Genome-Wide Transcriptome Profiles of Rice Hybrids and Their Parents

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Alignment of RNA-seq Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | # of Reads | Length of Reads | # of Bases | # of Uniquely Mapped Reads | Percentage of Uniquely Mapped Reads |
|---|---|---|---|---|---|
| NPB | 77,019,774 | 50 | 3,850,988,700 | 55,413,403 | 71.95% |
| 9311 | 74,384,043 | 50 | 3,719,202,150 | 58,116,517 | 78.13% |
| NPB × 93-11 | 81,735,674 | 50 | 4,086,783,700 | 68,473,811 | 83.77% |
| 93-11 × NPB | 78,026,146 | 50 | 3,901,307,300 | 66,122,696 | 84.74% |

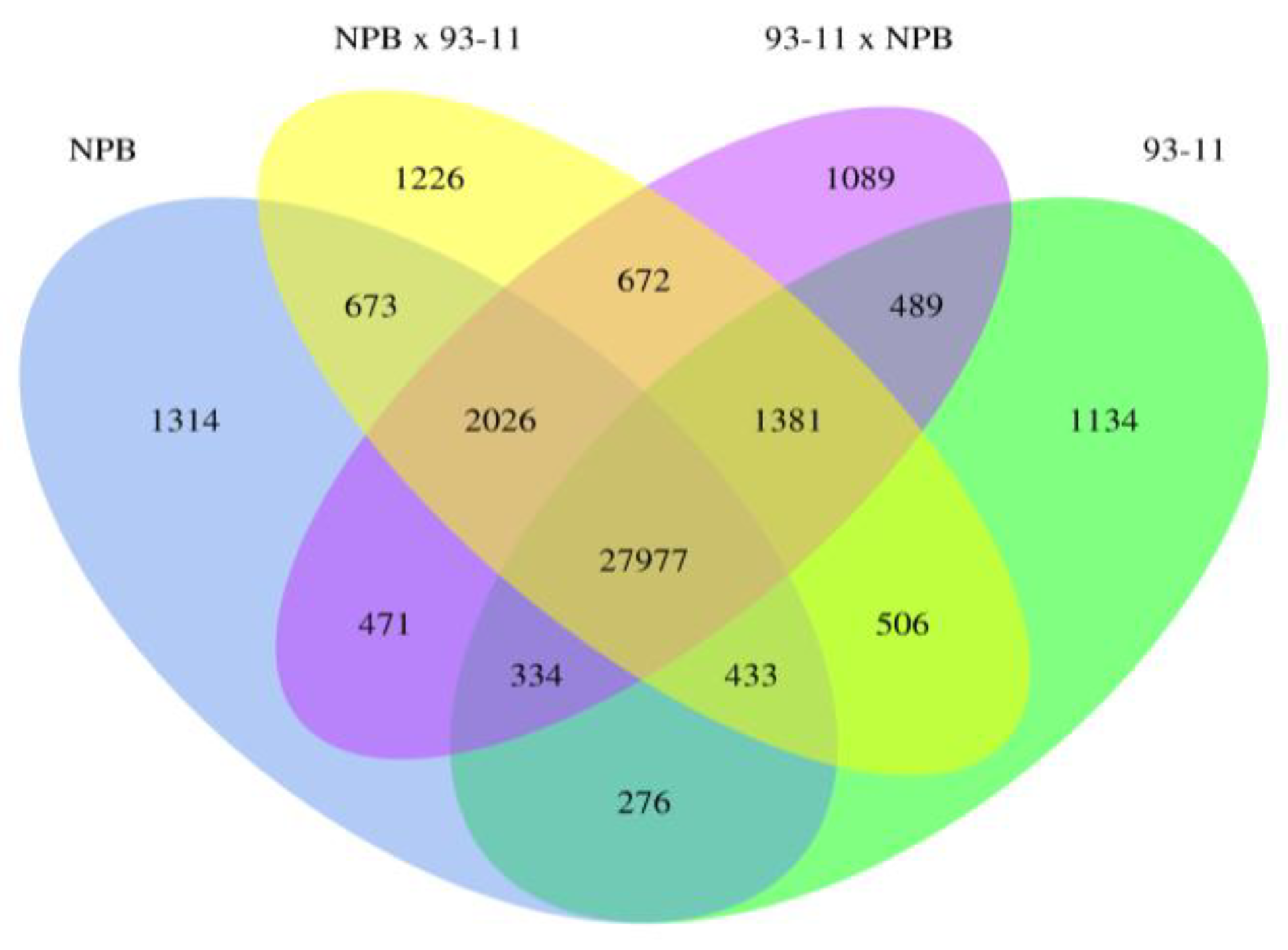
2.1.2. Global Analysis of Gene Expression

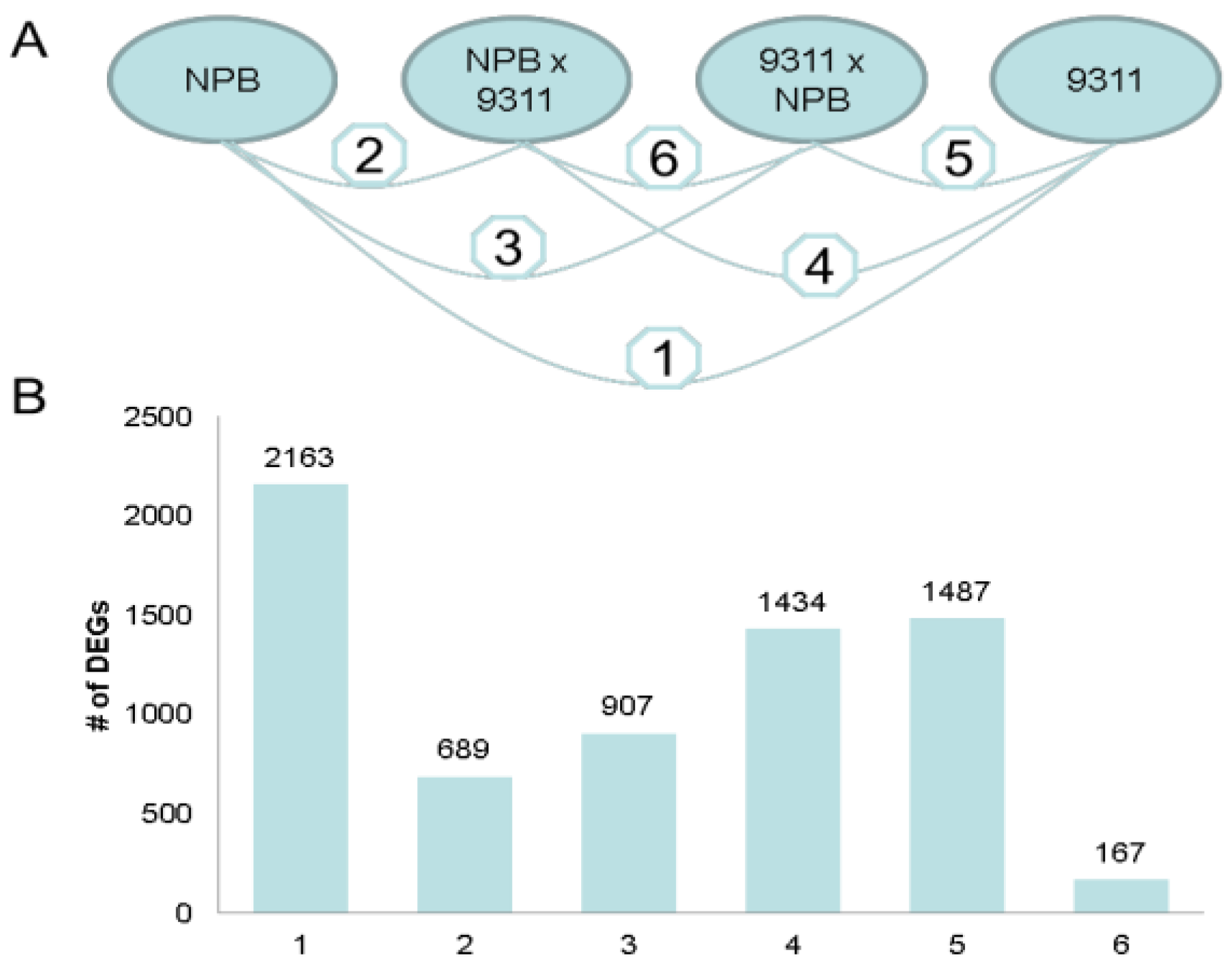
2.1.3. Parental Influence on Hybrids in Rice
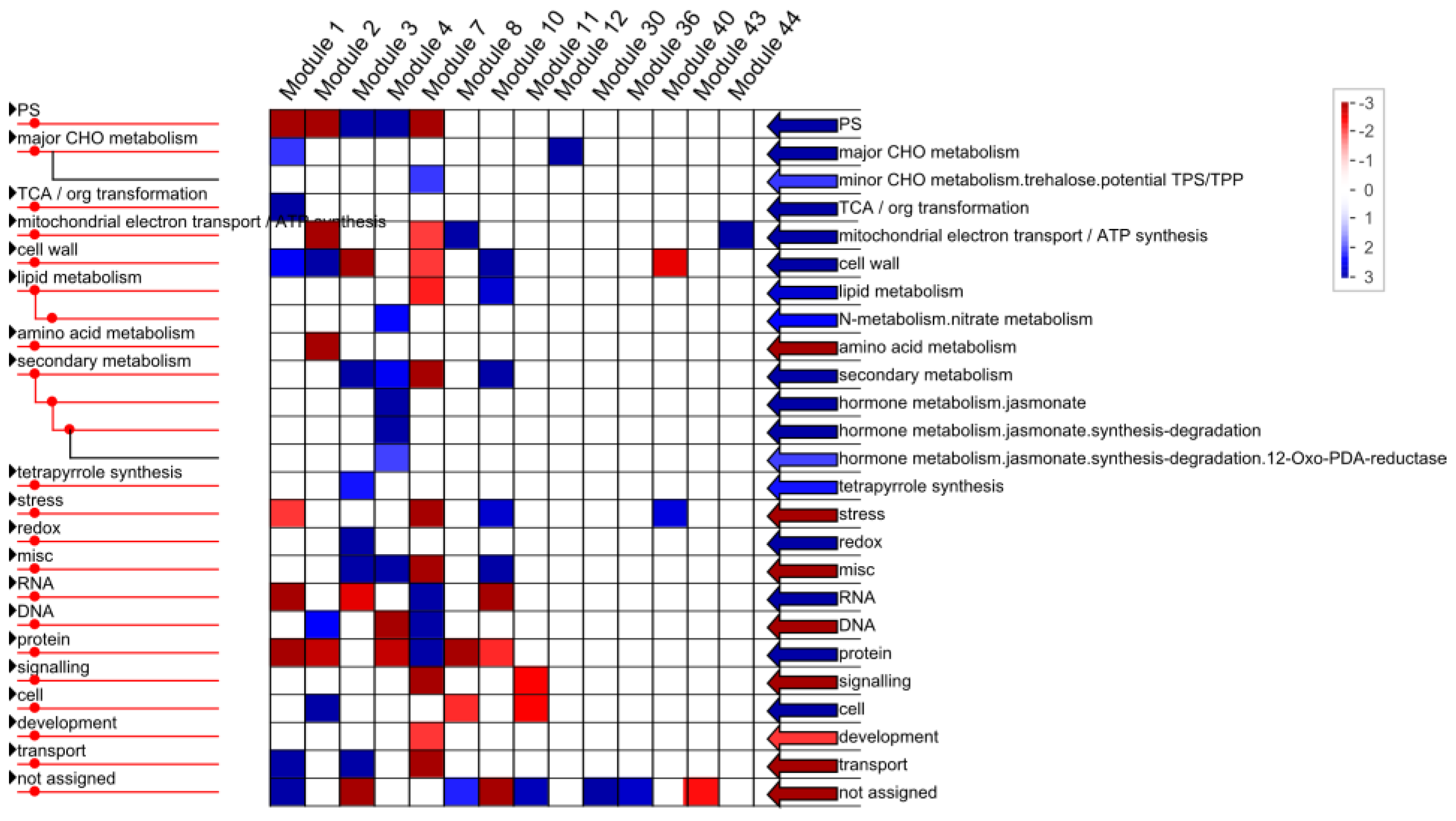
2.1.4. Functional Annotation of Differentially Expressed Genes (DEGs) by Pairwise Comparisons



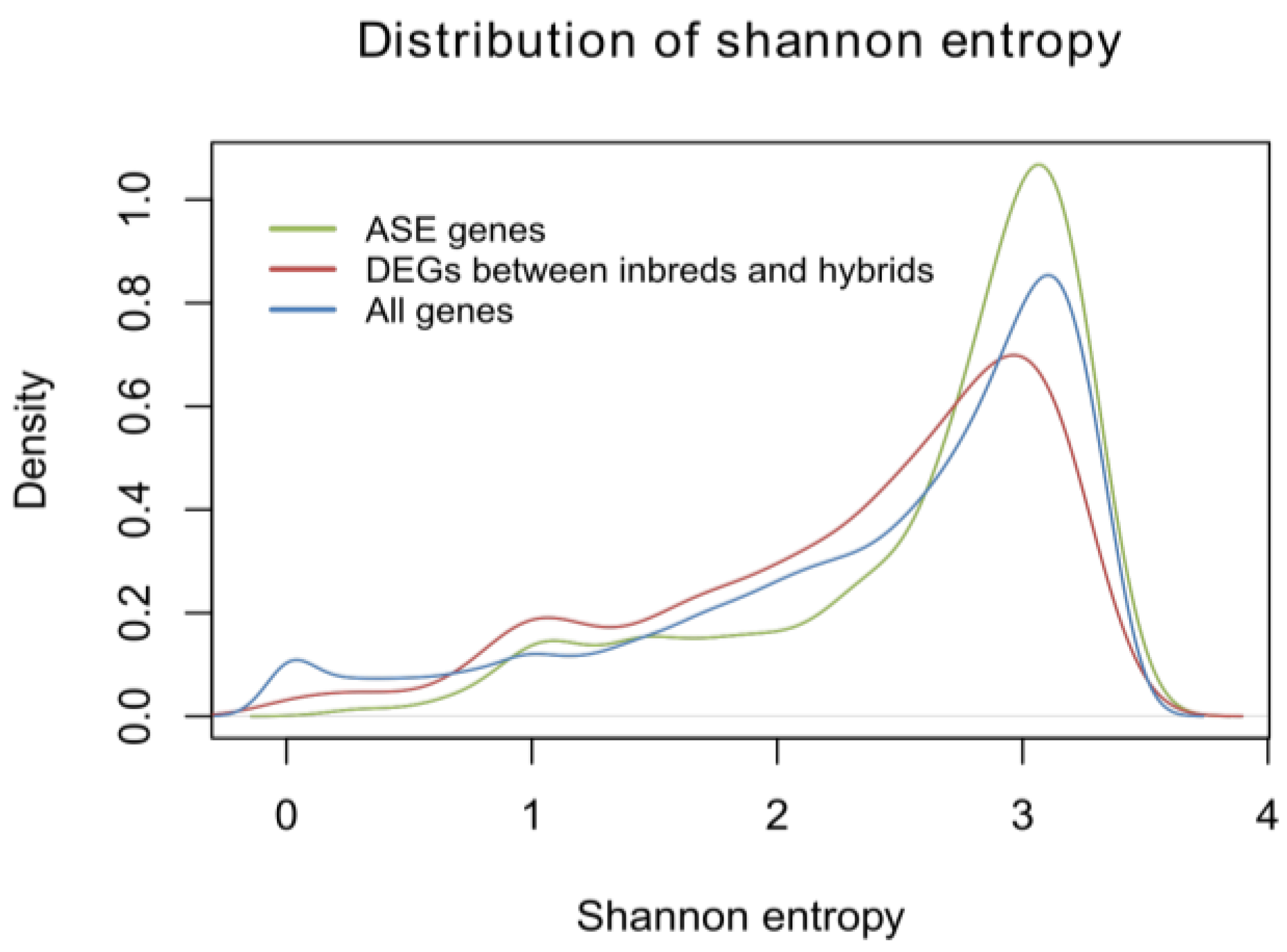
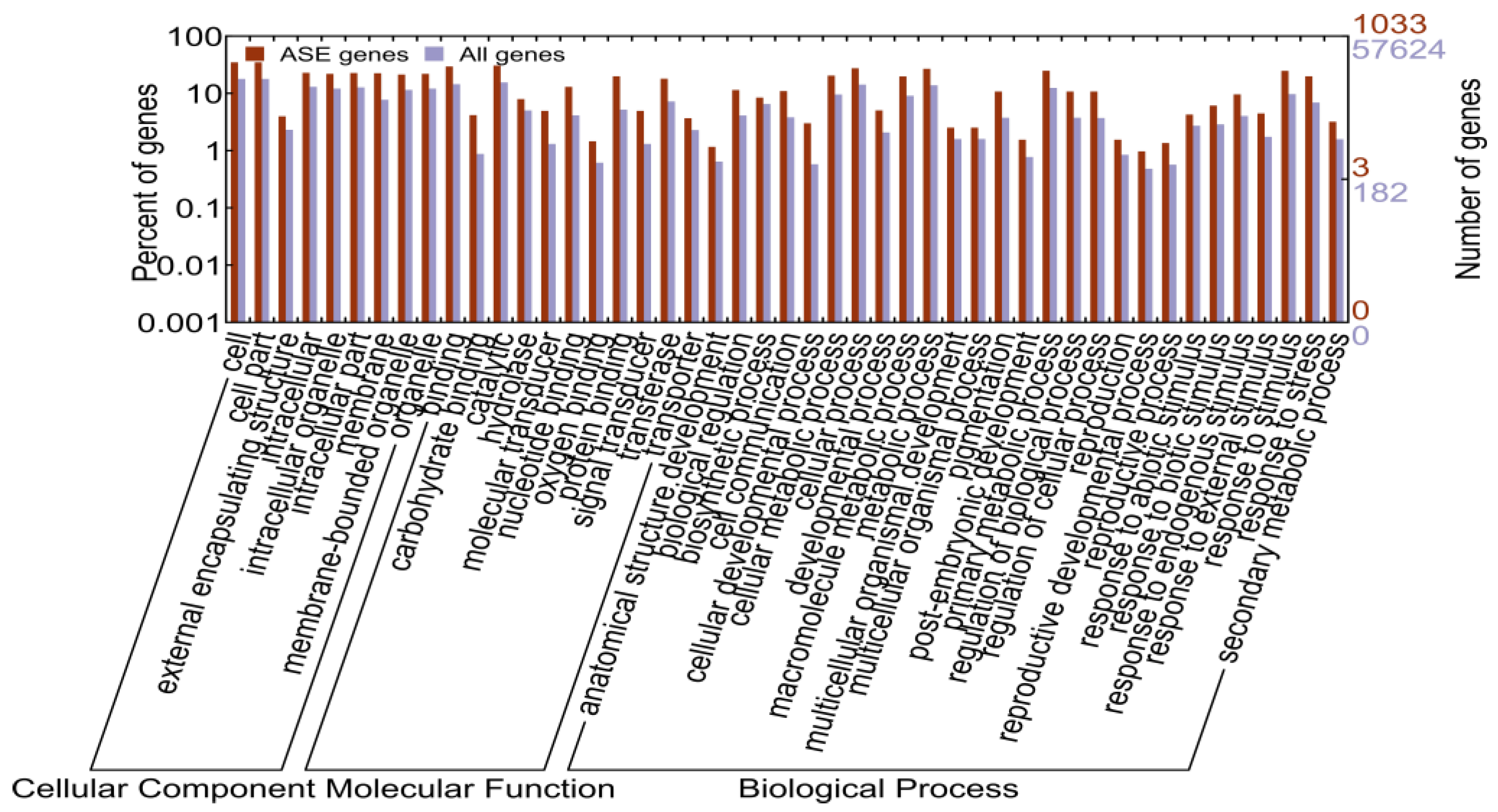
2.1.5. Identification of Allele-Specific Expressed Genes

2.1.6. Allele-Specific Expressed Transcription Factors
2.2. Discussion
3. Materials and Methods
3.1. RNA-seq Data
3.2. Alignment of RNA-seq Data
3.3. Identification of Differentially Expressed Genes
3.4. Identification of Allele-Specific Expressed Genes and Imprinted Genes
3.5. Calculation of Fragments per Kilobase of Transcripts per Million Mapped Reads (FPKM)
3.6. Construction of Co-Expression Network
3.7. Mapman
3.8. Calculation of Shannon Entropy
3.9. Gene Ontology Classification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar] [CrossRef]
- Song, G.-S.; Zhai, H.-L.; Peng, Y.-G.; Zhang, L.; Wei, G.; Chen, X.-Y.; Xiao, Y.-G.; Wang, L.; Chen, Y.-J.; Wu, B.; et al. Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol. Plant 2010, 3, 1012–1025. [Google Scholar] [CrossRef]
- Zhai, R.; Feng, Y.; Wang, H.; Zhan, X.; Shen, X.; Wu, W.; Zhang, Y.; Chen, D.; Dai, G.; Yang, Z.; et al. Transcriptome analysis of rice root heterosis by RNA-seq. BMC Genomics 2013, 14, 19. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar] [CrossRef]
- Scotti, C.; Carelli, M.; Calderini, O.; Panara, F.; Gaudenzi, P.; Biazzi, E.; May, S.; Graham, N.; Paolocci, F.; Arcioni, S. Agronomic and molecular analysis of heterosis in alfalfa. Plant Genet. Resour. 2011, 9, 288–290. [Google Scholar] [CrossRef]
- Paschold, A.; Jia, Y.; Marcon, C.; Lund, S.; Larson, N.B.; Yeh, C.-T.; Ossowski, S.; Lanz, C.; Nettleton, D.; Schnable, P.S.; et al. Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res. 2012, 22, 2445–2454. [Google Scholar] [CrossRef]
- Zhai, R.; Feng, Y.; Zhan, X.; Shen, X.; Wu, W.; Yu, P.; Zhang, Y.; Chen, D.; Wang, H.; Lin, Z.; et al. Identification of transcriptome SNPs for assessing allele-specific gene expression in a super-hybrid rice Xieyou9308. PLoS One 2013, 8, e60668. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar]
- Luo, M.; Taylor, J.M.; Spriggs, A.; Zhang, H.; Wu, X.; Russell, S.; Singh, M.; Koltunow, A. A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Schug, J.; Schuller, W.P.; Kappen, C.; Salbaum, J.M.; Bucan, M.; Stoeckert, C.J. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005, 6, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Sabar, M.; Gagliardi, D.; Balk, J.; Leaver, C.J. ORFB is a subunit of F1F(O)-ATP synthase: Insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003, 4, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; He, H.; Chen, L.-B.; Li, L.; Liang, M.-Z.; Wang, X.-F.; Liu, X.-G.; He, G.-M.; Chen, R.-S.; Ma, L.-G.; et al. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 2008, 1, 720–731. [Google Scholar] [CrossRef]
- Ni, Z.; Kim, E.-D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Rupe, M.A.; Danilevskaya, O.N.; Yang, X.; Hu, Z. Genome-wide mRNA profiling reveals heterochronic allelic variation and a new imprinted gene in hybrid maize endosperm. Plant J. 2003, 36, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Swanson-Wagner, R.A.; DeCook, R.; Jia, Y.; Bancroft, T.; Ji, T.; Zhao, X.; Nettleton, D.; Schnable, P.S. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 2009, 326, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010, 15, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR rice genome annotation resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, D1114–D1117. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M.; Gowda, M.; Moghe, G.; Lin, H.; Vaillancourt, B.; Shiu, S.-H.; Jiang, N.; Buell, C.R. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012, 71, 492–502. [Google Scholar] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
E, Z.; Huang, S.; Zhang, Y.; Ge, L.; Wang, L. Genome-Wide Transcriptome Profiles of Rice Hybrids and Their Parents. Int. J. Mol. Sci. 2014, 15, 20833-20845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151120833
E Z, Huang S, Zhang Y, Ge L, Wang L. Genome-Wide Transcriptome Profiles of Rice Hybrids and Their Parents. International Journal of Molecular Sciences. 2014; 15(11):20833-20845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151120833
Chicago/Turabian StyleE, Zhiguo, Shanshan Huang, Yuping Zhang, Lei Ge, and Lei Wang. 2014. "Genome-Wide Transcriptome Profiles of Rice Hybrids and Their Parents" International Journal of Molecular Sciences 15, no. 11: 20833-20845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151120833