
The Complete Chloroplast Genome Sequence of Eupatorium fortunei: Genome Organization and Comparison with Related Species
 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. DNA Sequencing and Assembly
2.3. Chloroplast Genome Annotation
2.4. SSRs, Long Repeat Sequence, and Codon Usage Analysis
2.5. Genome Comparison and Structural Analysis
2.6. Phylogenetic Analysis
3. Results
3.1. Genome Sequencing and Assembly
3.2. Basic Structural Properties of the Chloroplast Genome
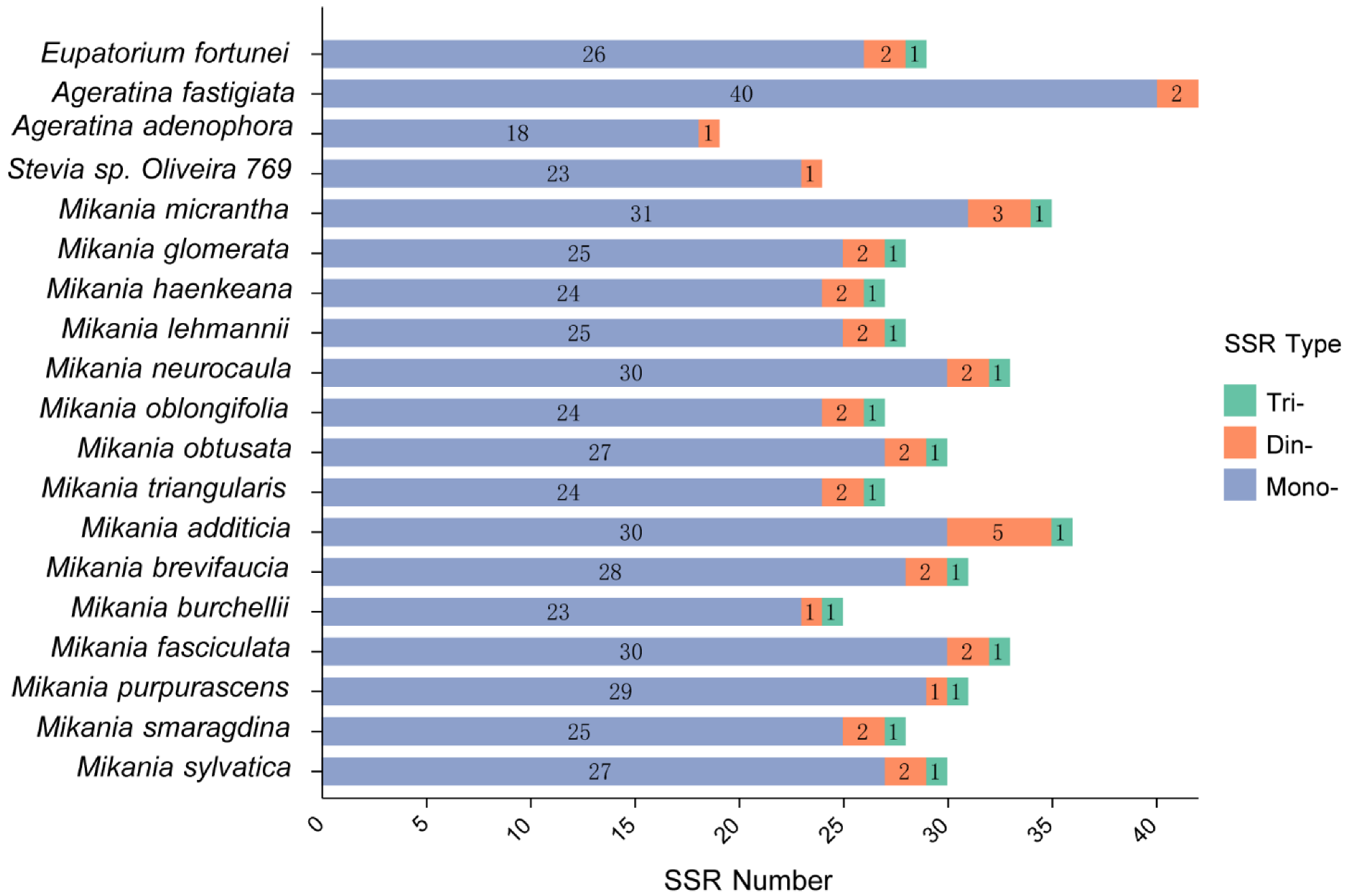
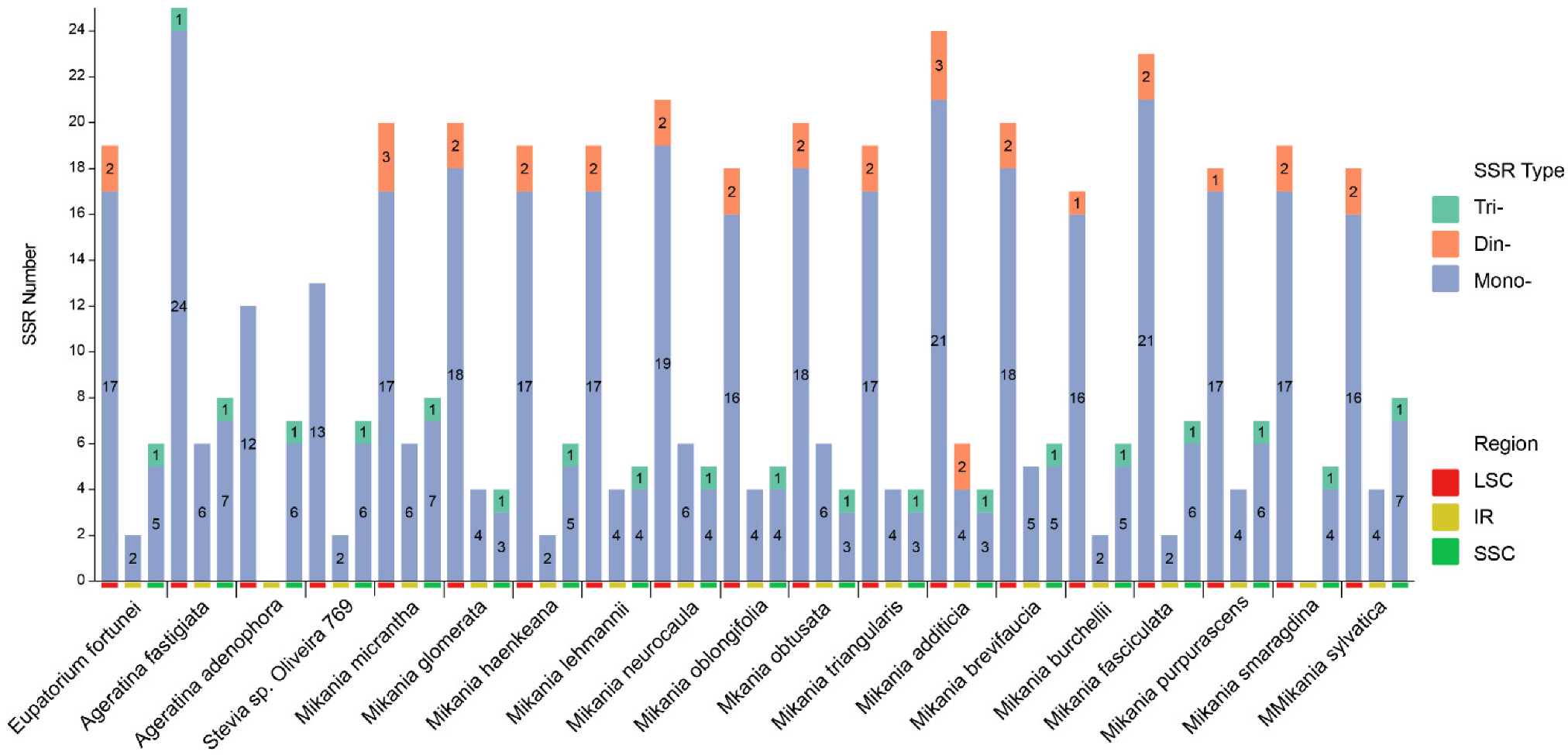
3.3. SSRs Analysis
3.4. Long Repeat Analysis
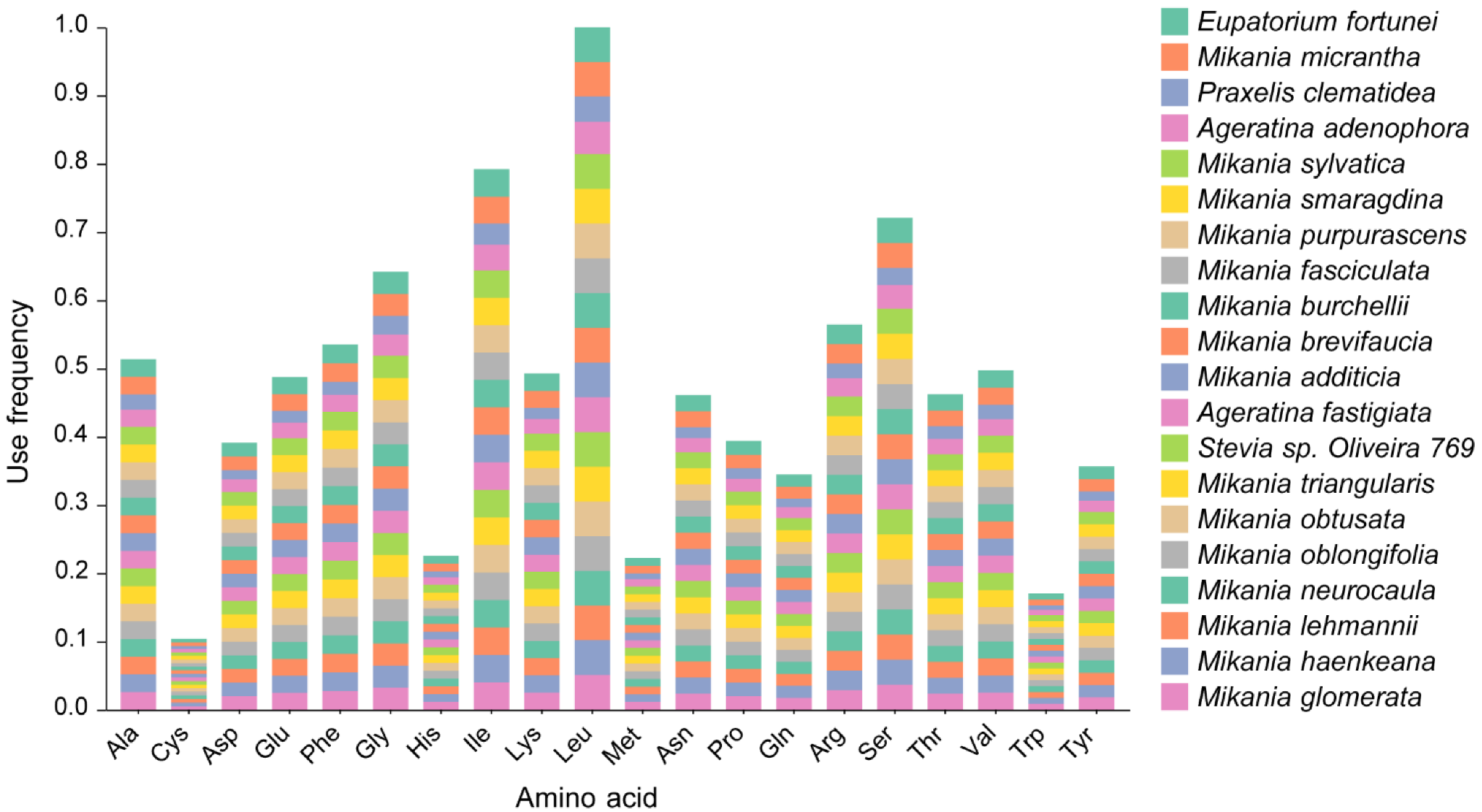
3.5. Analysis of Codon Preference
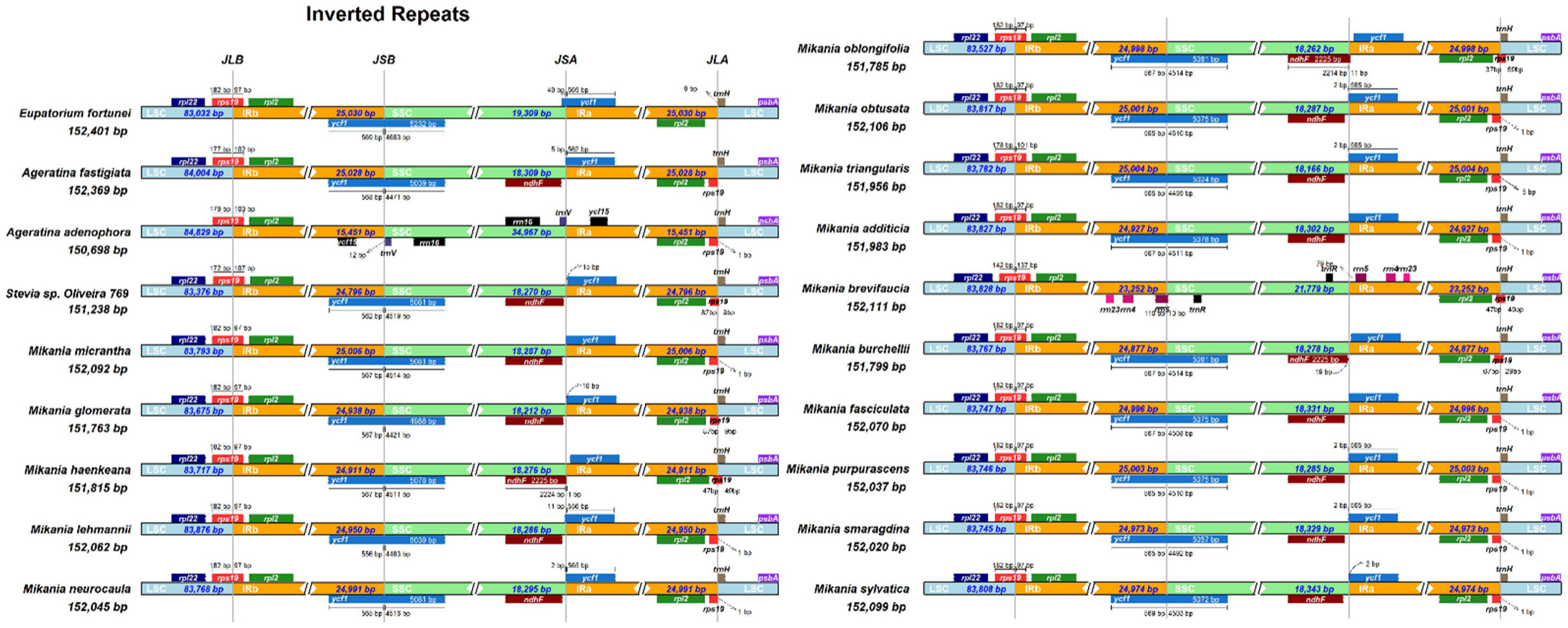
3.6. Expansion and Contraction of Border Regions
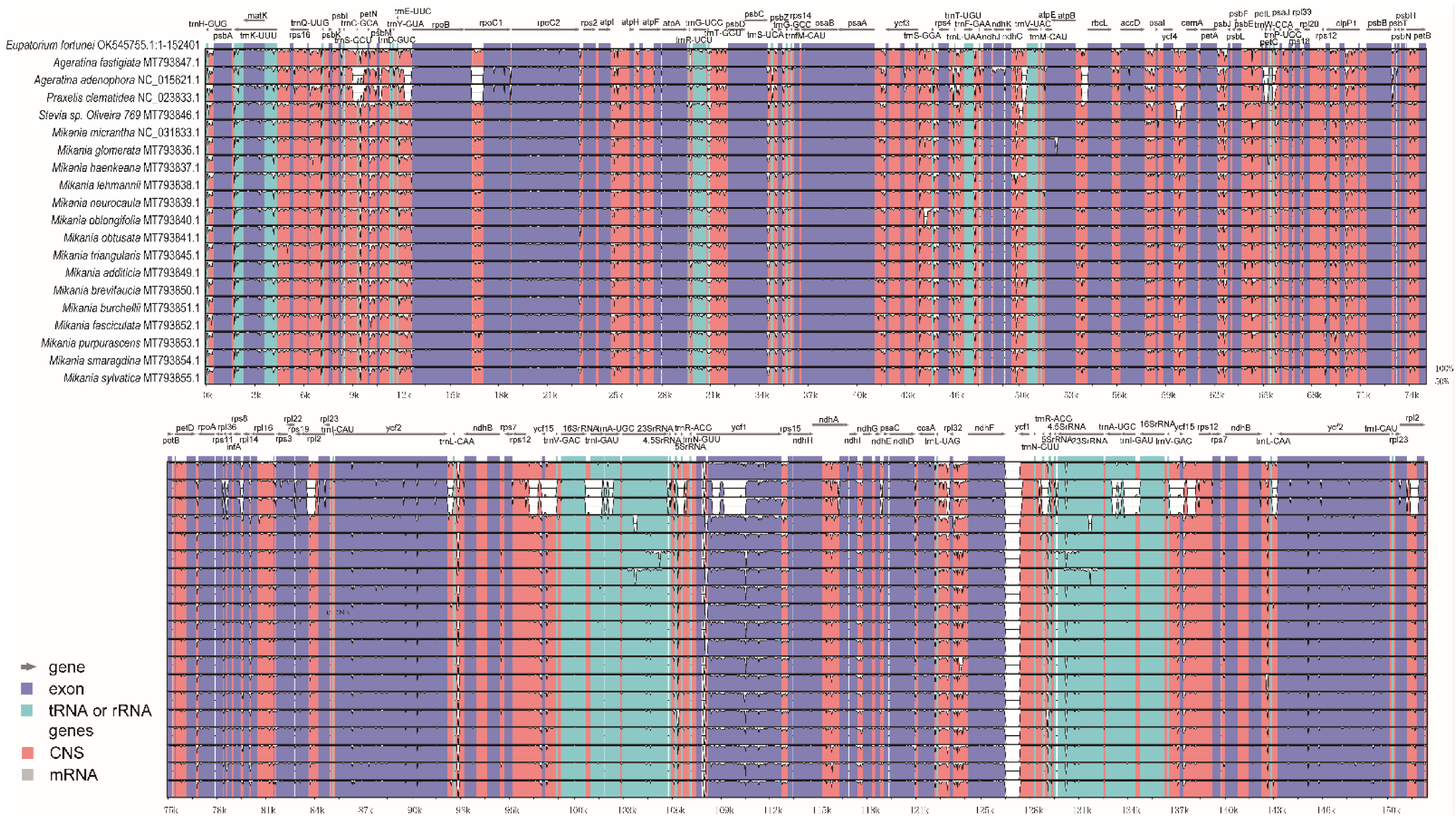
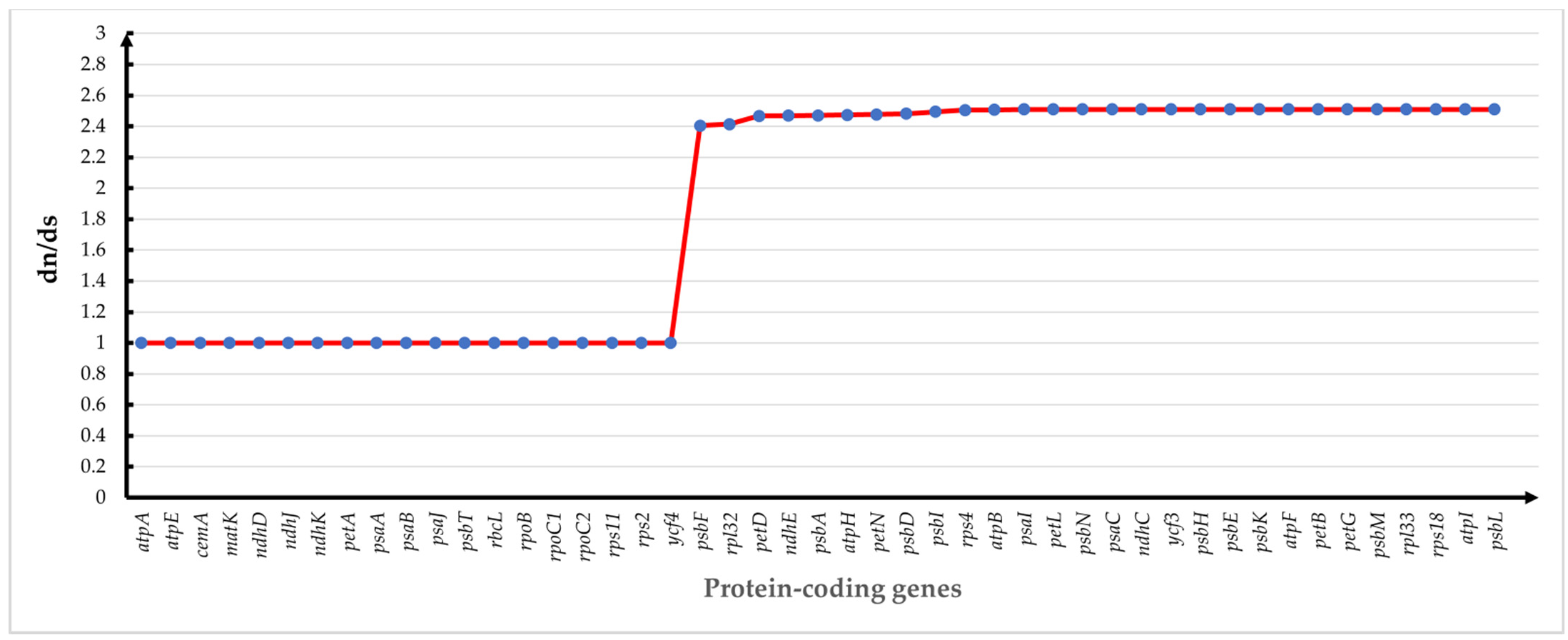
3.7. Sequence Diversity Analysis of Chloroplast Genomes
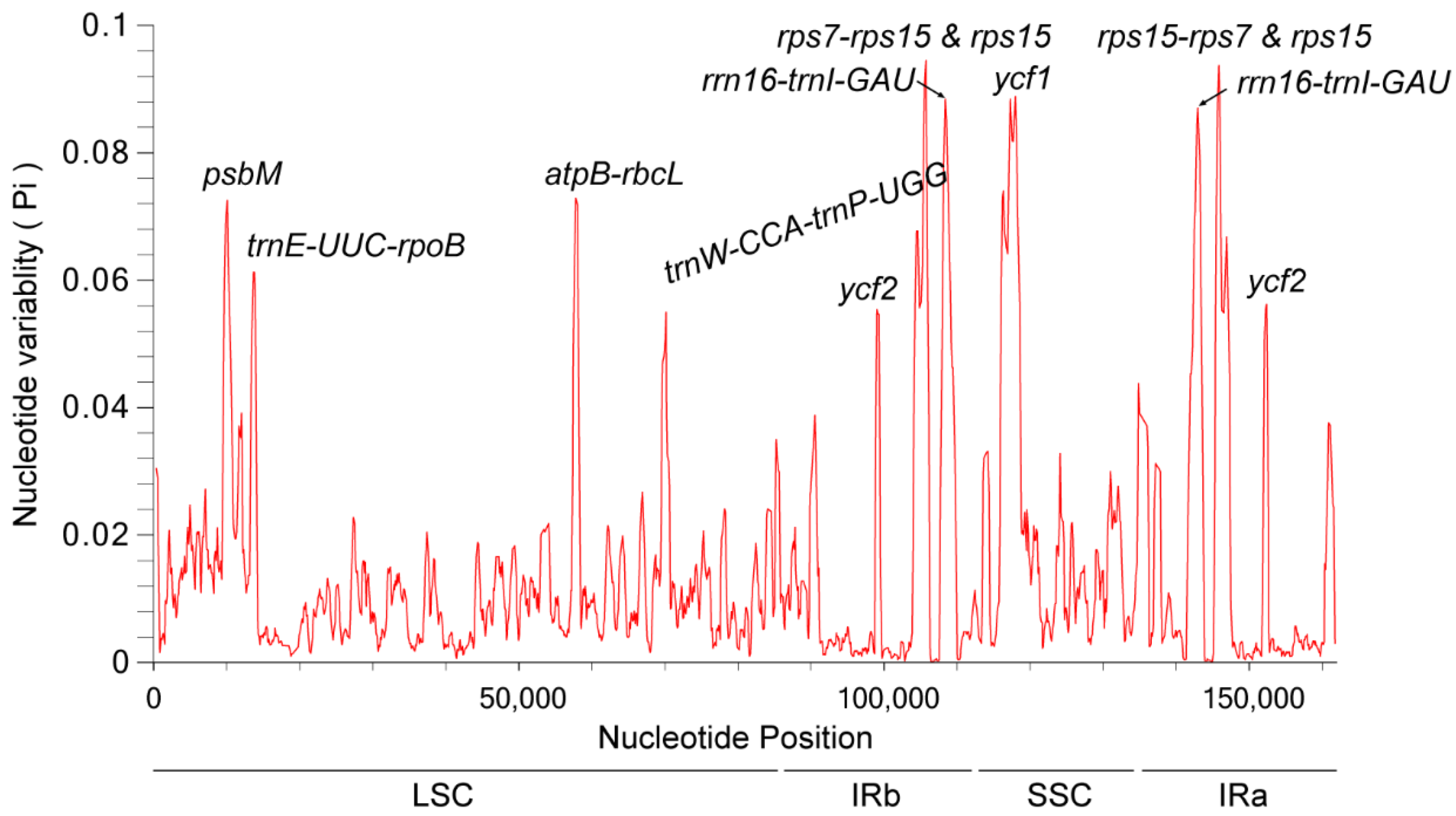
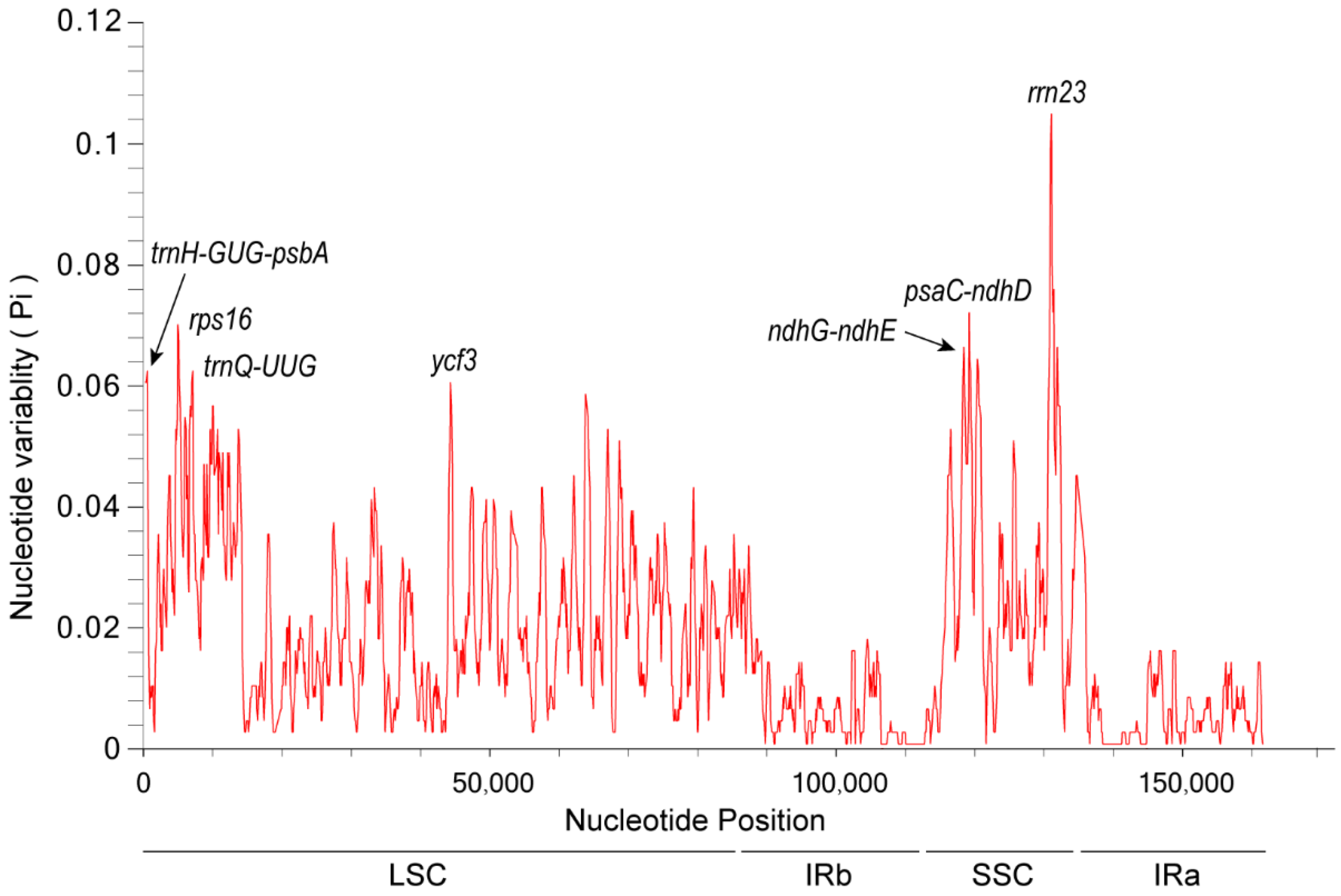
3.8. Nucleotide Polymorphism Analysis
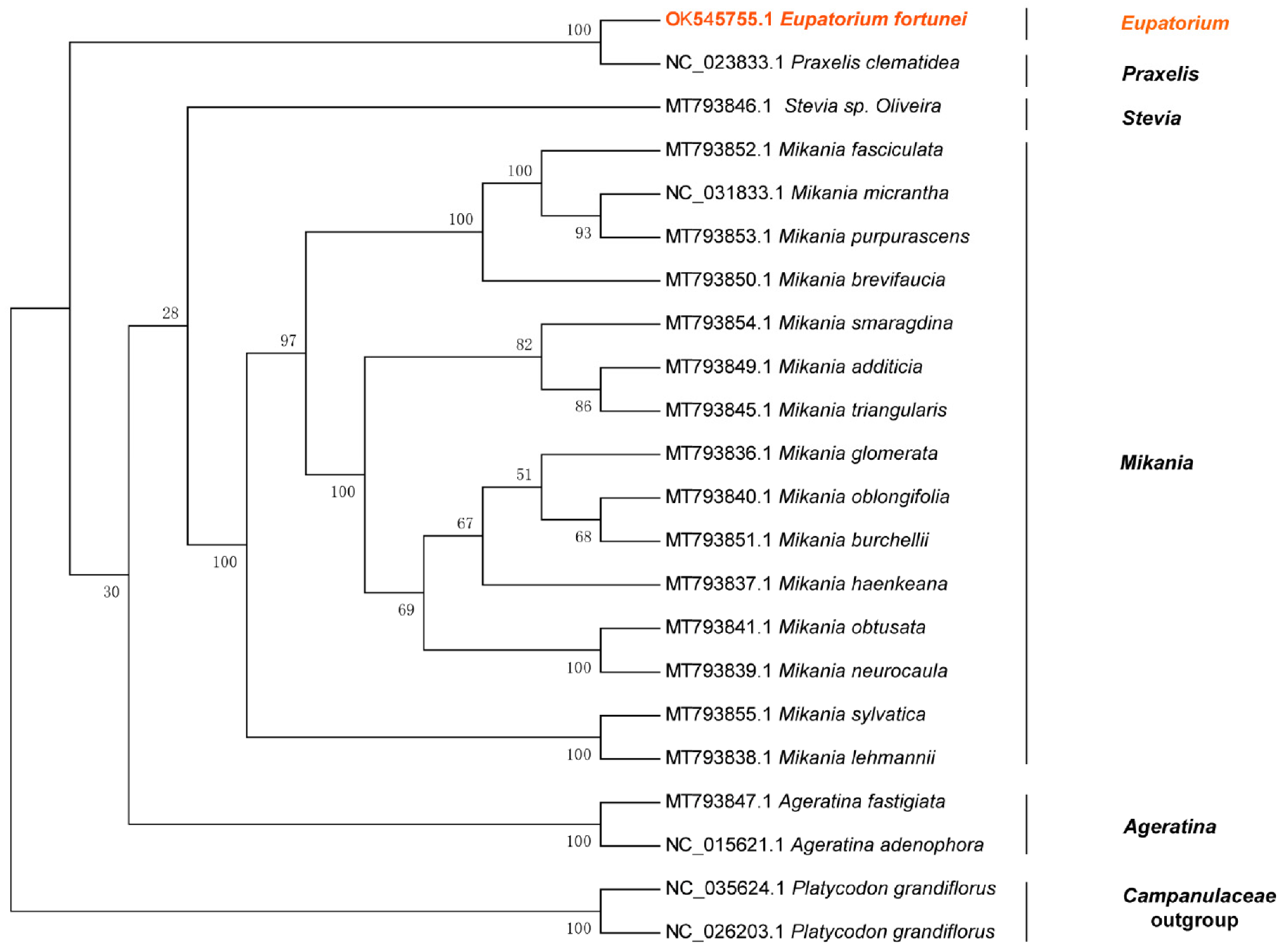
3.9. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete Chloroplast Genome of the Multifunctional Crop Globe Artichoke and Comparison with Other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, L.; Yan, T.L.; Liu, Q. Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene 2014, 549, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Lee, H.; Hwang, Y.H.; Lee, J.S.; Cho, W.K.; Ma, J.Y. Eupatorium fortunei and Its Components Increase Antiviral Immune Responses against RNA Viruses. Front. Pharmacol. 2017, 8, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatmaker, E.A.; Wadl, P.A.; Rinehart, T.A.; Carroll, J.L.; Thomas, S.T.; Robert, N.; Staton, M.E.; Schilling, E.E. Complete chloroplast genome comparisons for Pityopsis (Asteraceae). PLoS ONE 2020, 15, e0241391. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, Y.; Xu, Z.; Yang, G.; Peng, J.; Peng, X. Initial Characterization of the Chloroplast Genome of Vicia sepium, an Important Wild Resource Plant, and Related Inferences About Its Evolution. Front. Genet. 2020, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Li, D.M.; Zhu, G.F.; Xu, Y.C.; Ye, Y.J.; Liu, J.M. Complete Chloroplast Genomes of Three Medicinal Alpinia Species: Genome Organization, Comparative Analyses and Phylogenetic Relationships in Family Zingiberaceae. Plants 2020, 9, 286. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ge, X.; Cano, A.; Salazar, B.; Deng, Y. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): Molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ 2020, 8, e8450. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.X.; Xian, Y.F.; Xiang, L.; Zhang, D.; Shi, Y.H.; Wu, M.L.; Dong, G.Q.; Ip, S.P.; Lin, Z.X.; Wu, L.; et al. Complete Chloroplast Genomes from Sanguisorba: Identity and Variation Among Four Species. Molecules 2018, 23, 2137. [Google Scholar] [CrossRef] [Green Version]
- Mader, M.; Pakull, B.; Blanc-Jolivet, C.; Paulini-Drewes, M.; Bouda, Z.H.; Degen, B.; Small, I.; Kersten, B. Complete Chloroplast Genome Sequences of Four Meliaceae Species and Comparative Analyses. Int. J. Mol. Sci. 2018, 19, 701. [Google Scholar] [CrossRef] [Green Version]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Kazuhiko, U.; Yasuhiko, S.; Masayuki, T.; Zhen, C. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, J.; et al. The complete nucleotide sequence of the tobacco chloroplast genome its gene organization and expression. Plant Mol. Biol. Rep. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Wang, L.; Wuyun, T.-N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Fan, W.B.; Wu, Y.; Yang, J.; Shahzad, K.; Li, Z.H. Comparative Chloroplast Genomics of Dipsacales Species: Insights into Sequence Variation, Adaptive Evolution, and Phylogenetic Relationships. Front. Plant Sci. 2018, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, Y.; Cai, Q.; Lin, F.; Huang, P.; Zheng, Y. Development of chloroplast genomic resources for Akebia quinata (Lardizabalaceae). Conserv. Genet. Resour. 2016, 8, 447–449. [Google Scholar] [CrossRef]
- Li, J.; Liu, L.; Wang, H.; Li, C.; Zuo, W.; Zeng, Y. The complete chloroplast genome of a medical herb, Potentilla parvifolia Fisch. (Rosaceae), from Qinghai-Tibet Plateau in China. Mitochondrial DNA Part B 2021, 6, 349–350. [Google Scholar] [CrossRef]
- Tang, H.; Tang, L.; Shao, S.; Peng, Y.; Li, L.; Luo, Y. Chloroplast genomic diversity in Bulbophyllum section Macrocaulia (Orchidaceae, Epidendroideae, Malaxideae): Insights into species divergence and adaptive evolution. Plant Divers. 2021, 43, 350–361. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, B.; Guo, C.; Han, R.; Li, J.; Rong, F.; Ding, J. The complete mitochondrial genome of Holothuria fuscocinerea (Jaeger, 1833). Mitochondrial DNA Part B 2020, 5, 33–34. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Weng, Y.; Ye, D.; You, Y.; Shi, J.; Chen, J. The complete chloroplast genome sequence of Casuarina equisetifolia. Mitochondrial DNA Part B 2021, 6, 3046–3048. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, S.D.; Fitz-Gibbon, S.T.; Strenkert, D.; Purvine, S.O.; Pellegrini, M.; Merchant, S.S. High-throughput sequencing of the chloroplast and mitochondrion of Chlamydomonas reinhardtii to generate improved de novo assemblies, analyze expression patterns and transcript speciation, and evaluate diversity among laboratory strains and wild isolates. Plant J. 2018, 93, 545–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Stefan, K.; Choudhuri, J.; Enno, O.; Chris, S.; Jens, S.; Robert, G. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, X.; Liu, D.; Chen, X.; Li, F.; Qi, X.; Luo, Z.; Wang, C. Genetic diversity and population structure of the endangered medicinal plant Phellodendron amurense in China revealed by SSR markers. Biochem. Syst. Ecol. 2016, 66, 286–292. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Zheng, C.; Tian, X.; Zheng, M.; Xiang, B.; Wang, Y. Comparison of Four Complete Chloroplast Genomes of Medicinal and Ornamental Meconopsis Species: Genome Organization and Species Discrimination. Sci. Rep. 2019, 9, 10567. [Google Scholar] [CrossRef] [Green Version]
- Leonie, D.; Barbara, G.; Youri, L.; Yavuz, A.; Thomas, C.A.W.; Klaas, V. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Curci, P.L.; Sonnante, G. The complete chloroplast genome of Cynara humilis. Mitochondrial DNA Part A 2015, 27, 2345–2346. [Google Scholar] [CrossRef]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Han, L.J.; Yang, C.W.; Yin, Z.L.; Tian, X.; Qian, Z.G.; Li, G.D. Comparative chloroplast genome analysis of medicinally important Veratrum (Melanthiaceae) in China: Insights into genomic characterization and phylogenetic relationships. Plant Divers. 2022, 44, 70–82. [Google Scholar] [CrossRef]
- Ki-Joong, K.; Keung-Sun, C.; Jansen, R.K. Two Chloroplast DNA Inversions Originated Simultaneously During the Early Evolution of the Sunflower Family (Asteraceae). Mol. Biol. Evol. 2005, 22, 1783–1792. [Google Scholar] [CrossRef]
- Lu, R.-S.; Li, P.; Qiu, Y.-X. The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caixiang, W.; Juanjuan, L.; Yue, S.; Meili, L.; Xiaoyu, X.; Junji, S. Complete Chloroplast Genome Sequence of Sonchus brachyotus Helps to Elucidate Evolutionary Relationships with Related Species of Asteraceae. BioMed Res. Int. 2021, 2021, 9410496. [Google Scholar] [CrossRef]
- Tibihika, P.D.; Curto, M.; Dornstauder-Schrammel, E.; Winter, S.; Alemayehu, E.; Waidbacher, H.; Meimberg, H. Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv. Genet. 2019, 20, 357–372. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Names | E. fortunei OK545755.1 | A. fastigiata MT793847.1 | A. adenophora NC_015621.1 |
|---|---|---|---|
| Total length (bp) | 152,401 | 152,369 | 150,698 |
| LSC length (bp) | 83,032 | 84,004 | 84,829 |
| SSC length (bp) | 19,309 | 18,309 | 34,967 |
| IR length (bp) | 50,060 | 50,056 | 30,902 |
| Coding length (bp) | 78,396 | 78,621 | 73,554 |
| Noncoding length (bp) | 74,005 | 73,748 | 77,144 |
| Total number of genes | 133 | 134 | 136 |
| Protein-coding genes (duplicated) | 87 (7) | 87 (7) | 86 (7) |
| tRNA genes (duplicated) | 37 (7) | 37 (7) | 37 (10) |
| rRNA genes (duplicated) | 8 (4) | 8 (4) | 8 (4) |
| Pseudo genes | 1 | 2 | 5 |
| GC content of genome | 37.5884 | 37.5018 | 37.4617 |
| GC content of LSC | 35.7103 | 35.5852 | 35.7424 |
| GC content of SSC | 31.4775 | 31.1540 | 40.7298 |
| GC content of IR | 43.0604 | 43.0398 | 38.4830 |
| Function | Group of Genes | Name of Genes | Total Number |
|---|---|---|---|
| Self-replication | Large subunit of ribosome | rpl20,rpl22,rpl32,rpl23(X2),rpl14,rpl33,rpl16,rpl36,rpl2(X2) | 11 |
| Small subunit of ribosome | rps11,rps14,rps15,rps16,rps2,rps3,rps18,rps19,rps4,rps7(X2),rps8,rps12(X2) | 14 | |
| DNA-dependent RNA polymerase | rpoA,rpoB,rpoC1,rpoC2 | 4 | |
| rRNA gene | rrn5(X2),rrn4.5(X2),rrn16(X2),rrn23(X2), | 8 | |
| tRNA gene | trnR-UCU,trnE-UUC,trnI-CAU(X2),trnS-GGA,trnT-GGU,trnR-ACG(X2),trnV-GAC(X2),trnL-UAA,trnG-GCC,trnD-GUC,trnL-CAA(X2),trnP-UGG,trnM-CAU,trnY-GUA,trnS-GCU,trnW-CCA,trnF-GAA,trnT-UGU,trnS-UGA,trnV-UAC,trnG-UCC,trnL-UAG,trnI-GAU(X2),trnH-GUG,trnN-GUU(X2),trnA-UGC(X2),trnfM-CAU,trnQ-UUG,trnK-UUU,trnC-GCA | 37 | |
| Gene for photosynthesis | Subunits of photosystem I | psaA,psaB,psaC,psaI,psaJ | 5 |
| Subunits of photosystem II | psbL,psbZ,psbM,psbA,psbB,psbC,psbD,psbE,psbF,psbT,psbH,psbI,psbJ,psbK,psbN | 15 | |
| Subunits of NADH-dehydrogenase | ndhG,ndhH,ndhI,ndhJ,ndhK,ndhA,ndhB(X2),ndhC,ndhD,ndhE,ndhF | 12 | |
| Subunits of cytochrome b/f complex | petL,petA,petN,petB,petD,petG | 6 | |
| Subunit for ATP synthase | atpI,atpA,atpB,atpE,atpF,atpH | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Other genes | Translational initiation factor | infA | 1 |
| Maturase | matK | 1 | |
| Protease | clpP1 | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Subunit of Acetyl-carboxylase | accD | 1 | |
| C-type cytochrome synthesis gene | ccsA | 1 | |
| Unknown function | Open reading frames (ORF,ycf) | ycf1,ycf2(X2),ycf3,ycf4,ycf15(X2),ycf1-fragment | 8 |
| Number | SSR Type | Size | Start | End | Position |
|---|---|---|---|---|---|
| 1 | (T)10 | 10 | 2299 | 2308 | trnK-UUU |
| 2 | (C)12 | 12 | 5418 | 5429 | rps16 intron |
| 3 | (T)10 | 10 | 9543 | 9552 | IGS |
| 4 | (A)10 | 10 | 13,302 | 13,311 | rpoB |
| 5 | (T)12 | 12 | 16,571 | 16,582 | rpoC1 intron |
| 6 | (A)10 | 10 | 18,356 | 18,365 | rpoC1 |
| 7 | (T)13 | 13 | 24,915 | 24,927 | IGS |
| 8 | (TA)6 | 12 | 26,593 | 26,604 | IGS |
| 9 | (T)14 | 14 | 27,969 | 27,982 | IGS |
| 10 | (T)10 | 10 | 30,842 | 30,851 | trnT-GGU |
| 11 | (AT)7 | 14 | 43,871 | 43,884 | IGS |
| 12 | (T)14 | 14 | 46,306 | 46,319 | IGS |
| 13 | (T)12 | 12 | 53,844 | 53,855 | IGS |
| 14 | (T)11 | 11 | 58,539 | 58,549 | IGS |
| 15 | (T)10 | 10 | 69,445 | 69,454 | rps12 intron |
| 16 | (T)10 | 10 | 69,481 | 69,490 | clpP1 intron |
| 17 | (T)10 | 10 | 70,461 | 70,470 | clpP1 intron |
| 18 | (T)10 | 10 | 70,625 | 70,634 | clpP1 intron |
| 19 | (T)10 | 10 | 77,060 | 77,069 | rpoA |
| 20 | (T)10 | 10 | 79,989 | 79,998 | IGS |
| 21 | (A)11 | 11 | 106,342 | 106,352 | IGS |
| 22 | (GAA)5 | 15 | 108,410 | 108,424 | ycf1 |
| 23 | (T)12 | 12 | 108,521 | 108,532 | ycf1 |
| 24 | (T)10 | 10 | 108,649 | 108,658 | ycf1 |
| 25 | (A)11 | 11 | 109,134 | 109,144 | ycf1 |
| 26 | (A)10 | 10 | 110,236 | 110,245 | ycf1 |
| 27 | (A)13 | 13 | 120,967 | 120,979 | IGS |
| 28 | (T)13 | 13 | 122,925 | 122,937 | IGS |
| 29 | (T)11 | 11 | 129,082 | 129,092 | IGS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, K.; Ran, J.; Bao, S.; Li, Y.; Islam, R.; Zhang, N.; Zhao, W.; Ma, Y.; Sun, C. The Complete Chloroplast Genome Sequence of Eupatorium fortunei: Genome Organization and Comparison with Related Species. Genes 2023, 14, 64. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010064
Yan K, Ran J, Bao S, Li Y, Islam R, Zhang N, Zhao W, Ma Y, Sun C. The Complete Chloroplast Genome Sequence of Eupatorium fortunei: Genome Organization and Comparison with Related Species. Genes. 2023; 14(1):64. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010064
Chicago/Turabian StyleYan, Kan, Juan Ran, Songming Bao, Yimeng Li, Rehmat Islam, Nai Zhang, Wei Zhao, Yanni Ma, and Chao Sun. 2023. "The Complete Chloroplast Genome Sequence of Eupatorium fortunei: Genome Organization and Comparison with Related Species" Genes 14, no. 1: 64. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010064