Triterpenoid Saponin Biosynthetic Pathway Profiling and Candidate Gene Mining of the Ilex asprella Root Using RNA-Seq

Abstract
:1. Introduction
2. Results and Discussion
2.1. RNA-Seq Output, Sequence Assembly and Gene Annotation
2.1.1. Transcriptome Sequencing Output and Sequence Assembly
2.1.2. Gene Expression Overview
2.1.3. Functional Annotation
2.2. Candidate Genes Involved in the Biosynthesis of Triterpenoid Saponins
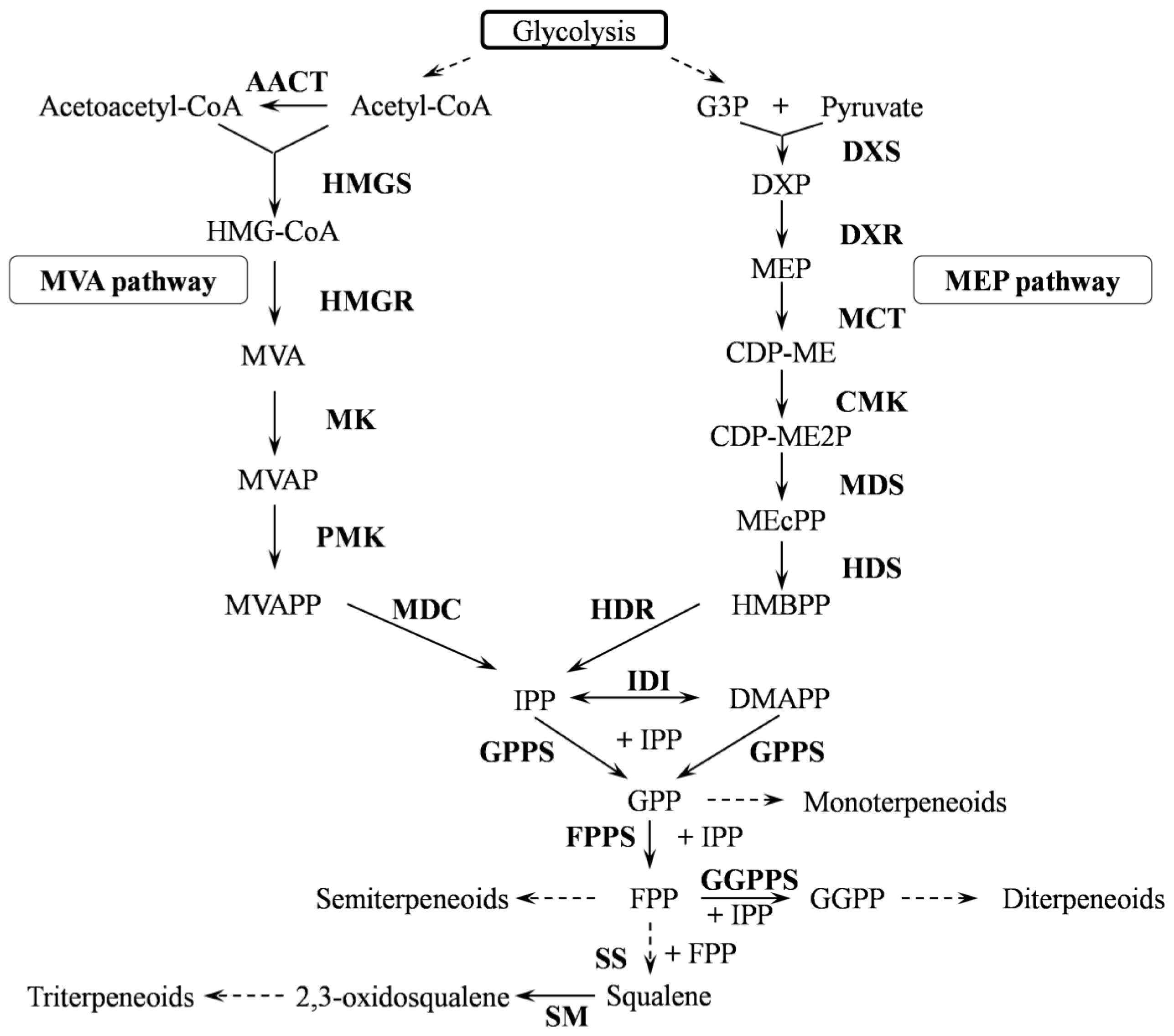
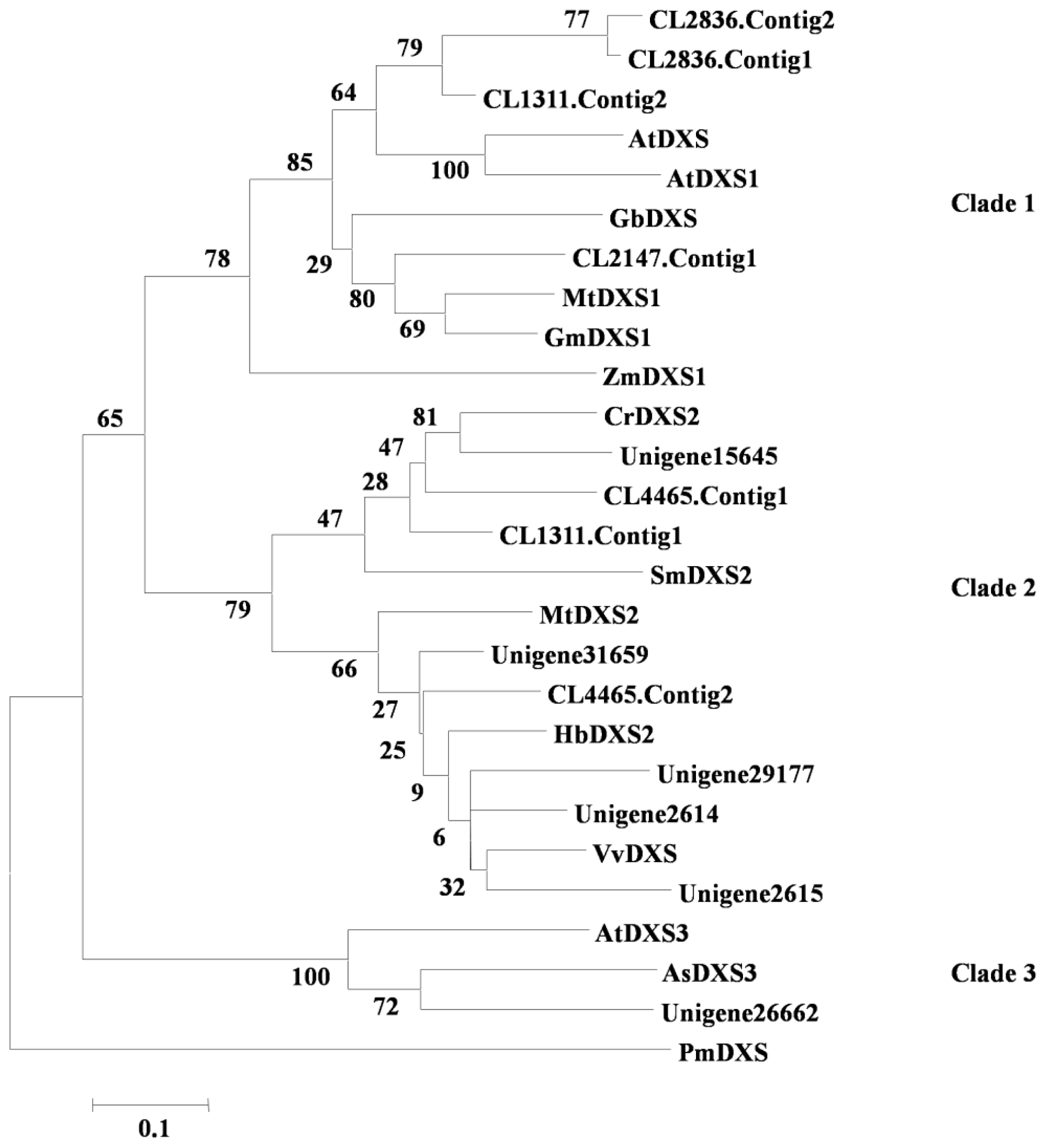
2.2.1. Terpenoid Backbone Biosynthesis
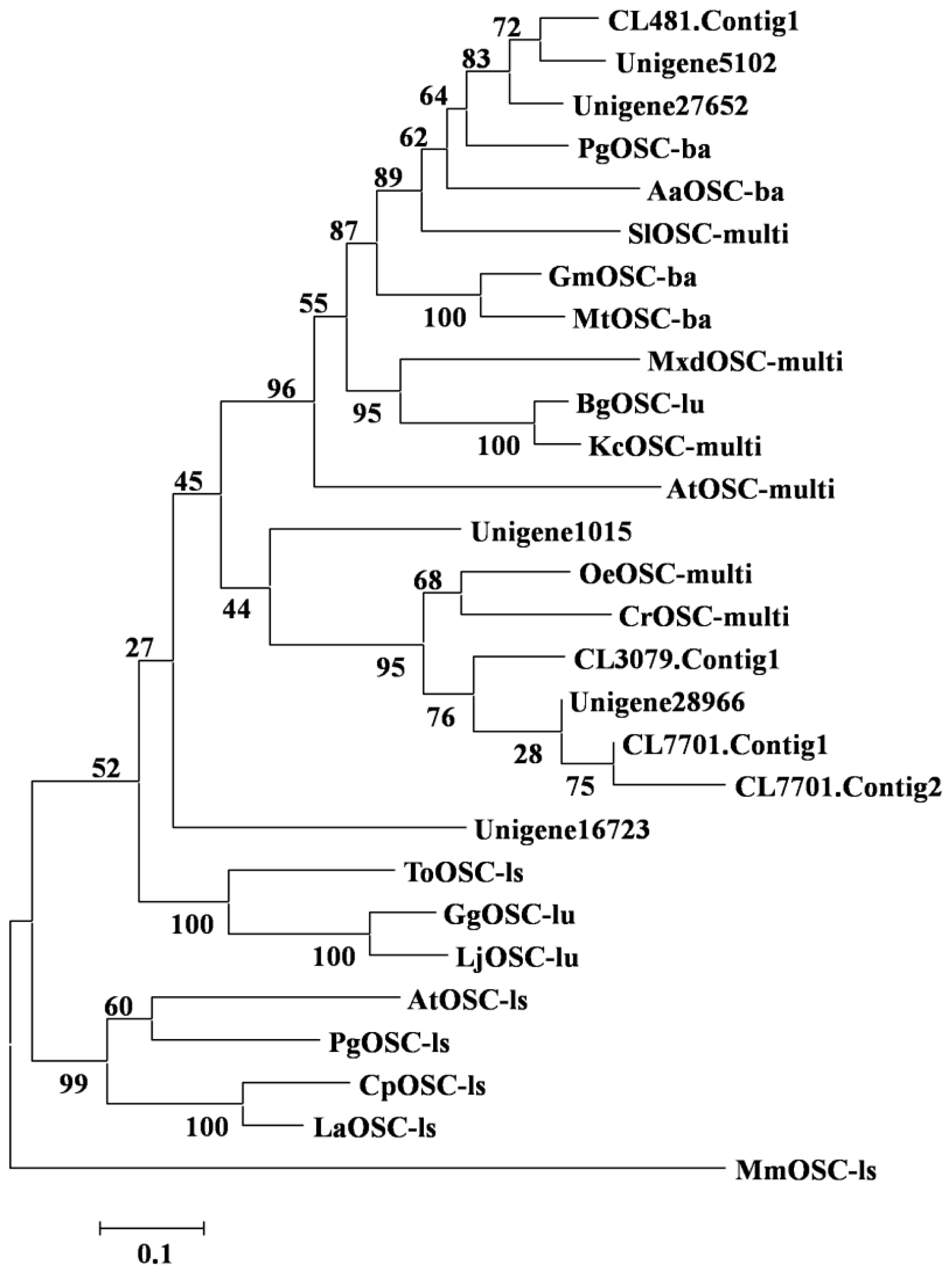
2.2.2. Amyrin Synthaes
2.2.3. P450s
2.2.4. UGTs
2.3. Discussion
3. Experimental Section
3.1. Plant Material and RNA Preparation
3.2. cDNA Synthesis and Sequencing
3.3. Sequence Quality Control and Cleaning
3.4. Sequence Assembly
3.5. Gene Annotation
3.6. Gene Expression Analysis
3.7. Homology-Based Gene Discovery
3.8. Phylogenetic Analysis
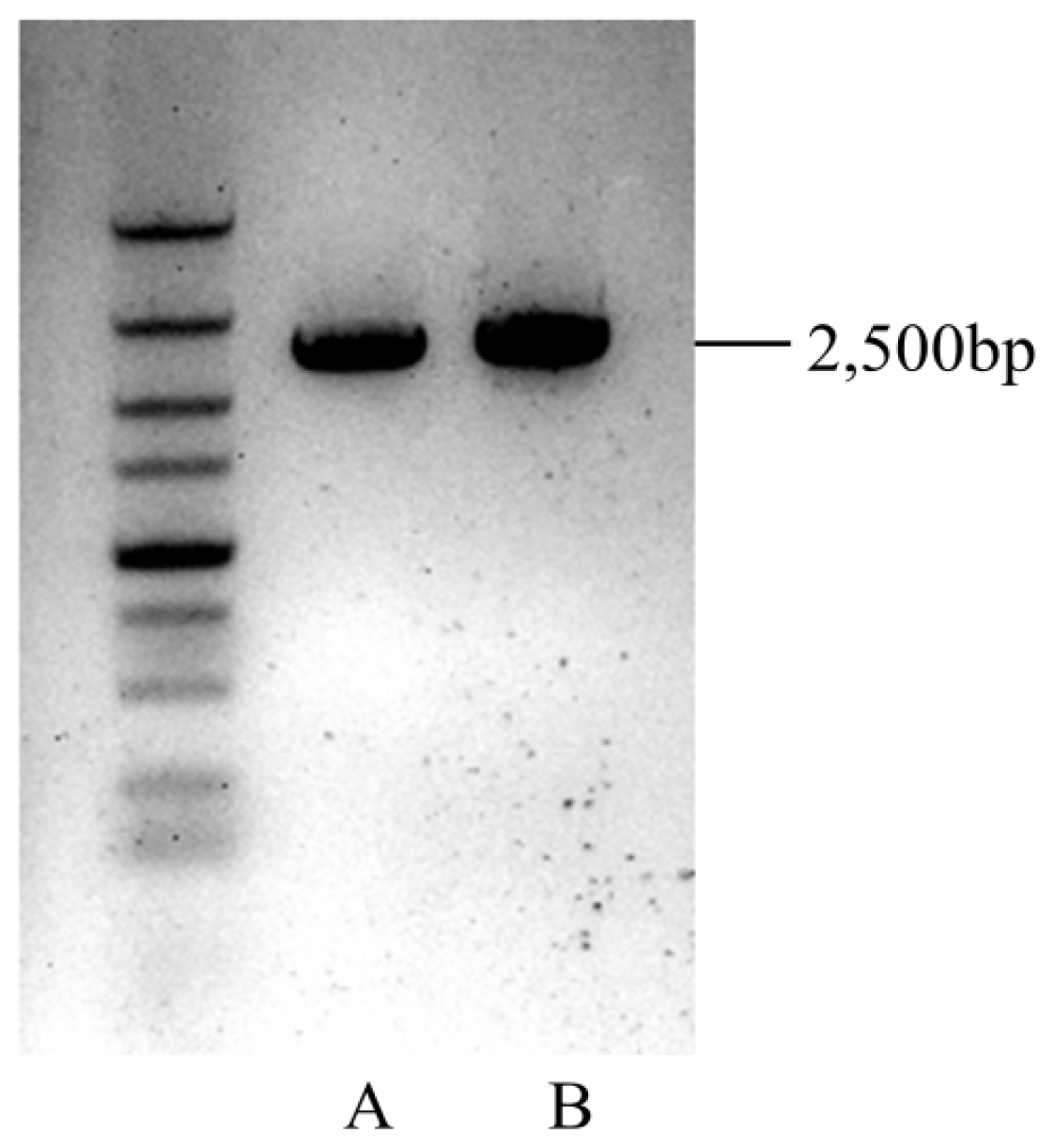
3.9. Amplification of 2 OSC Genes
- for CL3079.contig1, Forward: TCTCTCTGTGTTTATGGGTA (5′→3′) and reverse: GAACACTGAAGGATACAAAC (5′→3′).
- for CL481.contig1, Forward: GCCACAGTTATCTTCGTATT (5′→3′), and reverse: CATACTTCAAGGACCTCAAA (5′→3′).
4. Conclusions
Supplementary Information
ijms-15-05970-s001.pdfAcknowledgments
Conflicts of Interest
- Author ContributionsXiasheng Zheng contributed to the tissue sample collection, RNA extraction, data analysis and writing of this manuscript. Hui Xu carried out the construction of the cDNA library and helped with phylogenetic analysis and conserved motif analysis of ASs. Xinye Ma participated in the PCR and sequence alignment. Ruoting Zhan and Weiwen Chen conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- Sun, S.X.; Li, Y.M.; Fang, W.R.; Cheng, P.; Liu, L.; Li, F. Effect and mechanism of AR-6 in experimental rheumatoid arthritis. Clin. Exp. Med 2010, 10, 113–121. [Google Scholar]
- Man, S.; Gao, W.; Zhang, Y.; Huang, L.; Liu, C. Chemical study and medical application of saponins as anti-cancer agents. Fitoterapia 2010, 81, 703–714. [Google Scholar]
- Saleem, M.; Nazir, M.; Ali, M.S.; Hussain, H.; Lee, Y.S.; Riaz, N.; Jabbar, A. Antimicrobial natural products: An update on future antibiotic drug candidates. Nat. Prod. Rep 2010, 27, 238–254. [Google Scholar]
- Suzuki, H.; Achnine, L.; Xu, R.; Matsuda, S.P.; Dixon, R.A. A genomics approach to the early stages of triterpene saponin biosynthesis inMedicago truncatula. Plant J 2002, 32, 1033–1048. [Google Scholar]
- Sparg, S.; Light, M.; van Staden, S.J. Biological activities and distribution of plant saponis. J. Ethnopharmacol 2004, 94, 219–243. [Google Scholar]
- Kashiwada, Y.; Zhang, D.C.; Chen, Y.P.; Cheng, C.M.; Chen, H.T.; Chang, H.C.; Chang, J.J.; Lee, K.H. Antitumor agents, 145. Cytotoxic asprellic acids A and C and asprellic acid B. new p-coumaroyl triterpenes, from Ilex asprella. J. Nat. Prod 1993, 56, 2077–2082. [Google Scholar]
- Zhou, M.; Xu, M.; Ma, X.X.; Zheng, K.; Yang, K.; Yang, C.R.; Wang, Y.F.; Zhang, Y.J. Antiviral triterpenoid saponins from the roots ofIlex asprella. Planta Med 2012, 78, 1702–1705. [Google Scholar]
- Cai, Y.; Zhang, Q.; Li, Z.; Fan, C.; Wang, L.; Zhang, X.; Ye, W. Chemical constituents from roots ofIlex asprella. Chin. Tradit. Herb. Drugs 2010, 41, 1426–1429. [Google Scholar]
- Wang, L.; Cai, Y.; Zhang, X.Q.; Fan, C.L.; Zhang, Q.W.; Lai, X.P.; Ye, W.C. New triterpenoid glycosides from the roots ofIlex asprella. Carbohydr. Res 2012, 349, 39–43. [Google Scholar]
- Huang, J.; Chen, F.; Chen, H.; Zeng, Y.; Xu, H. Chemical constituents in roots ofIlex asprella. Chin. Tradit. Herb. Drugs 2012, 43, 1475–1478. [Google Scholar]
- Zhao, Z.X.; Lin, C.Z.; Zhu, C.C.; He, W.J. A new triterpenoid glycoside from the roots ofIlex asprella. Chin. J. Nat. Med 2013, 11, 415–418. [Google Scholar]
- Sun, H.; Fang, W.-S.; Wang, W.-Z.; Hu, C. Structure-activity relationships of oleanane- and ursane-type triterpenoids. Bot. Stud 2006, 47, 339–368. [Google Scholar]
- Chung, E.; Cho, C.-W.; Kim, K.-Y.; Chung, J.; Kim, J.-I.; Chung, Y.-S.; Fukui, K.; Lee, J.-H. Molecular characterization of the GmAMS1 gene encoding β-amyrin synthase in soybean plants. Russ. J. Plant Physiol 2007, 54, 518–523. [Google Scholar]
- Lee, M.H.; Jeong, J.H.; Seo, J.W.; Shin, C.G.; Kim, Y.S.; In, J.G.; Yang, D.C.; Yi, J.S.; Choi, Y.E. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 2004, 45, 976–984. [Google Scholar]
- Laule, O.; Furholz, A.; Chang, H.S.; Zhu, T.; Wang, X.; Heifetz, P.B.; Gruissem, W.; Lange, M. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis inArabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2003, 100, 6866–6871. [Google Scholar]
- Strickler, S.R.; Bombarely, A.; Mueller, L.A. Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am. J. Bot 2012, 99, 257–266. [Google Scholar]
- Schliesky, S.; Gowik, U.; Weber, A.P.; Brautigam, A. RNA-seq assembly—Are we there yet? Front. Plant Sci 2012, 3, 220. [Google Scholar]
- Dewick, P.W. Medicinal Natural Products: A Biosynthetic Approach, 2nd ed; John Wiley & Sons, Ltd: Chichester, UK, 2002; p. 169. [Google Scholar]
- Cordoba, E.; Porta, H.; Arroyo, A.; San Roman, C.; Medina, L.; Rodriguez-Concepcion, M.; Leon, P. Functional characterization of the three genes encoding 1-deoxy-d-xylulose-5-phosphate synthase in maize. J. Exp. Bot 2011, 62, 2023–2038. [Google Scholar]
- Abe, I. Enzymatic synthesis of cyclic triterpenes. Nat. Prod. Rep 2007, 24, 1311–1331. [Google Scholar]
- Xu, R.; Fazio, G.C.; Matsuda, S.P. On the origins of triterpenoid skeletal diversity. Phytochemistry 2004, 65, 261–291. [Google Scholar]
- Phillips, D.R.; Rasbery, J.M.; Bartel, B.; Matsuda, S.P. Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Biol 2006, 9, 305–314. [Google Scholar]
- Wu, Y.; Zou, H.D.; Cheng, H.; Zhao, C.Y.; Sun, L.F.; Su, S.Z.; Li, S.P.; Yuan, Y.P. Cloning and characterization of a β-amyrin synthase gene from the medicinal tree Aralia elata (Araliaceae). Genet. Mol. Res 2012, 11, 2301–2314. [Google Scholar]
- Poralla, K.; Hewelt, A.; Prestwich, G.D.; Abe, I.; Reipen, I.; Sprenger, G. A specific amino acid repeat in squalene and oxidosqualene cyclases. Trends Biochem. Sci 1994, 19, 157–158. [Google Scholar]
- Abe, I.; Prestwich, G.D. Active site mapping of affinity labeled rat oxidosqualene cyclase. J. Biol. Chem 1994, 269, 802–804. [Google Scholar]
- Kushiro, T.; Shibuya, M.; Masuda, K.; Ebizuka, Y. Mutational studies on triterpene synthases: Engineering lupeol synthase into β-amyrin synthase. J. Am. Chem. Soc 2000, 122, 6816–6824. [Google Scholar]
- Fukushima, E.O.; Seki, H.; Ohyama, K.; Ono, E.; Umemoto, N.; Mizutani, M.; Saito, K.; Muranaka, T. CYP716A subfamily members are multifunctional oxidases in triterpenoid biosynthesis. Plant Cell Physiol 2011, 52, 2050–2061. [Google Scholar]
- Carelli, M.; Biazzi, E.; Panara, F.; Tava, A.; Scaramelli, L.; Porceddu, A.; Graham, N.; Odoardi, M.; Piano, E.; Arcioni, S.; et al. Medicago truncatula CYP716A12 is a multifunctional oxidase involved in the biosynthesis of hemolytic saponins. Plant Cell 2011, 23, 3070–3081. [Google Scholar]
- Paquette, S.; Moller, B.L.; Bak, S. On the origin of family 1 plant glycosyltransferases. Phytochemistry 2003, 62, 399–413. [Google Scholar]
- Wortman, J.R.; Haas, B.J.; Hannick, L.I.; Smith, R.K., Jr.; Maiti, R.; Ronning, C.M.; Chan, A.P.; Yu, C.; Ayele, M.; Whitelaw, C.A.; et al. Annotation of the Arabidopsis genome. Plant Physiol 2003, 132, 461–468. [Google Scholar]
- Shibuya, M.; Hoshino, M.; Katsube, Y.; Hayashi, H.; Kushiro, T.; Ebizuka, Y. Identification of beta-amyrin and sophoradiol 24-hydroxylase by expressed sequence tag mining and functional expression assay. FEBS J 2006, 273, 948–959. [Google Scholar]
- Seki, H.; Ohyama, K.; Sawai, S.; Mizutani, M.; Ohnishi, T.; Sudo, H.; Akashi, T.; Aoki, T.; Saito, K.; Muranaka, T. Licorice β-amyrin 11-oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc. Natl. Acad. Sci. USA 2008, 105, 14204–14209. [Google Scholar]
- Huang, L.; Li, J.; Ye, H.; Li, C.; Wang, H.; Liu, B.; Zhang, Y. Molecular characterization of the pentacyclic triterpenoid biosynthetic pathway in Catharanthus roseus. Planta 2012, 236, 1571–1581. [Google Scholar]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genomics 2009, 4, 59–65. [Google Scholar]
- Mackenzie, P.I.; Owens, I.S.; Burchell, B.; Bock, K.W.; Bairoch, A.; Belanger, A.; Fournel-Gigleux, S.; Green, M.; Hum, D.W.; Iyanagi, T.; et al. The UDP glycosyltransferase gene superfamily: Recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 1997, 7, 255–269. [Google Scholar]
- Augustin, J.M.; Drok, S.; Shinoda, T.; Sanmiya, K.; Nielsen, J.K.; Khakimov, B.; Olsen, C.E.; Hansen, E.H.; Kuzina, V.; Ekstrom, C.T.; et al. UDP-glycosyltransferases from the UGT73C subfamily in Barbarea vulgaris catalyze sapogenin 3-O-glucosylation in saponin-mediated insect resistance. Plant Physiol 2012, 160, 1881–1895. [Google Scholar]
- Naoumkina, M.A.; Modolo, L.V.; Huhman, D.V.; Urbanczyk-Wochniak, E.; Tang, Y.; Sumner, L.W.; Dixon, R.A. Genomic and coexpression analyses predict multiple genes involved in triterpene saponin biosynthesis in Medicago truncatula. Plant Cell 2010, 22, 850–866. [Google Scholar]
- Meessapyodsuk, D.; Balsevich, J.; Reed, D.W.; Covello, P.S. Saponin biosynthesis in Saponaria vaccaria. cDNAs encoding beta-amyrin synthase and a triterpene carboxylic acid glucosyltransferase. Plant Physiol 2007, 143, 959–969. [Google Scholar]
- Augustin, J.M.; Kuzina, V.; Andersen, S.B.; Bak, S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry 2011, 72, 435–457. [Google Scholar]
- Achnine, L.; Huhman, D.V.; Farag, M.A.; Sumner, L.W.; Blount, J.W.; Dixon, R.A. Genomics-based selection and functional characterization of triterpene glycosyltransferases from the model legumeMedicago truncatula. Plant J 2005, 41, 875–887. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 2011, 29, 644–652. [Google Scholar]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol 1999, 138–148. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res 2006, 34, W293–W297. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol 2013, 30, 2725–2729. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triterpene skeleton | R1 | R2 | R3 | R4 | R5 | R6 | References |
|---|---|---|---|---|---|---|---|
| A * | H | H | CH3 | H | - | - | [8] |
| Xyl | Glc | CH3 | H | - | - | [8] | |
| Xyl | Glc | H | CH3 | - | - | [8] | |
| Glucuronic | Glc | H | CH3 | - | - | [9] | |
| Glucuronic | Glc | CH3 | H | - | - | [9] | |
| B | H | H | - | - | - | - | [8] |
| Xyl | Glc | - | - | - | - | [8] | |
| Glucuronic | Glc | - | - | - | - | [9] | |
| C | Glucuronic acid methyl ester | Glucuronic | - | - | - | - | [9] |
| D | H | H | - | - | CH3 | CH3 | [10] |
| H | Glc | - | - | CH3 | CH3 | [8] | |
| SO3Na | Glc | - | - | CH3 | CH3 | [8] | |
| Xyl | H | - | - | CH3 | CH3 | [8] | |
| Xyl | Glc | - | - | CH3 | CH3 | [8] | |
| Glucuronic | H | - | - | CH3 | CH3 | [9] | |
| Glucuronic-3-OSO3Na | Glc | - | - | CH3 | CH3 | [9] | |
| Glucuronic-3-OSO3Na | H | - | - | CH3 | CH3 | [9] | |
| Glucuronic | Glc | - | - | CH3 | CH3 | [9] | |
| Xyl-3-OSO3H | Glc | - | - | CH3 | CH3 | [7] | |
| Xyl-3-OSO3H | H | - | - | CH3 | CH3 | [7] | |
| Xyl(2-1)Glc(2-1)Rha | H | - | - | CH3 | CH3 | [7] | |
| Ara | Glc | - | - | CH3 | CH3 | [7] | |
| Xyl | Glc | - | - | CH3 | CH3 | [7] | |
| Ara(2-1)Glc | H | - | - | CH3 | CH3 | [7] | |
| H | Glc | - | - | CH3 | COOH | [7] | |
| H | H | - | - | CH3 | COOH | [7] | |
| Xyl | Glc | - | - | CH2OH | CH3 | [7] | |
| E | H | - | - | - | - | - | [10] |
| Xyl | - | - | - | - | - | [10] | |
| Value of FPKM | Count | Proportion/% |
|---|---|---|
| >0 | 50,879 | 98.10 |
| >1 | 46,426 | 89.51 |
| >10 | 12,650 | 24.39 |
| >100 | 1475 | 2.84 |
| >1000 | 102 | 0.20 |
| Public database | No. of matched unigenes | Annotation percentage/% |
|---|---|---|
| Nr | 37,674 | 72.64 |
| Nt | 32,994 | 63.62 |
| Swiss-Prot | 22,661 | 43.69 |
| KEGG | 20,752 | 40.01 |
| COG | 12,860 | 24.80 |
| GO | 29,375 | 56.64 |
| Total | 39,269 | 75.71 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zheng, X.; Xu, H.; Ma, X.; Zhan, R.; Chen, W. Triterpenoid Saponin Biosynthetic Pathway Profiling and Candidate Gene Mining of the Ilex asprella Root Using RNA-Seq. Int. J. Mol. Sci. 2014, 15, 5970-5987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms15045970
Zheng X, Xu H, Ma X, Zhan R, Chen W. Triterpenoid Saponin Biosynthetic Pathway Profiling and Candidate Gene Mining of the Ilex asprella Root Using RNA-Seq. International Journal of Molecular Sciences. 2014; 15(4):5970-5987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms15045970
Chicago/Turabian StyleZheng, Xiasheng, Hui Xu, Xinye Ma, Ruoting Zhan, and Weiwen Chen. 2014. "Triterpenoid Saponin Biosynthetic Pathway Profiling and Candidate Gene Mining of the Ilex asprella Root Using RNA-Seq" International Journal of Molecular Sciences 15, no. 4: 5970-5987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms15045970