
Identification of Candidate Genes for Seed Glucosinolate Content Using Association Mapping in Brassica napus L.


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Variations of GS Content


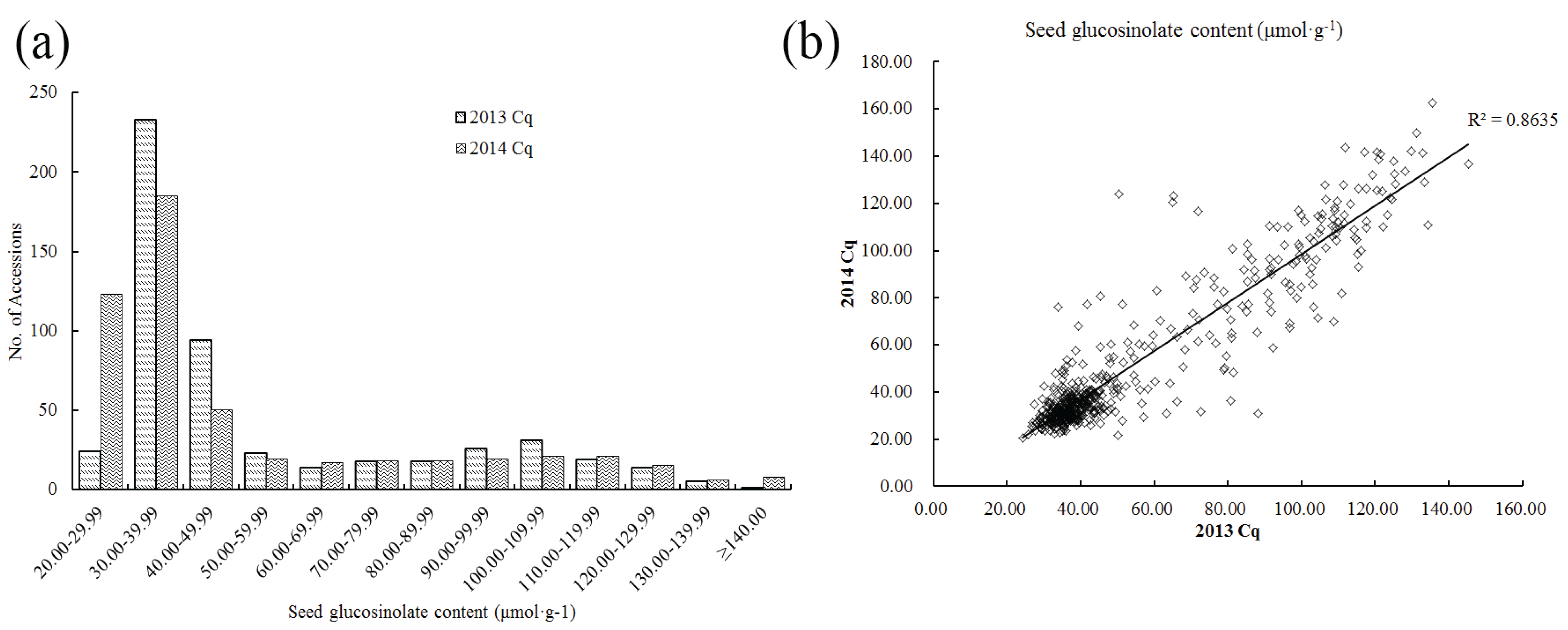{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Range (μmol·g−1) | Average (μmol·g−1) | Standard Deviation | Coefficient of Variation (%) | Skewness | Kurtosis |
|---|---|---|---|---|---|---|
| 2013 Cq (Chongqing) | 24.22–145.24 | 54.90 ± 1.28 | 29.11 | 53.02 | 1.24 | 0.15 |
| 2014 Cq (Chongqing) | 20.53–162.51 | 52.12 ± 1.41 | 32.24 | 61.86 | 1.34 | 0.56 |
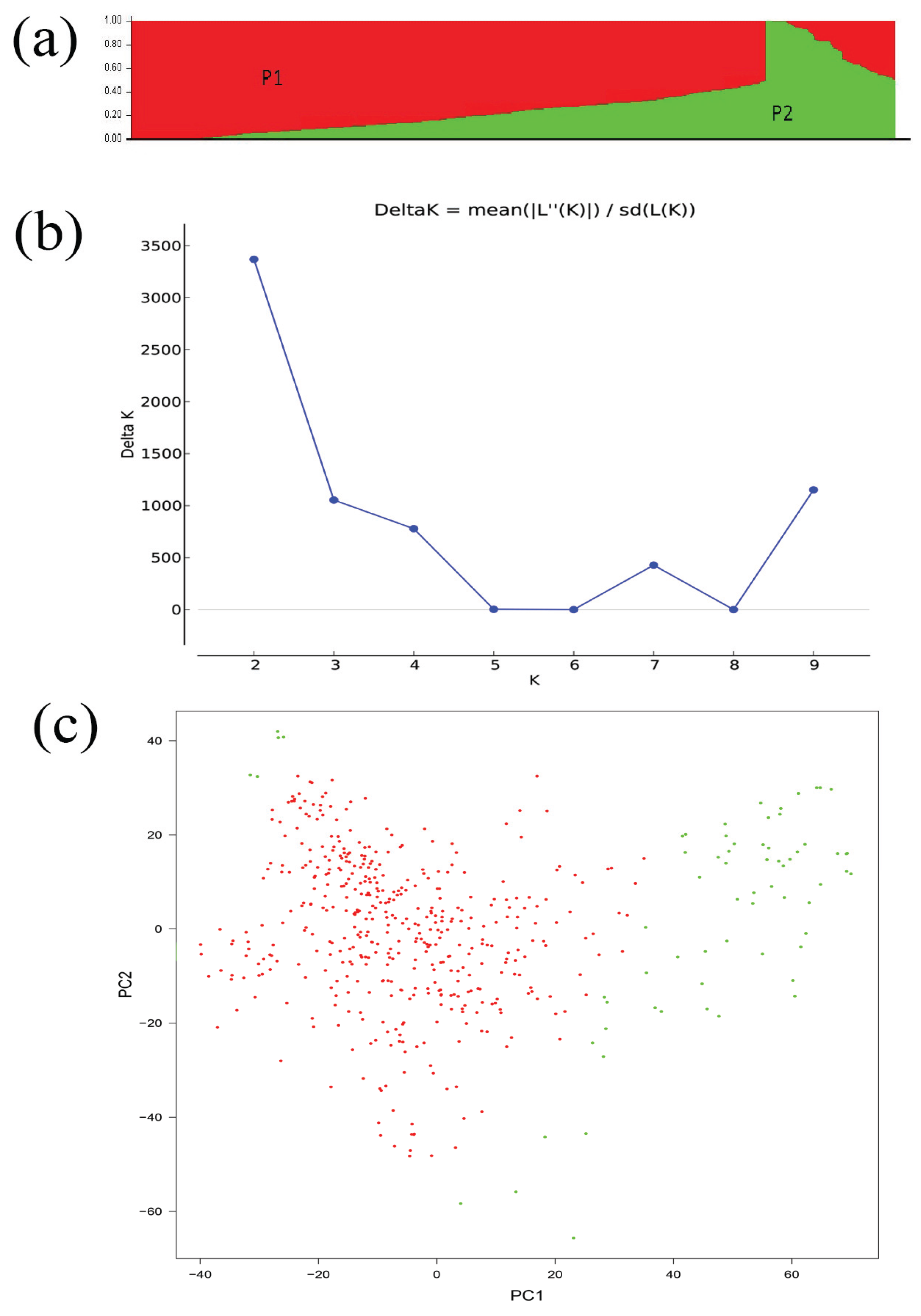
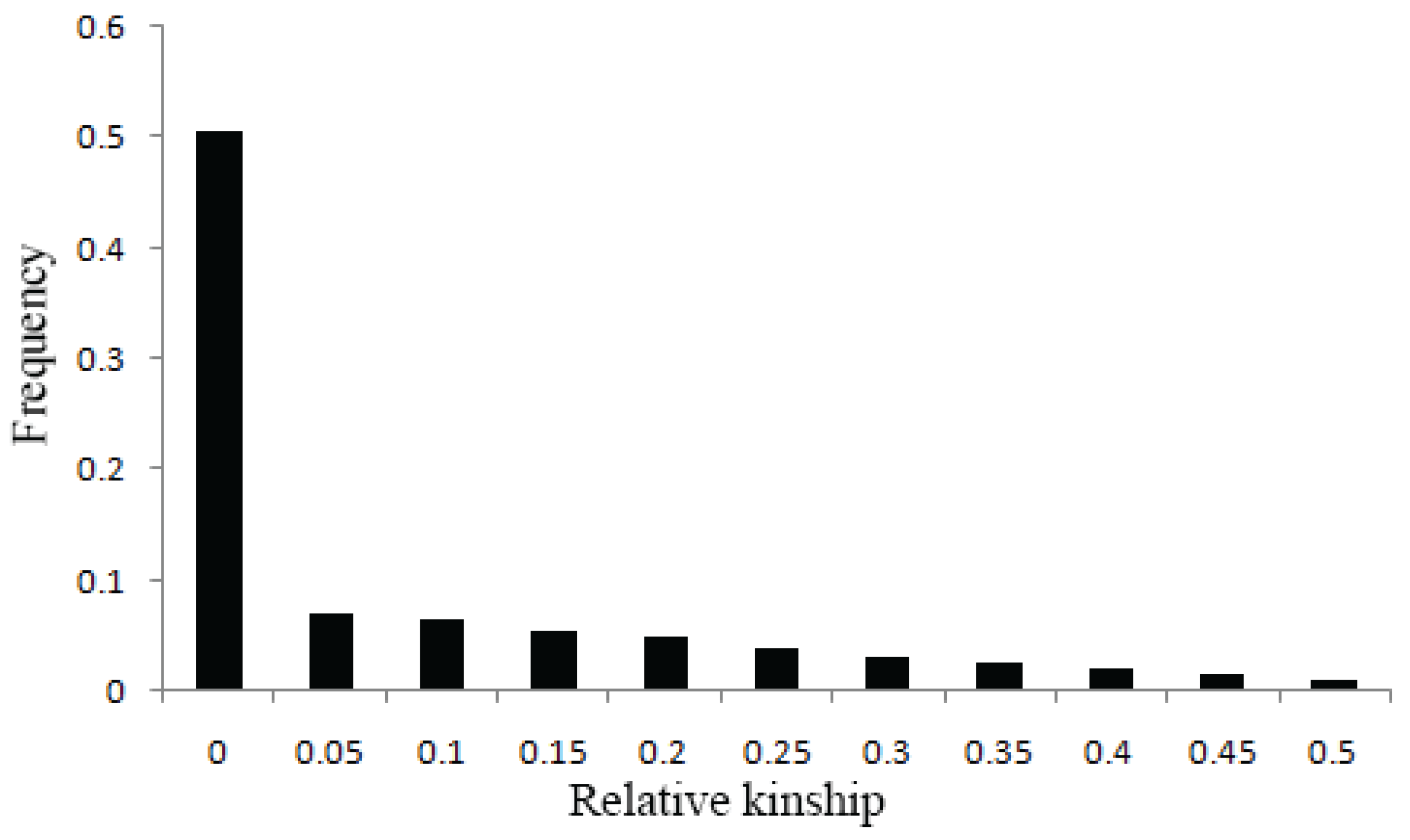
2.2. Population Structure, Relative Kinship and Diversity Panel Analysis

| K | Mean LnP(K) | Stdev LnP(K) | Delta K |
|---|---|---|---|
| 1 | −5591550.233 | 14.608331 | — |
| 2 | −5240293.4 | 49.596068 | 3368.547254 |
| 3 | −5056103.267 | 48.066863 | 1053.798744 |
| 4 | −4922565.933 | 57.973902 | 777.935099 |
| 5 | −4834128.533 | 3719.934136 | 2.630127 |
| 6 | −4755475.033 | 8254.854459 | 0.036322 |
| 7 | −4677121.367 | 90.730884 | 427.251798 |
| 8 | −4637532.633 | 2440.407122 | 0.025228 |
| 9 | −4597882.333 | 1000.043131 | 1151.624163 |
| 10 | −5709905.867 | 1989293.383 | — |

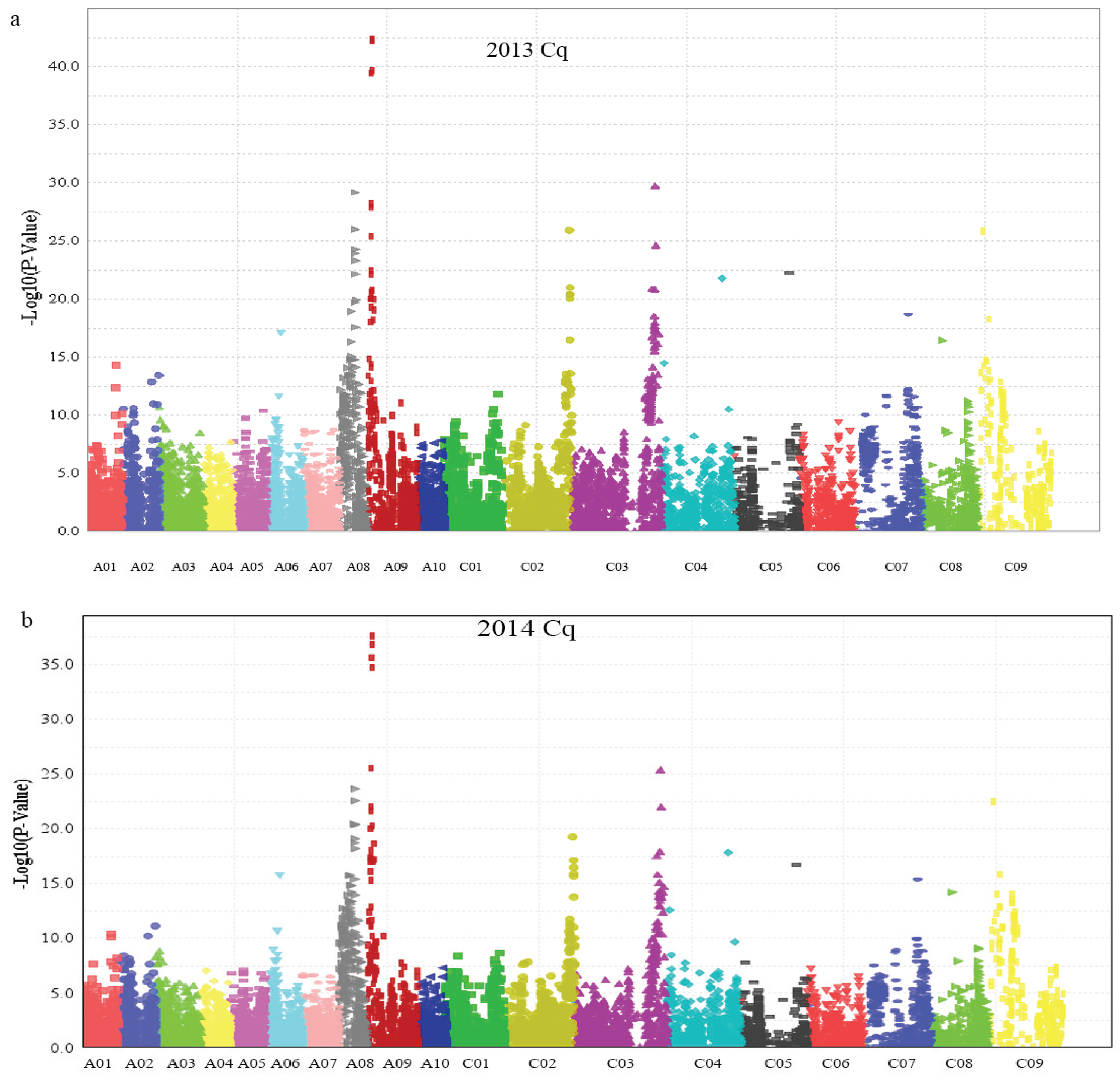
2.3. Association Mapping Analysis
| SNP | p-Value | Phenotypic Variation (%) | Chr. | Physical Interval (bp) | ||
|---|---|---|---|---|---|---|
| 2013 | 2014 | 2013 | 2014 | |||
| Bn-A09-p3029767 | 7.76 × 10−37 | 8.16 × 10−33 | 33.50 | 31.59 | A09 | 2,949,846–3,135,091 |
| Bn-A09-p3116738 | 2.61 × 10−36 | 4.05 × 10−32 | 32.94 | 30.82 | A09 | |
| Bn-A09-p3053532 | 1.79 × 10−33 | 4.94 × 10−30 | 29.96 | 28.54 | A09 | |
| Bn-A09-p3234323 | 3.15 × 10−18 | 15.19 | A09 | |||
| Bn-A01-p9125819 | 2.86 × 10−32 | 9.68 × 10−30 | 28.73 | 28.23 | A09 | 2,450,781–2,472,858 |
| Bn-A01-p9149601 | 4.04 × 10−22 | 3.93 × 10−20 | 18.78 | 18.27 | A09 | |
| Bn-A08-p12660208 | 3.41 × 10−23 | 5.80 × 10−21 | 18.95 | 18.22 | C03 | 56,050,681–56,466,188 |
| Bn-A08-p12905848 | 6.81 × 10−20 | 4.05 × 10−18 | 16.71 | 16.28 | C03 | |
| Bn-A09-p1832760 | 8.94 × 10−21 | 1.89 × 10−20 | 17.52 | 18.58 | A09 | 2,101,520–2,206,660 |
| Bn-A09-p1727915 | 5.27 × 10−20 | 16.81 | A09 | |||
| Bn-scaff_19783_1-p327775 | 1.91 × 10−20 | 3.39 × 10−19 | 17.22 | 17.34 | C09 | 2,815,377–2,815,426 |
| Bn-A08-p12913949 | 1.31 × 10−20 | 1.08 × 10−18 | 16.57 | 16.01 | A08 | 10,587,677–10,694,560 |
| Bn-A08-p12820786 | 1.46 × 10−21 | 1.99 × 10−18 | 18.26 | 16.58 | A08 | |
| Bn-scaff_17119_1-p84986 | 6.12 × 10−20 | 15.96 | A08 | |||
| Bn-scaff_17177_1-p546184 | 6.61 × 10−19 | 15.81 | C02 | 44,655,688–44,655,731 | ||


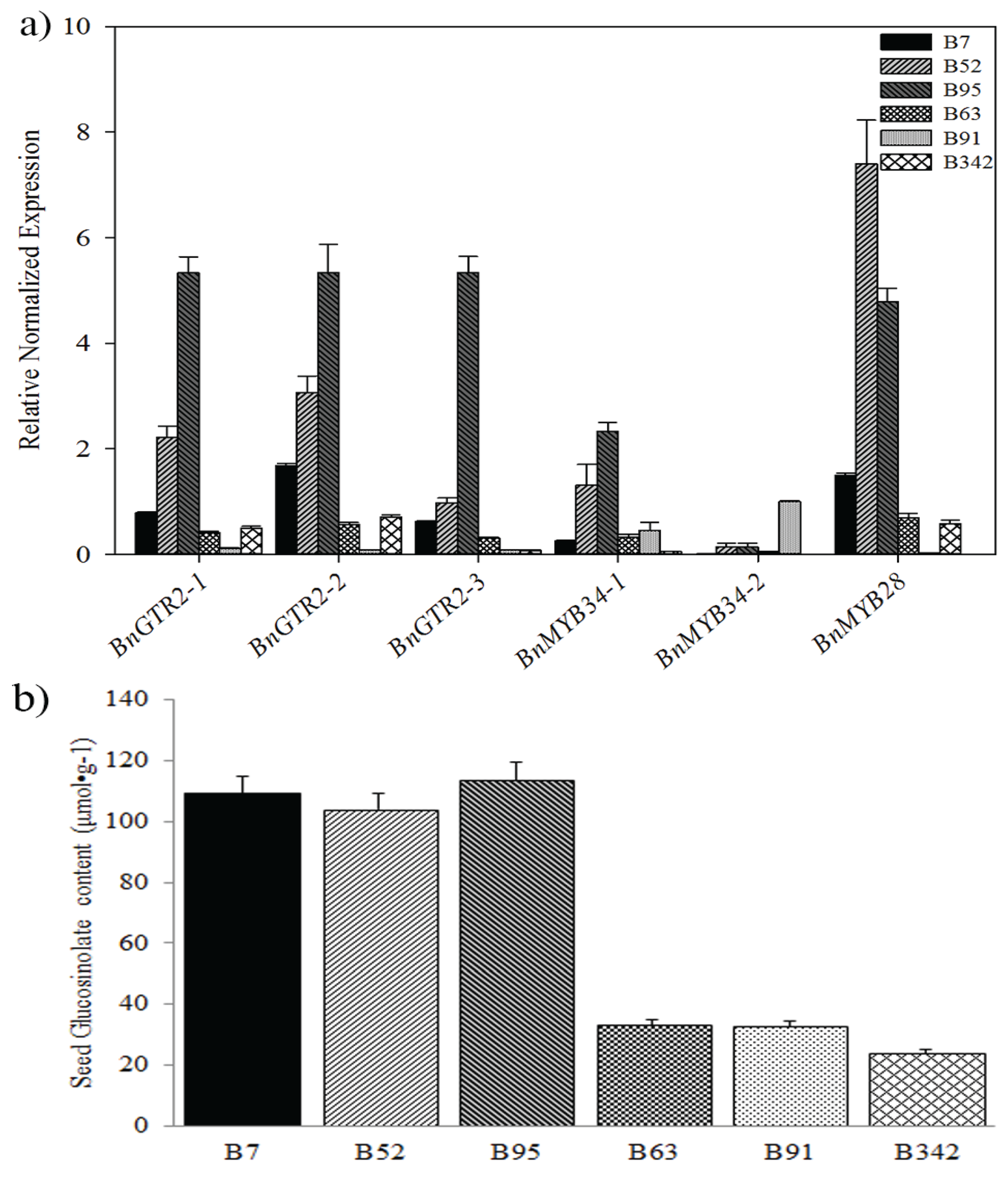
2.4. Identification and Validation of Candidate Genes

| Species | Lf a | MF1 b | MF2 c | Non-Genome Triplication d | AGI NO. | Description |
|---|---|---|---|---|---|---|
| Bra | Bra010111 | Bra029248 | Bra035886 | Bra035885 | AT5G62680 | GLUCOSINOLATE TRANSPORTER-2 |
| Bol | Bol019440 | Bol020699 | Bol019185 | |||
| BnaA | BnaA06g22160D | BnaA02g33530D | BnaA09g06190D | |||
| BnaC | BnaC03g51560D | BnaC02g42260D | BnaC09g05810D | BnaC02g42280D | ||
| Bra | Bra013000 | Bra029350 | Bra035954 | AT5G60890 | myb domain protein 34 (MYB34) | |
| Bol | Bol017062 | Bol007760 | Bol036264 | |||
| BnaA | BnaA03g39790D | BnaA09g05480D | BnaAnng06630D | |||
| BnaC | BnaC02g41860D | BnaC09g05060D | BnaCnng21270D | |||
| Bra | Bra012961 | Bra029311 | Bra035929 | AT5G61420 | myb domain protein 28 (MYB28) | |
| Bol | Bol017019 | Bol007795 | Bol036286 | |||
| BnaA | BnaA03g40190D | |||||
| BnaC | BnaC09g05300D | BnaCnng43220D |
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Phenotypic Data
4.2. DNA Extraction and SNP Analysis
4.3. Population Structure and Relative Kinship Analysis
4.4. Genome-Wide Association Study (GWAS) and Candidate Genes Identification
4.5. Real-time Quantitative PCR (qRT-PCR) Verification of Candidate Genes
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walker, K.C.; Booth, E.J. Agricultural aspects of rape and other Brassica products. Eur. J. Lipid Sci. Technol. 2001, 103, 441–446. [Google Scholar] [CrossRef]
- Naczk, M.; Amarowicz, R.; Sullivan, A.; Shahidi, F. Current research developments on polyphenolics of rapeseed/canola: A review. Food Chem. 1998, 62, 489–502. [Google Scholar] [CrossRef]
- Zhao, J.; Meng, J. Detection of loci controlling seed glucosinolate content and their association with Sclerotinia resistance in Brassica napus. Plant Breed. 2003, 122, 19–23. [Google Scholar] [CrossRef]
- Howell, P.; Sharpe, A.; Lydiate, D. Homoeologous loci control the accumulation of seed glucosinolates in oilseed rape (Brassica napus). Genome 2003, 46, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, M.; Ecke, W.; Weissleder, K.; R bbelen, G. Mapping the genome of rapeseed (Brassica napus L.). I. Construction of an RFLP linkage map and localization of QTLs for seed glucosinolate content. Theor. Appl. Genet. 1995, 90, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.J.; Wei, L.J.; Li, J.N.; Xu, X.F.; Chen, L.; Liu, L.Z. Quantitative traits loci analysis of seed glucosinolate content in Brassica napus using high-density SNP map (In Chinese). Acta Agron. Sin. 2014, 40, 1386–1391. [Google Scholar]
- Xu, J.; Long, Y.; Wu, J.; Xu, H.; Zhao, Z.; Wen, J.; Meng, J.; Shi, C. QTL identification on two genetic systems for rapeseed glucosinolate and erucic acid contents over two seasons. Euphytica 2015, 205, 647–657. [Google Scholar] [CrossRef]
- Basunanda, P.; Spiller, T.; Hasan, M.; Gehringer, A.; Schondelmaier, J.; Lühs, W.; Friedt, W.; Snowdon, R. Marker-assisted increase of genetic diversity in a double-low seed quality winter oilseed rape genetic background. Plant Breed. 2007, 126, 581–587. [Google Scholar] [CrossRef]
- Quijada, P.A.; Udall, J.A.; Lambert, B.; Osborn, T.C. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 1. Identification of genomic regions from winter germplasm. Theor. Appl. Genet. 2006, 113, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, A.T.W.; Niks, R.E.; van den Berg, P.M.M.M.; Stam, P.; van Eeuwijk, F.A. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 2004, 168, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Warburton, M.; Crouch, J. Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci. 2011, 51, 433–449. [Google Scholar] [CrossRef]
- Yan, J.B.; Shah, T.; Warburton, M.L.; Buckler, E.S.; McMullen, M.D.; Crouch, J. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 2009, 4, e8451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrama, H.; Eizenga, G.; Yan, W. Association mapping of yield and its components in rice cultivars. Mol. Breed. 2007, 19, 341–356. [Google Scholar] [CrossRef]
- Meuwissen, T.; Goddard, M. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 2000, 155, 421–430. [Google Scholar] [PubMed]
- Rezaeizad, A.; Wittkop, B.; Snowdon, R.; Hasan, M.; Mohammadi, V.; Zali, A.; Friedt, W. Identification of QTLs for phenolic compounds in oilseed rape (Brassica napus L.) by association mapping using SSR markers. Euphytica 2011, 177, 335–342. [Google Scholar] [CrossRef]
- Zou, J.; Jiang, C.; Cao, Z.; Li, R.; Long, Y.; Chen, S.; Meng, J. Association mapping of seed oil content in Brassica napus and comparison with quantitative trait loci identified from linkage mapping. Genome 2010, 53, 908–916. [Google Scholar] [PubMed]
- Lu, G.; Harper, A.L.; Trick, M.; Morgan, C.; Fraser, F.; O’Neill, C.; Bancroft, I. Associative transcriptomics study dissects the genetic architecture of seed glucosinolate content in Brassica napus. DNA Res. 2014, 21, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, B.; Xu, K.; Wu, J.; Song, W.; Bancroft, I.; Harper, A.L.; Trick, M.; Liu, S.; Gao, G. Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. 2014, 21, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Gajardo, H.A.; Wittkop, B.; Soto-Cerda, B.; Higgins, E.E.; Parkin, I.A.; Snowdon, R.J.; Federico, M.L.; Iniguez-Luy, F.L. Association mapping of seed quality traits in Brassica napus L. using GWAS and candidate QTL approaches. Mol. Breed. 2015, 35, 1–19. [Google Scholar] [CrossRef]
- The Multinational Brassica Genome Project. Available online: http://www.brassica.info/ (accessed on 22 September 2015).
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Fu, F.; Lu, K.; Zhang, K.; Wang, R.; Xu, X.; Wang, M.; Lu, J.; Wan, H.; Zhanglin, T.; et al. Differential accumulation of phenolic compounds and expression of related genes in black-and yellow-seeded Brassica napus. J. Exp. Bot. 2013, 64, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.; Eales, D.; Marshall, D.; Imelfort, M.; Stiller, J.; Berkman, P.J.; Clark, T.; McKenzie, M.; Appleby, N.; Batley, J. Future tools for association mapping in crop plants. Genome 2010, 53, 1017–1023. [Google Scholar] [PubMed]
- Li, F.; Chen, B.; Xu, K.; Gao, G.; Yan, G.; Qiao, J.; Li, J.; Li, H.; Li, L.; Xiao, X. A genome-wide association study of plant height and primary branch number in Rapeseed (Brassica napus). Plant Sci. 2015. [Google Scholar] [CrossRef]
- Luo, X.; Ma, C.; Yue, Y.; Hu, K.; Li, Y.; Duan, Z.; Wu, M.; Tu, J.; Shen, J.; Yi, B. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics 2015. [Google Scholar] [CrossRef] [PubMed]
- Hatzig, S.V.; Frisch, M.; Breuer, F.; Nesi, N.; Ducournau, S.; Wagner, M.-H.; Leckband, G.; Abbadi, A.; Snowdon, R.J. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front. Plant sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jørgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Madsen, S.R.; Olsen, C.E.; Nour-Eldin, H.H.; Halkier, B.A. Elucidating the role of transport processes in leaf glucosinolate distribution. Plant Physiol. 2014, 166, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.G.; Nour-Eldin, H.H.; Fuller, V.L.; Olsen, C.E.; Burow, M.; Halkier, B.A. Integration of biosynthesis and long-distance transport establish organ-specific glucosinolate profiles in vegetative Arabidopsis. Plant Cell Online 2013, 25, 3133–3145. [Google Scholar] [CrossRef] [PubMed]
- Celenza, J.L.; Quiel, J.A.; Smolen, G.A.; Merrikh, H.; Silvestro, A.R.; Normanly, J.; Bender, J. The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 2005, 137, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Gigolashvili, T.; Berger, B.; Mock, H.P.; Müller, C.; Weisshaar, B.; Flügge, U.I. The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 2007, 50, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Skirycz, A.; Reichelt, M.; Burow, M.; Birkemeyer, C.; Rolcik, J.; Kopka, J.; Zanor, M.I.; Gershenzon, J.; Strnad, M.; Szopa, J. DOF transcription factor AtDof1.1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 2006, 47, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Frerigmann, H.; Gigolashvili, T. MYB34, MYB51, and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol. Plant 2014, 7, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Li, X.; Kim, S.-J.; Kim, H.H.; Lee, J.; Kim, H.; Park, S.U. MYB transcription factors regulate glucosinolate biosynthesis in different organs of Chinese cabbage (Brassica rapa ssp. pekinensis). Molecules 2013, 18, 8682–8695. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.Y.; Sugiyama, K.; Sawada, Y.; Tohge, T.; Obayashi, T.; Suzuki, A.; Araki, R.; Sakurai, N.; Suzuki, H.; Aoki, K. Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. 2007, 104, 6478–6483. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Mukhopadhyay, A.; Bisht, N.C. Targeted silencing of BjMYB28 transcription factor gene directs development of low glucosinolate lines in oilseed Brassica juncea. Plant Biotechnol. J. 2013, 11, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Qian, H.; Shen, W.; Liu, L.; Zhang, M.; Cai, C.; Zhao, Y.; Qiao, J.; Wang, Q. BZR1 and BES1 participate in regulation of glucosinolate biosynthesis by brassinosteroids in Arabidopsis. J. Exp. Bot. 2013, 64, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- The Manufacture’s Protocol of BeadChips. Available online: http://www.illumina.com/technology/infinium_hd_assay.ilmn (accessed on 22 September 2015).
- Mackay, I.; Powell, W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007, 12, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- The Arabidopsis Information Resource (TAIR). Available online: http://www.arabidopsis.org/index.jsp (accessed on 22 September 2015).
- Wu, G.; Li, Z.; Yuhua, W.; Yinglong, C.; Changming, L. Comparison of five endogenous reference genes for specific PCR detection and quantification of Brassica napus. J. Agric. Food Chem. 2010, 58, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, C.-M.; Li, S.-M.; Duan, X.-J.; Fan, J.-H.; Jia, L.-D.; Zhao, H.-Y.; Lu, K.; Li, J.-N.; Xu, X.-F.; Wang, R. Identification of Candidate Genes for Seed Glucosinolate Content Using Association Mapping in Brassica napus L. Genes 2015, 6, 1215-1229. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6041215
Qu C-M, Li S-M, Duan X-J, Fan J-H, Jia L-D, Zhao H-Y, Lu K, Li J-N, Xu X-F, Wang R. Identification of Candidate Genes for Seed Glucosinolate Content Using Association Mapping in Brassica napus L. Genes. 2015; 6(4):1215-1229. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6041215
Chicago/Turabian StyleQu, Cun-Min, Shi-Meng Li, Xiu-Jian Duan, Jin-Hua Fan, Le-Dong Jia, Hui-Yan Zhao, Kun Lu, Jia-Na Li, Xin-Fu Xu, and Rui Wang. 2015. "Identification of Candidate Genes for Seed Glucosinolate Content Using Association Mapping in Brassica napus L." Genes 6, no. 4: 1215-1229. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6041215