 (375.0; 15) | 276.3 ± 34.1 (270.0; 20) | 288.7 ± 41.8 (232.0; 16) | 150–450 | 0.05 | 0.08 | 1.00 | 0.09 |
| ARC | × 103/µL | 164.5 ± 17.9 (145.9; 33) | 238.3 ± 45.9 (234.6; 16) | 217.5 ± 50.5 (144.5; 13) | 20–90 | 0.49 | - | - | - |
| Biochemical data and iron balance | |||||||||
| Total bilirubin | mg/dL | 3.7 ± 0.6 (3.9; 21) | 2.9 ± 0.6 (2.4; 16) | 3.0 ± 0.5 (3.5; 12) | 0.3–1.0 | 0.59 | - | - | - |
| Ferritin | ng/mL | 491.3 ± 82.1 (363.0; 19) | 319.1 ± 117.8 (150.2; 14) | 409.8 ± 154.4 (128.5; 13) | 22–275 | 0.18 | - | - | - |
| Ferritin level/dosage age † | - | 38.9 ± 6.5 (30.3; 18) | 10.9 ± 2.9 (7.9; 14) | 36.2 ± 16.1 (11.0; 13) | - | <0.001 | 0.004 | 1.00 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andolfo, I.; Martone, S.; Rosato, B.E.; Marra, R.; Gambale, A.; Forni, G.L.; Pinto, V.; Göransson, M.; Papadopoulou, V.; Gavillet, M.; et al. Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A Case Series Study of 155 Patients. Genes 2021, 12, 958. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070958
Andolfo I, Martone S, Rosato BE, Marra R, Gambale A, Forni GL, Pinto V, Göransson M, Papadopoulou V, Gavillet M, et al. Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A Case Series Study of 155 Patients. Genes. 2021; 12(7):958. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070958
Chicago/Turabian StyleAndolfo, Immacolata, Stefania Martone, Barbara Eleni Rosato, Roberta Marra, Antonella Gambale, Gian Luca Forni, Valeria Pinto, Magnus Göransson, Vasiliki Papadopoulou, Mathilde Gavillet, and et al. 2021. "Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A Case Series Study of 155 Patients" Genes 12, no. 7: 958. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070958