
FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP

Abstract
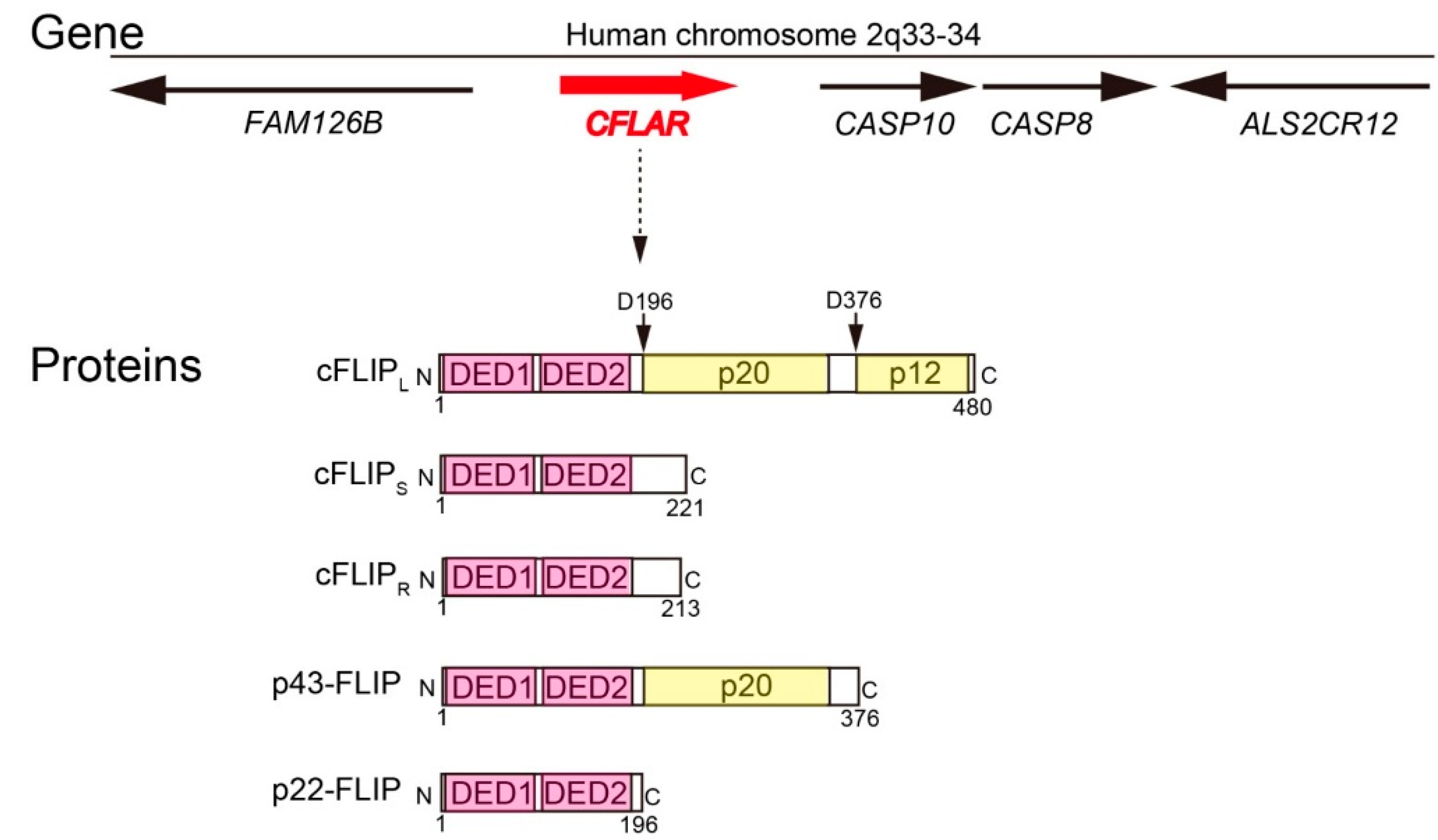
:1. Introduction

2. Molecular Functions of cFLIP in Death Receptor-Mediated Apoptosis Pathway, Ripoptosome Formation, and Necroptosis
2.1. Molecular Function of cFLIP in Death Receptor-Dependent Apoptosis Pathway

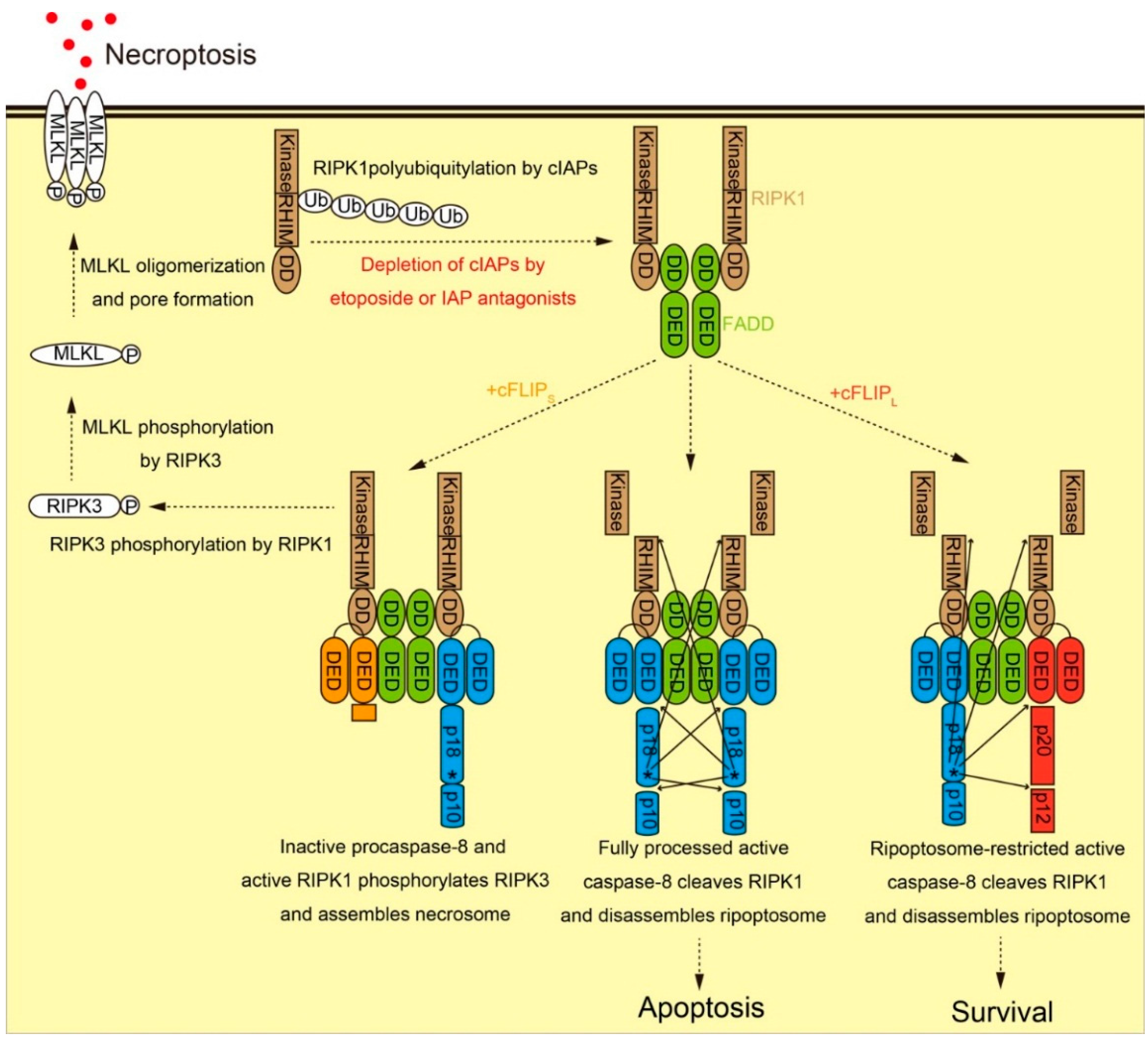
2.2. Molecular Function of cFLIP in Ripoptosome Formation and Necroptosis

3. Quantitative Regulation of cFLIP by Ubiquitin-Proteasome System
3.1. cFLIP Concentration as a Critical Parameter for Cell Fate Determination Revealed by Mathematical Modeling
3.2. Regulation of cFLIP Concentration by Proteolysis

{kind=link}
{kind=link}
{kind=link}
3.3. Itch as an E3 Ubiquitin Ligase for cFLIP Polyubiquitylation
3.4. Other Candidate E3 Ubiquitin Ligases for cFLIP Polyubiquitylation
3.5. Post-Translational Modifications Regulating cFLIP Stability
3.6. Physiological Significance of cFLIP Polyubiquitylation
4. Physiological Roles of cFLIP in Maintaining Tissue Homeostasis in Mammals
4.1. Embryonic Lethality of Cflip-Deficient Mice
| Genotype/Organ | Phenotype | References | |
|---|---|---|---|
| Whole body KO mice | Cflip−/− | Embryonic lethality | [82] |
| Cflip-l+46 mutant | Embryonic lethality | [83] | |
| Cflip−/−Ripk3−/− | Embryonic lethality | [84] | |
| Cflip−/−Fadd−/−Ripk3−/− | Normal development | [84] | |
| Conditional KO mice | T cells | Increased cell death | [85,86,87,88] |
| B cells | Increased cell death | [89,90] | |
| Myeloid lineage | Growth retardation, Splenomegaly | [91,92] | |
| Dendritic cells | Increased inflammation | [93,94] | |
| Liver | Increased liver failure | [95,96,97,98,99,100] | |
| Intestine | Increased cell death and inflammation | [95,101] | |
| Skin epidermis | Increased cell death and inflammation | [102,103] | |
| Transgenic mice | cFLIPR in Cflip-deficient T cells | Similar to Cflip-deficient T cells | [104,105] |
| cFLIPL in T cells | Increased survival | [106,107,108,109,110,111,112,113,114,115] | |
| cFLIPS in T cells | Increased survival | [116,117] | |
| cFLIPL in neuron | Increased survival | [118] | |
| cFLIPL in thyroid | Better resolution from autoimmune disease | [119,120,121,122] | |
| cFLIPL in heart | Reduced cardiac hypertrophy, prevention of cardiac remodeling | [123,124,125] | |
| cFLIPL in eosinophil | Increased survival | [126] | |
| cFLIPL in testis | Testis atrophy | [127] | |
| cFLIPL in muscle | Muscle aging | [128] | |
| cFLIPR in hematopoietic cells | Better bacterial clearance, increased autoimmune disease | [129,130] |
4.2. Rescue of Cflip-Deficient Mice by Ablating Apoptosis and Necroptosis
4.3. Conditional Knockout Mice Lacking cFLIP in T Cells
4.4. Conditional Knockout Mice Lacking cFLIP in Other Lineages of Blood Cells
4.5. Conditional Knockout Mice Lacking cFLIP in Liver
4.6. Conditional Knockout Mice Lacking cFLIP in Epithelial Cells
4.7. cFLIP Isoform-Specific Transgenic Mice
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schröter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [PubMed]
- Shu, H.B.; Halpin, D.; Goeddel, D.V. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 1997, 6, 751–763. [Google Scholar] [CrossRef]
- Hu, S.; Vincenz, C.; Ni, J.; Gentz, R.; Dixit, V.M. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J. Biol. Chem. 1997, 272, 17255–17257. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ahmad, M.; Ottilie, S.; Bullrich, F.; Banks, S.; Wang, Y.; Fernandes-Alnemri, T.; Croce, C.M.; Litwack, G.; Tomaselli, K.J.; et al. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem. 1997, 272, 18542–18545. [Google Scholar] [CrossRef] [PubMed]
- Goltsev, Y.V.; Kovalenko, A.V.; Arnold, E.; Varfolomeev, E.E.; Brodianskii, V.M.; Wallach, D. CASH, a novel caspase homologue with death effector domains. J. Biol. Chem. 1997, 272, 19641–19644. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; Hu, Y.; Chen, S.; Núñez, G. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 10717–10722. [Google Scholar] [CrossRef] [PubMed]
- Han, D.K.; Chaudhary, P.M.; Wright, M.E.; Friedman, C.; Trask, B.J.; Riedel, R.T.; Baskin, D.G.; Schwartz, S.M.; Hood, L. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc. Natl. Acad. Sci. USA 1997, 94, 11333–11338. [Google Scholar] [CrossRef] [PubMed]
- Rasper, D.M.; Vaillancourt, J.P.; Hadano, S.; Houtzager, V.M.; Seiden, I.; Keen, S.L.; Tawa, P.; Xanthoudakis, S.; Nasir, J.; Martindale, D.; et al. Cell death attenuation by “Usurpin”, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998, 5, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Golks, A.; Brenner, D.; Fritsch, C.; Krammer, P.H.; Lavrik, I.N. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 2005, 280, 14507–14513. [Google Scholar] [CrossRef] [PubMed]
- Ueffing, N.; Keil, E.; Freund, C.; Kühne, R.; Schulze-Osthoff, K.; Schmitz, I. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. 2008, 15, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ueffing, N.; Singh, K.K.; Christians, A.; Thorns, C.; Feller, A.C.; Nagl, F.; Fend, F.; Heikaus, S.; Marx, A.; Zotz, R.B.; et al. A single nucleotide polymorphism determines protein isoform production of the human c-FLIP protein. Blood 2009, 114, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Walsh, C.M. Functions of caspase 8: The identified and the mysterious. Semin. Immunol. 2014, 26, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Piao, X.; Shindo, R.; Komazawa-Sakon, S. Cellular FLICE-Inhibitory protein regulates tissue homeostasis. Curr. Top. Microbiol. Immunol. 2015. [Google Scholar] [CrossRef]
- Safa, A.R. Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J. Carcinog. Mutagen. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001, 276, 20633–20640. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002, 21, 3704–3714. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grütter, M.G. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Deis, C.; Denault, J.B.; Sutherlin, D.P.; Salvesen, G.S. Activation of caspases-8 and -10 by FLIPL. Biochem. J. 2004, 382, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Oberst, A.; Drag, M.; van Raam, B.J.; Riedl, S.J.; Green, D.R.; Salvesen, G.S. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 2011, 433, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA 2009, 106, 8169–8174. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Warnken, U.; Fricker, N.; Oztürk, S.; Richter, P.; Kammerer, K.; Schnölzer, M.; Krammer, P.H.; Lavrik, I.N. Stoichiometry of the CD95 death-inducing signaling complex: Experimental and modeling evidence for a death effector domain chain model. Mol. Cell 2012, 47, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.M.; Martin, D.A.; Zheng, L.; Ng, S.Y.; Bertin, J.; Cohen, J.; Lenardo, M.J. Death-effector filaments: Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 1998, 141, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Majkut, J.; Sgobba, M.; Holohan, C.; Crawford, N.; Logan, A.E.; Kerr, E.; Higgins, C.A.; Redmond, K.L.; Riley, J.S.; Stasik, I.; et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallenberger, S.M.; Beaudouin, J.; Claus, J.; Fischer, C.; Sorger, P.K.; Legewie, S.; Eils, R. Intra- and interdimeric caspase-8 self-cleavage controls strength and timing of CD95-induced apoptosis. Sci. Signal. 2014, 7, ra23. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Buchbinder, J.H.; Pietkiewicz, S.; Kähne, T.; Warnken, U.; Öztürk, S.; Schnölzer, M.; Naumann, M.; Krammer, P.H.; Lavrik, I.N. Molecular architecture of the DED chains at the DISC: Regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Boldin, M.P.; Goncharov, T.M.; Wallach, D. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: Proteins binding to the death domains of the two receptors also bind to each other. J. Exp. Med. 1996, 183, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Huang, J.; Shu, H.-B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-a signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Weinlich, R.; Green, D.R. The two faces of receptor interacting protein kinase-1. Mol. Cell 2014, 56, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Schilling, R.; Geserick, P.; Leverkus, M. Characterization of the ripoptosome and its components: Implications for anti-inflammatory and cancer therapy. Methods Enzymol. 2014, 545, 83–102. [Google Scholar] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Panayotova-Dimitrova, D.; Leverkus, M. Pick your poison: The ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 2012, 11, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Day, T.W.; Huang, S.; Safa, A.R. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8, and caspase-9-dependent apoptosis in breast cancer cells. Biochem. Pharmacol. 2008, 76, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Toscano, F.; Virard, F.; Jacquemin, G.; Pierrot, A.; Vanbervliet, B.; Bonnin, M.; Lalaoui, N.; Mercier-Gouy, P.; Pachéco, Y.; et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012, 19, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Bentele, M.; Lavrik, I.; Ulrich, M.; Stösser, S.; Heermann, D.W.; Kalthoff, H.; Krammer, P.H.; Eils, R. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J. Cell Biol. 2004, 166, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N.; Golks, A.; Riess, D.; Bentele, M.; Eils, R.; Krammer, P.H. Analysis of CD95 threshold signaling: Triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. J. Biol. Chem. 2007, 282, 13664–13671. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N. Systems biology of death receptor networks: Live and let die. Cell Death Dis. 2014, 5, e1259. [Google Scholar] [CrossRef] [PubMed]
- Fricker, N.; Beaudouin, J.; Richter, P.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J. Cell Biol. 2010, 190, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhao, Y.; Jia, X. Mathematical modeling identified c-FLIP as an apoptotic switch in death receptor induced apoptosis. Apoptosis 2008, 13, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.; Pforr, C.; Beaudouin, J.; Pappa, A.; Fricker, N.; Krammer, P.H.; Lavrik, I.N.; Eils, R. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol. Syst. Biol. 2010, 6, 352. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Toivonen, H.T.; Meinander, A.; Asaoka, T.; Westerlund, M.; Pettersson, F.; Mikhailov, A.; Eriksson, J.E.; Saxén, H. Modeling reveals that dynamic regulation of c-FLIP levels determines cell-to-cell distribution of CD95-mediated apoptosis. J. Biol. Chem. 2011, 286, 18375–18382. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Hafner, M.; Bandara, S.; Sims, J.J.; Hudson, H.; Chai, D.; Sorger, P.K. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 2015, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Pathak, S.K.; Kumawat, K.; Basu, S.; Chatterjee, G.; Pathak, S.; Noguchi, T.; Takeda, K.; Ichijo, H.; Thien, C.B.; et al. A TNF- and c-Cbl-dependent FLIPS-degradation pathway and its function in Mycobacterium tuberculosis-induced macrophage apoptosis. Nat. Immunol. 2009, 10, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gallagher, P.J. Mind bomb 1 regulation of cFLIP interactions. Am. J. Physiol. Cell Physiol. 2009, 297, C1275–C1283. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, I.; Zotti, T.; Ferravante, A.; Vessichelli, M.; Reale, C.; Masone, M.C.; Leonardi, A.; Vito, P.; Stilo, R. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFα-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J. Biol. Chem. 2012, 287, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, W.; Hao, X.; Li, T.; Su, L.; Liu, X. Down-regulation of cellular FLICE-inhibitory protein (Long Form) contributes to apoptosis induced by Hsp90 inhibition in human lung cancer cells. Cancer Cell Int. 2012, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Komazawa-Sakon, S.; Takekawa, M.; Sasazuki, T.; Yeh, W.C.; Yagita, H.; Okumura, K.; Nakano, H. An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J. 2006, 25, 5549–5559. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Kojima, Y.; Nakayama, M.; Yagita, H.; Okumura, K.; Nakano, H. Downregulation of c-FLIP promotes caspase-dependent JNK activation and reactive oxygen species accumulation in tumor cells. Oncogene 2008, 27, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Murata, E.; Hashimoto, M.; Aoki, T. Interaction between cFLIP and Itch, a ubiquitin ligase, is obstructed in Trypanosoma. cruzi-infected human cells. Microbiol. Immunol. 2008, 52, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Demange, C.; Ferrand, N.; Prunier, C.; Bourgeade, M.F.; Atfi, A. A model of partnership co-opted by the homeodomain protein TGIF and the Itch/AIP4 ubiquitin ligase for effective execution of TNF-α cytotoxicity. Mol. Cell 2009, 36, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Parsa, A.T.; Pieper, R.O. A novel PTEN-dependent link to ubiquitination controls FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2009, 69, 7911–7916. [Google Scholar] [CrossRef] [PubMed]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Fang, S.; Parsa, A.T.; Pieper, R.O. Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2010, 70, 5046–5053. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yue, P.; Khuri, F.R.; Sun, S.Y. mTOR complex 2 is involved in regulation of Cbl-dependent c-FLIP degradation and sensitivity of TRAIL-induced apoptosis. Cancer Res. 2013, 73, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, J.; Zhang, Y.; Qu, J.; Xu, L.; Zheng, H.; Liu, Y.; Qu, X. Cbl-b-dependent degradation of FLIPL is involved in ATO-induced autophagy in leukemic K562 and gastric cancer cells. FEBS Lett. 2012, 586, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Haimerl, F.; Erhardt, A.; Sass, G.; Tiegs, G. Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific protease 2 contributes to tumor necrosis factor-α-induced hepatocyte survival. J. Biol. Chem. 2009, 284, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Abedini, M.R.; Muller, E.J.; Brun, J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 2008, 68, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Abedini, M.R.; Muller, E.J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein. Oncogene 2010, 29, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Abedini, M.R.; Wang, P.W.; Huang, Y.F.; Cao, M.; Chou, C.Y.; Shieh, D.B.; Tsang, B.K. Cell fate regulation by gelsolin in human gynecologic cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 14442–14447. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Stagni, V.; Giambruno, R.; Fianco, G.; Di Benedetto, A.; Mottolese, M.; Pellegrini, M.; Barilà, D. ATM kinase activity modulates ITCH E3-ubiquitin ligase activity. Oncogene 2014, 33, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tay, K.H.; Dong, L.; Thorne, R.F.; Jiang, C.C.; Yang, E.; Tseng, H.Y.; Liu, H.; Christopherson, R.; Hersey, P.; et al. Cystatin B inhibition of TRAIL-induced apoptosis is associated with the protection of FLIPL from degradation by the E3 ligase itch in human melanoma cells. Cell Death Differ. 2010, 17, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Tran, T.; Sobkoviak, R.; Pope, R.M. Activation-induced degradation of FLIPL is mediated via the phosphatidylinositol 3-kinase/Akt signaling pathway in macrophages. J. Biol. Chem. 2009, 284, 14513–14523. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pérez, T.; Ortiz-Ferrón, G.; López-Rivas, A. Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key events in the sensitization of breast tumor cells to TRAIL by antimicrotubule agents. Cell Death Differ. 2010, 17, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Pérez, T.; Medema, R.H.; López-Rivas, A. Delaying mitotic exit downregulates FLIP expression and strongly sensitizes tumor cells to TRAIL. Oncogene 2015, 34, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Yerbes, R.; López-Rivas, A. Itch/AIP4-independent proteasomal degradation of cFLIP induced by the histone deacetylase inhibitor SAHA sensitizes breast tumour cells to TRAIL. Investig. New Drugs 2012, 30, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Kaunisto, A.; Kochin, V.; Asaoka, T.; Mikhailov, A.; Poukkula, M.; Meinander, A.; Eriksson, J.E. PKC-mediated phosphorylation regulates c-FLIP ubiquitylation and stability. Cell Death Differ. 2009, 13, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Wilkie-Grantham, R.P.; Matsuzawa, S.; Reed, J.C. Novel phosphorylation and ubiquitination sites regulates reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. J. Biol. Chem. 2013, 288, 12777–12790. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Nimmannit, U.; Wang, L.; Stehlik, C.; Lu, B.; Azad, N.; Rojanasakul, Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. J. Biol. Chem. 2005, 280, 42044–42050. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.E.; Meinander, A.; Holmström, T.H.; Rivero-Müller, A.; Heiskanen, K.M.; Linnau, E.K.; Courtney, M.J.; Mosser, D.D.; Sistonen, L.; Eriksson, J.E. Heat stress downregulates FLIP and sensitizes cells to Fas receptor-mediated apoptosis. Cell Death Differ. 2003, 10, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Meinander, A.; Söderström, T.S.; Kaunisto, A.; Poukkula, M.; Sistonen, L.; Eriksson, J.E. Fever-like hyperthermia controls T Lymphocyte persistence by inducing degradation of cellular FLIPshort. J. Immunol. 2007, 178, 3944–3953. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Kim, S.Y.; Zhou, Z.; Lagasse, E.; Kwon, Y.T.; Lee, Y.J. Hyperthermia enhances mapatumumab-induced apoptotic death through ubiquitin-mediated degradation of cellular FLIP(long) in human colon cancer cells. Cell Death Dis. 2013, 4, e577. [Google Scholar] [CrossRef] [PubMed]
- Morlé, A.; Garrido, C.; Micheau, O. Hyperthermia restores apoptosis induced by death receptors through aggregation-induced c-FLIP cytosolic depletion. Cell Death Dis. 2015, 6, e1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishioka, T.; Katayama, R.; Kikuchi, R.; Nishimoto, M.; Takada, S.; Takada, R.; Matsuzawa, S.; Reed, J.C.; Tsuruo, T.; Naito, M. Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells 2007, 12, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Jückstock, J. Misfolded proteins: From little villains to little helpers in the fight against cancer. Front. Oncol. 2015, 5, 47. [Google Scholar] [PubMed]
- Yeh, W.C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef]
- Shibata, N.; Ohoka, N.; Sugaki, Y.; Onodera, C.; Inoue, M.; Sakuraba, Y.; Takakura, D.; Hashii, N.; Kawasaki, N.; Gondo, Y.; et al. Degradation of stop codon read-through mutant proteins via the ubiquitin-proteasome system causes hereditary disorders. J. Biol. Chem. 2015, 190, 28428–28437. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIPL complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Chau, H.; Wong, V.; Chen, N.J.; Huang, H.L.; Lin, W.J.; Mirtsos, C.; Elford, A.R.; Bonnard, M.; Wakeham, A.; You-Ten, A.I.; et al. Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J. Exp. Med. 2005, 202, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; He, Y.W. An essential role for c-FLIP in the efficient development of mature T lymphocytes. J. Exp. Med. 2005, 202, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Hopkins, K.; He, Y.W. c-FLIP protects mature T lymphocytes from TCR-mediated killing. J. Immunol. 2008, 181, 5368–5373. [Google Scholar] [CrossRef]
- He, M.X.; He, Y.W. c-FLIP protects T lymphocytes from apoptosis in the intrinsic pathway. J. Immunol. 2015, 194, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rosenberg, S.; Coffey, F.J.; He, Y.W.; Manser, T.; Hardy, R.R.; Zhang, J. A role for cFLIP in B cell proliferation and stress MAPK regulation. J. Immunol. 2009, 182, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Coffey, F.; Manser, T. Expression of cellular FLIP by B cells is required for their participation in an immune response. J. Immunol. 2010, 184, 4871–4879. [Google Scholar] [CrossRef] [PubMed]
- Gordy, C.; Pua, H.; Sempowski, G.D.; He, Y.W. Regulation of steady-state neutrophil homeostasis by macrophages. Blood 2011, 117, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Perlman, H.; Huang, Z.; Birkett, R.; Kan, L.; Agrawal, H.; Misharin, A.; Gurbuxani, S.; Crispino, J.D.; Pope, R.M. FLIP: A novel regulator of macrophage differentiation and granulocyte homeostasis. Blood 2010, 116, 4968–4977. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Perlman, H.; Birkett, R.; Doyle, R.; Fang, D.; Haines, G.K.; Robinson, W.; Datta, S.; Huang, Z.; Li, Q.Z.; et al. CD11c-mediated deletion of Flip promotes autoreactivity and inflammatory arthritis. Nat. Commun. 2015, 6, 7086. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Wu, Y.H.; Mo, S.T.; Hsiao, H.W.; He, Y.W.; Lai, M.Z. Cellular FLIP inhibits myeloid cell activation by suppressing selective innate signaling. J. Immunol. 2015, 195, 2612–2623. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Komazawa-Sakon, S.; Nishina, T.; Koike, M.; Piao, J.H.; Ehlken, H.; Kurihara, H.; Hara, M.; van Rooijen, N.; Schütz, G.; et al. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Sci. Signal. 2012, 5, ra93. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Zimmermann, T.; Wörns, M.; Sprinzl, M.F.; Kreft, A.; Kohl, T.; Nagel, M.; Siebler, J.; Schulze Bergkamen, H.; He, Y.W.; et al. Ablation of c-FLIP in hepatocytes enhances death-receptor mediated apoptosis and toxic liver injury in vivo. J. Hepatol. 2011, 55, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Nagel, M.; Kim, Y.O.; Kohl, T.; Wörns, M.A.; Zimmermann, T.; Schad, A.; Longerich, T.; Schuppan, D.; He, Y.W.; et al. Increased hepatic fibrosis and JNK2-dependent liver injury in mice exhibiting hepatocyte-specific deletion of cFLIP. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G498–G506. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Wörns, M.A.; Zimmermann, T.; He, Y.W.; Galle, P.R.; Schuchmann, M. The role of death effector domain-containing proteins in acute oxidative cell injury in hepatocytes. Free Radic. Biol. Med. 2012, 52, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.; Gehrke, N.; Schad, A.; Nagel, M.; Wörns, M.A.; Sprinzl, M.F.; Zimmermann, T.; He, Y.W.; Galle, P.R.; Schuchmann, M.; et al. Diabetic liver injury from streptozotocin is regulated through the caspase-8 homolog cFLIP involving activation of JNK2 and intrahepatic immunocompetent cells. Cell Death Dis. 2013, 4, e712. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Garcia-Bardon, D.; Mann, A.; Schad, A.; Alt, Y.; Wörns, M.A.; Sprinzl, M.F.; Zimmermann, T.; Menke, J.; Engstler, A.J.; et al. Acute organ failure following the loss of anti-apoptotic cellular FLICE-inhibitory protein involves activation of innate immune receptors. Cell Death Differ. 2015, 22, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Wittkopf, N.; Günther, C.; Martini, E.; He, G.; Amann, K.; He, Y.W.; Schuchmann, M.; Neurath, M.F.; Becker, C. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology 2013, 145, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Dillon, C.P.; Janke, L.J.; Milasta, S.; Lukens, J.R.; Rodriguez, D.A.; Gurung, P.; Savage, C.; Kanneganti, T.D.; et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013, 5, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Panayotova-Dimitrova, D.; Feoktistova, M.; Ploesser, M.; Kellert, B.; Hupe, M.; Horn, S.; Makarov, R.; Jensen, F.; Porubsky, S.; Schmieder, A.; et al. cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 2013, 5, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Hopkins, K.; He, Y.W. The long isoform of cellular FLIP is essential for T lymphocyte proliferation through an NF-κB-independent pathway. J. Immunol. 2008, 180, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- He, M.X.; He, Y.W. A role for c-FLIPL in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013, 20, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.; Kataoka, T.; Fortner, K.A.; Tinel, A.; Ferrero, I.; MacDonald, R.H.; Hahne, M.; Beermann, F.; Attinger, A.; Orbea, H.A.; et al. The caspase 8 inhibitor c-FLIPL modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol. Cell. Biol. 2002, 22, 5419–5433. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.S.; Fang, L.W.; Lai, M.Z. c-FLICE inhibitory protein expression inhibits T-cell activation. Cell. Death Differ. 2004, 11, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Dohrman, A.; Kataoka, T.; Cuenin, S.; Russell, J.Q.; Tschopp, J.; Budd, R.C. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-κB activation. J. Immunol. 2005, 174, 5270–5278. [Google Scholar] [CrossRef] [PubMed]
- Dohrman, A.; Russell, J.Q.; Cuenin, S.; Fortner, K.; Tschopp, J.; Budd, R.C. Cellular FLIP long form augments caspase activity and death of T cells through heterodimerization with and activation of caspase-8. J. Immunol. 2005, 175, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Rinaldi, L.; Fortner, K.A.; Russell, J.Q.; Tschopp, J.; Irvin, C.; Budd, R.C. Cellular FLIP long form-transgenic mice manifest a Th2 cytokine bias and enhanced allergic airway inflammation. J. Immunol. 2004, 172, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Tseveleki, V.; Bauer, J.; Taoufik, E.; Ruan, C.; Leondiadis, L.; Haralambous, S.; Lassmann, H.; Probert, L. Cellular FLIP (long isoform) overexpression in T cells drives Th2 effector responses and promotes immunoregulation in experimental autoimmune encephalomyelitis. J. Immunol. 2004, 173, 6619–6626. [Google Scholar] [CrossRef] [PubMed]
- Tseveleki, V.; Tsagozis, P.; Koutsoni, O.; Dotsika, E.; Probert, L. Cellular FLIP long isoform transgenic mice overcome inherent Th2-biased immune responses to efficiently resolve Leishmania. major infection. Int. Immunol. 2007, 19, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Dohrman, A.; Sartini, D.; Budd, R.C. Reduced myocarditis following Coxsackievirus infection in cellular FLICE inhibitory protein—Long form-transgenic mice. Immunology 2006, 119, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Li, Z.; Minto, A.W.; Shia, J.; Yang, L.; Bao, L.; Tschopp, J.; Gao, J.X.; Wang, J.; Quigg, R.J.; et al. Altered thymic selection by overexpressing cellular FLICE inhibitory protein in T cells causes lupus-like syndrome in a BALB/c but not C57BL/6 strain. Cell Death Differ. 2010, 17, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Sharp, G.C.; Braley-Mullen, H. Effect of transgenic overexpression of FLIP on lymphocytes on development and resolution of experimental autoimmune thyroiditis. Am. J. Pathol. 2011, 179, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Neumann, F.; Bösser, S.; Zörnig, M. Transgenic overexpression of the Caspase-8 inhibitor FLIPshort leads to impaired T cell proliferation and an increased memory T cell pool after Staphylococcal enterotoxin B injection. Eur. J. Immunol. 2005, 35, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Buskiewicz, I.A.; Koenig, A.; Roberts, B.; Russell, J.; Shi, C.; Lee, S.H.; Jung, J.U.; Huber, S.A.; Budd, R.C. c-FLIP-Short reduces type I interferon production and increases viremia with coxsackievirus B3. PLoS ONE 2014, 9, e96156. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Valable, S.; Müller, G.J.; Roberts, M.L.; Divoux, D.; Tinel, A.; Voulgari-Kokota, A.; Tseveleki, V.; Altruda, F.; Lassmann, H.; et al. FLIPL protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J. Neurosci. 2007, 27, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Braley-Mullen, H. Cultured murine thyroid epithelial cells expressing transgenic Fas-associated death domain-like interleukin-1β converting enzyme inhibitory protein are protected from Fas-mediated apoptosis. Endocrinology 2008, 149, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Arscott, P.; Wu, P.; Baker, J.R., Jr. No apparent damage in the thyroid of transgenic mice expressing antiapoptotic FLIP. Thyroid 2006, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wei, Y.; Demarco, V.; Chen, K.; Sharp, G.C.; Braley-Mullen, H. Murine FLIP transgene expressed on thyroid epithelial cells promotes resolution of granulomatous experimental autoimmune thyroiditis in DBA/1 mice. Am. J. Pathol. 2007, 170, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; DeMarco, V.G.; Sharp, G.C.; Braley-Mullen, H. Expression of transgenic FLIP on thyroid epithelial cells inhibits induction and promotes resolution of granulomatous experimental autoimmune thyroiditis in CBA/J mice. Endocrinology 2007, 148, 5734–5745. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tang, Q.Z.; Liu, C.; Moon, M.; Chen, M.; Yan, L.; Bian, Z.Y.; Zhang, Y.; Wang, A.B.; Nghiem, M.P.; et al. Cellular FLICE-inhibitory protein protects against cardiac remodeling induced by angiotensin II in mice. Hypertension 2010, 56, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Moon, M.; Yan, L.; Nian, M.; Zhang, Y.; Liu, C.; Lu, J.; Guan, H.; Chen, M.; Jiang, D.; et al. Cellular FLICE-inhibitory protein protects against cardiac remodelling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 239. [Google Scholar] [CrossRef] [PubMed]
- Gordy, C.; Liang, J.; Pua, H.; He, Y.W. c-FLIP protects eosinophils from TNF-α-mediated cell death in vivo. PLoS ONE 2014, 9, e107724. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Petrungaro, S.; Coluccia, P.; Filippini, A.; Ziparo, E.; Giampietri, C. Testis atrophy and reduced sperm motility in transgenic mice overexpressing c-FLIPL. Fertil. Steril. 2010, 93, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Musumeci, M.; Coluccia, P.; Antonangeli, F.; De Cesaris, P.; Filippini, A.; Marano, G.; Ziparo, E. c-Flip overexpression reduces cardiac hypertrophy in response to pressure overload. J. Hypertens. 2008, 26, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Coluccia, P.; Antonangeli, F.; Giannakakis, K.; Faraggiana, T.; Filippini, A.; Cossu, G.; Ziparo, E. c-Flip overexpression affects satellite cell proliferation and promotes skeletal muscle aging. Cell Death Dis. 2010, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Telieps, T.; Ewald, F.; Gereke, M.; Annemann, M.; Rauter, Y.; Schuster, M.; Ueffing, N.; von Smolinski, D.; Gruber, A.D.; Bruder, D.; et al. Cellular-FLIP, Raji isoform (c-FLIPR) modulates cell death induction upon T-cell activation and infection. Eur. J. Immunol. 2013, 43, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Ewald, F.; Annemann, M.; Pils, M.C.; Plaza-Sirvent, C.; Neff, F.; Erck, C.; Reinhold, D.; Schmitz, I. Constitutive expression of murine c-FLIPR causes autoimmunity in aged mice. Cell Death Dis. 2014, 5, e1168. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Giampietri, C.; Petrungaro, S.; Filippini, A.; Ziparo, E. Expression profile of a 400-bp Stra8 promoter region during spermatogenesis. Microsc. Res. Tech. 2009, 72, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, L.; Jenkins, G.D.; Bhise, N.; Feldberg, T.; Mitra-Ghosh, T.; Fridley, B.L.; Lamba, J.K. Genome-wide association analysis identified splicing single nucleotide polymorphism in CFLAR predictive of triptolide chemo-sensitivity. BMC Genom. 2015, 16, 483. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Pollok, K.E. Targeting the Anti-apoptotic protein c-FLIP for cancer therapy. Cancers 2011, 2, 1639–1671. [Google Scholar] [CrossRef] [PubMed]
- Urbano, P.C.; Soccol, V.T.; Azevedo, V.F. Apoptosis and the FLIP and NF-κB proteins as pharmacodynamic criteria for biosimilar TNF-α antagonists. Biologics 2014, 8, 211–220. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321-30341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226232
Tsuchiya Y, Nakabayashi O, Nakano H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. International Journal of Molecular Sciences. 2015; 16(12):30321-30341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226232
Chicago/Turabian StyleTsuchiya, Yuichi, Osamu Nakabayashi, and Hiroyasu Nakano. 2015. "FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP" International Journal of Molecular Sciences 16, no. 12: 30321-30341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226232