
A Personalized Approach in Progressive Multiple Sclerosis: The Current Status of Disease Modifying Therapies (DMTs) and Future Perspectives


Abstract
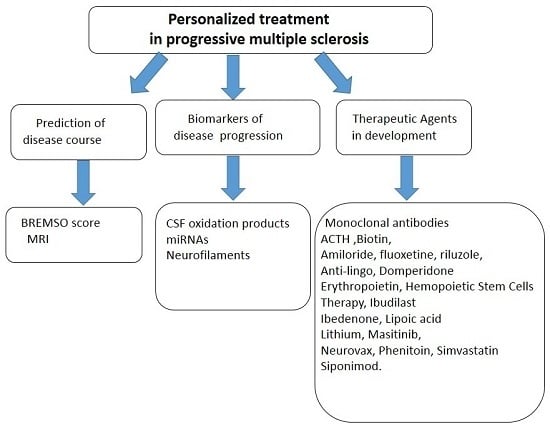
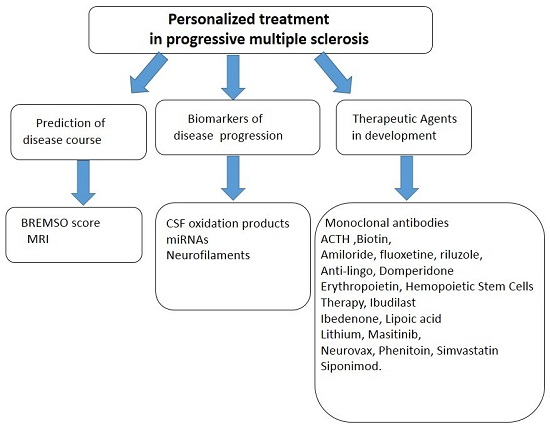
:
{kind=link}
1. Introduction
2. Insights into Pathogenesis of Progressive Forms
3. Is the Prediction of Progressive Disease Course Possible?
4. Biological Markers in Progressive Multiple Sclerosis (PMS)
5. Therapeutic Advances and Future Prospects in Progressive Multiple Sclerosis (PMS): State of the Art
5.1. Therapeutic Agents under Investigation
5.1.1. Adrenocorticotropic Hormone
5.1.2. Biotin
5.1.3. Amiloride, Fluoxetine, and Riluzole
5.1.4. Remyelination Agents
5.1.5. Domperidone
5.1.6. Erythropoietin
5.1.7. Hematopoetic Stem Cell Transplantation
5.1.8. Ibudilast
5.1.9. Idebenone
5.1.10. Lipoic Acid
5.1.11. Lithium
5.1.12. Masitinib
5.1.13. MIS416
5.1.14. NeuroVax
5.1.15. Oxcarbazepine
5.1.16. Simvastatin
5.1.17. Siponimod
5.1.18. Sunphenon Epigallocatechin-3-gallate
5.2. Monoclonal Antibodies
5.2.1. Natalizumab
5.2.2. Anti-CD20 Monoclonal Antibodies
6. Future Research Directions and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lugaresi, A.; di Ioia, M.; Travaglini, D.; Pietrolongo, E.; Pucci, E.; Onofrj, M. Risk-benefit considerations in the treatment of relapsing-remitting multiple sclerosis. Neuropsychiatr. Dis. Treat. 2013, 9, 893–914. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- Afsaneh, S.; Darin, T.; Okuda, O.S. Therapeutic advances and future prospects in progressive forms of multiple sclerosis. Neurotherapeutics 2016, 13, 58–69. [Google Scholar]
- Ontaneda, D.; Fox, R.J.; Chataway, J. Clinical trials in progressive multiple sclerosis: Lessons learned and future perspectives. Lancet Neurol. 2015, 14, 208–223. [Google Scholar] [CrossRef]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Howell, O.W.; Carassiti, D.; Magliozzi, R.; Gveric, D.; Muraro, P.A.; Nicholas, R.; Roncaroli, F.; Reynolds, R. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012, 135, 2925–2937. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.A.; Maghzal, G.J.; Khademi, M.; Piehl, F.; Ratzer, R.; Christensen, J.R.; Sellebjerg, F.T.; Olsson, T.; Stocker, R. Absence of systemic oxidative stress and increased CSF prostaglandin F2α in progressive MS. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e256. [Google Scholar] [CrossRef] [PubMed]
- Drulović, J.; Dujmović, I.; Mesaros, S. Raised cerebrospinal fluid nitrite and nitrate levels in patients with multiple sclerosis: No correlation with disease activity. Mult. Scler. 2001, 7, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S.; Adeleine, P. Christian confavreux, sandra vukusic, patrice adeleine. Early clinical predictors and progression of irreversible disability in multiple sclerosis: An amnesic process. Brain 2003, 126, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, R.; Quaglini, S.; Trojano, M.; Amato, M.P.; Tavazzi, E.; Paolicelli, D.; Zipoli, V.; Romani, A.; Fuiani, A.; Portaccio, E.; et al. Early prediction of the long term evolution of multiple sclerosis: The Bayesian Risk Estimate for Multiple Sclerosis (BREMS) score. J. Neurol. Neurosurg. Psychiatry 2007, 78, 757–759. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, N.; Giorgio, A.; Battaglini, M. Assessing brain atrophy rates in a large population of untreated multiple sclerosis subtypes. Neurology 2010, 74, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garriga, J.; Ingle, G.T.; Rovaris, M. Long-term clinical outcome of primary progressive MS: Predictive value of clinical and MRI data. Neurology 2005, 65, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Tiberio, M.; Chard, D.T.; Altmann, D.R. Gray and white matter volume changes in early RRMS: A 2-year longitudinal study. Neurology 2005, 64, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Nair, G.; Shea, C.D.; Crainiceanu, C.M.; Cortese, I.C.; Reich, D.S. Quantification of multiple-sclerosis-related brain atrophy in two heterogeneous MRI datasets using mixed-effects modeling. NeuroImage Clin. 2013, 3, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Grassiot, B.; Desgranges, B.; Eustache, F.; Defer, G. Quantification and clinical relevance of brain atrophy in multiple sclerosis: A review. J. Neurol. 2009, 256, 1397–1412. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.; Lavorgna, L.; Russo, P. Brain atrophy and lesion load in a large population of patients with multiple sclerosis. Neurology 2005, 65, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Pagani, E.; Rocca, M.A.; Gallo, A. Regional brain atrophy evolves differently in patients with multiple sclerosis according to clinical phenotype. AJNR Am. J. Neuroradiol. 2005, 26, 341–346. [Google Scholar] [PubMed]
- Bieniek, M.; Altmann, D.R.; Davies, G.R. Cord atrophy separates early primary progressive and relapsing remitting multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Sombekke, M.H.; Bellenberg, B. Relevance of spinal cord abnormalities to clinical disability in multiple sclerosis: MR imaging findings in a large cohort of patients. Radiology 2013, 269, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Chard, D.T.; Griffin, C.M.; Rashid, W. Progressive grey matter atrophy in clinically early relapsing-remitting multiple sclerosis. Mult. Scler. 2004, 10, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Laule, C.; Vavasour, I.M.; Zhao, Y. Two-year study of cervical cord volume and myelin water in primary progressive multiple sclerosis. Mult. Scler. 2010, 16, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Narayana, P.A. Magnetic resonance spectroscopy in the monitoring of multiple sclerosis. J. Neuroimaging 2005, 15, 46S–57S. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Paty, D.W.; Kappos, L.; Barkhof, F.; Compston, D.A.; Thompson, A.J.; Zhao, G.J.; Wiles, C.M.; McDonald, W.I.; Miller, D.H. Correlations between changes in disability and T2-weighted brain MRI activity in multiple sclerosis: A follow-up study. Neurology 1995, 45, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Moeri, D.; Radue, E.W.; Schoetzau, A.; Schweikert, K.; Barkhof, F.; Miller, D.; Guttmann, C.R.; Weiner, H.L.; Gasperini, C.; et al. Predictive value of gadolinium-enhanced magnetic resonance imaging for relapse rate and changes in disability or impairment in multiple sclerosis: A meta-analysis. Lancet 1999, 353, 964–969. [Google Scholar] [CrossRef]
- Pérez-Miralles, F.; Sastre-Garriga, J.; Tintoré, M.; Arrambide, G.; Nos, C.; Perkal, H.; Río, J.; Edo, M.C.; Horga, A.; Castilló, J.; et al. Clinical impact of early brain atrophy in clinically isolated syndromes. Mult. Scler. 2013, 19, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Minneboo, A.; Jasperse, B.; Barkhof, F.; Knol, D.L.; de Groot, V.; Polman, C.H.; Castelijns, J.A. Predicting short-term disability progression in early multiple sclerosis: Added value of MRI parameters. J. Neurol. Neurosurg. Psychiatry 2008, 79, 917–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.; Roncaroli, F.; Nicholas, R.; Radotra, B.; Gveric, D.; Howell, O. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011, 122, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Khaleeli, Z.; Ciccarelli, O.; Manfredonia, F.; Barkhof, F.; Brochet, B.; Cercignani, M.; Dousset, V.; Filippi, M.; Montalban, X.; Polman, C.; et al. Predicting progression in primary progressive multiple sclerosis: A 10-year multicenter study. Ann. Neurol. 2008, 63, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Popescu, V.; Agosta, F.; Hulst, H.E.; Sluimer, I.C.; Knol, D.L.; Sormani, M.P.; Enzinger, C.; Ropele, S.; Alonso, J.; Sastre-Garriga, J.; et al. Brain atrophy and lesion load predict long term disability in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Sailer, M.; Fischl, B.; Salat, D.; Tempelmann, C.; Schönfeld, M.A.; Busa, E.; Bodammer, N.; Heinze, H.J.; Dale, A. Focal thinning of the cerebral cortex in multiple sclerosis. Brain 2003, 126, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Audoin, B.; Davies, G.R.; Finisku, L.; Chard, D.T.; Thompson, A.J.; Miller, D.H. Localization of grey matter atrophy in early RRMS: A longitudinal study. J. Neurol. 2006, 253, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Charil, A.; Dagher, A.; Lerch, J.P.; Zijdenbos, A.P.; Worsley, K.J.; Evans, A.C. Focal cortical atrophy in multiple sclerosis: Relation to lesion load and disability. NeuroImage 2007, 34, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Atzori, M.; Bernardi, V.; Morra, A.; Romualdi, C.; Rinaldi, L.; McAuliffe, M.J.; Barachino, L.; Perini, P.; Fischl, B.; et al. Cortical atrophy is relevant in multiple sclerosis at clinical onset. J. Neurol. 2007, 254, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.; Ramasamy, D.; Munschauer, F.; Weinstock-Guttman, B.; Zivadinov, R. Memory impairment in multiple sclerosis: Correlation with deep grey matter and mesial temporal atrophy. J. Neurol. Neurosurg. Psychiatry 2009, 80, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Sicotte, N.L.; Kern, K.C.; Giesser, B.S.; Arshanapalli, A.; Schultz, A.; Montag, M.; Wang, H.; Bookheimer, S.Y. Regional hippocampal atrophy in multiple sclerosis. Brain 2008, 131, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.A.; Mesaros, S.; Pagani, E.; Sormani, M.P.; Comi, G.; Filippi, M. Thalamic damage and long-term progression of disability in multiple sclerosis. Radiology 2010, 257, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Faber-Rod, J.C.; Bauer, J.; Lucchinetti, C.F.; Sorensen, P.S.; Laursen, H.; Stadelmann, C.; Brück, W.; Rauschka, H.; Schmidbauer, M.; et al. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. 2007, 17, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Mattisi, I.; Rinaldi, F.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Romualdi, C.; Rinaldi, L.; Perini, P.; et al. Magnetic resonance evidence of cerebellar cortical pathology in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Grossi, P.; Favaretto, A.; Romualdi, C.; Atzori, M.; Rinaldi, F.; Perini, P.; Saladini, M.; Gallo, P. Cortical pathology in multiple sclerosis patients with epilepsy: A 3-year longitudinal study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Poretto, V.; Favaretto, A.; Alessio, S.; Bernardi, V.; Romualdi, C.; Rinaldi, F.; Perini, P.; Gallo, P. Cortical lesion load associates with progression of disability in multiple sclerosis. Brain 2012, 135, 2952–2961. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Rovaris, M.; Pagani, E.; Sormani, M.P.; Comi, G.; Filippi, M. Magnetization transfer MRI metrics predict the accumulation of disability 8 years later in patients with multiple sclerosis. Brain 2006, 129, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Martin, R. Development of biomarkers in multiple sclerosis. Brain 2004, 127, 1463–1478. [Google Scholar] [CrossRef] [PubMed]
- Malmeström, C.; Haghighi, S.; Rosengren, L.; Andersen, O.; Lycke, J. Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology 2003, 61, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Trentini, A.; Comabella, M.; Tintoré, M.; Koel-Simmelink, M.J.; Killestein, J.; Roos, B.; Rovira, A.; Korth, C.; Ottis, P.; Blankenstein, M.A.; et al. N-acetylaspartate and neurofilaments as biomarkers of axonal damage in patients with progressive forms of multiple sclerosis. J. Neurol. 2014, 261, 2338–2343. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Csurhes, P.A.; Wolfe, N.P.; Hooper, K.D.; Good, M.F.; McCombe, P.A.; Greer, J.M. Increased circulating T cell reactivity to GM3 and GQ1b gangliosides in primary progressive multiple sclerosis. J. Clin. Neurosci. 2003, 10, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Belogurov, A.A.; Kurkova, I.N.; Friboulet, A.; Thomas, D.; Misikov, V.K.; Zakharova, M.Y.; Suchkov, S.V.; Kotov, S.V.; Alehin, A.I.; Avalle, B.; et al. Recognition and degradation of myelin basic protein peptides by serum autoantibodies: Novel biomarker for multiple sclerosis. J. Immunol. 2008, 180, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.R.; Börnsen, L.; Khademi, M.; Olsson, T.; Jensen, P.E.; Sørensen, P.S.; Sellebjerg, F. CSF inflammation and axonal damage are increased and correlate in progressive multiple sclerosis. Mult. Scler. J. 2013, 19, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Xiao, B.; Ma, X.; Qu, M.; Li, Y.; Nagarkatti, P.; Nagarkatti, M.; Zhou, J. MicroRNAs associated with the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2016, 295, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Vistbakka, J.; Elovaara, I.; Lehtimäki, T.; Hagman, S. Circulating microRNAs as biomarkers in progressive multiple sclerosis. Mult. Scler. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Okuda, D.T. Immunosuppressive treatments in multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 503–511. [Google Scholar] [PubMed]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis—A quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Arnason, B.G.; Berkovich, R.; Catania, A.; Lisak, R.P.; Zaidi, M. Mechanisms of action of adrenocorticotropic hormone and other melanocortins relevant to the clinicalmanagement of patients with multiple sclerosis. Mult. Scler. 2013, 19, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Catania, A. Neuroprotective actions of melanocortins: A therapeutic opportunity. Trends Neurosci. 2008, 31, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F.; Papeix, C.; Bellanger, A.; Touitou, V.; Lebrun-Frenay, C.; Galanaud, D.; Gout, O.; Lyon-Caen, O.; Tourbah, A. High doses of biotin in chronic progressive multiple sclerosis: A pilot study. Mult. Scler. Relat. Disord. 2015, 4, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Frenay, C.L.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; de Seze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. Effect of MD1003 (high doses of biotin) in progressive multiple sclerosis: Results of a pivotal phase III randomized double blind placebo controlled study. In Proceedings of the 67th Annual Meeting of the American Academy of Neurology, Wasgington, DC, USA, 18–25 April 2015.
- Vergo, S.; Craner, M.J.; Etzensperger, R.; Attfield, K.; Friese, M.A.; Newcombe, J.; Esiri, M.; Fugger, L. Acid-sensing ion canne is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain 2011, 134, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Craner, M.J.; Etzensperger, R.; Vergo, S.; Wemmie, J.A.; Welsh, M.J.; Vincent, A.; Fugger, L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat. Med. 2007, 13, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Kiernan, M.C. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhou, H.; Wilson, B.C.; Shi, J.S.; Hong, J.S.; Gao, H.M. Fluoxetine protects neurons against microglial activation mediated neurotoxicity. Park. Relat. Disord. 2012, 18, S213–S217. [Google Scholar] [CrossRef]
- Rudick, R.A.; Mi, S.; Sandrock, A.W., Jr. LINGO-1 antagonists as therapy for multiple sclerosis: In vitro and in vivo evidence. Expert Opin. Biol. Ther. 2008, 8, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Miller, R.H.; Lee, X.; Scott, M.L.; Shulag-Morskaya, S.; Shao, Z.; Chang, J.; Thill, G.; Levesque, M.; Zhang, M.; et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Butzkueven, H.; Ziemssen, F.; Massacesi, L.; Chai, Y.; et al. Evidence of remyelination with the anti-LINGO-1 monoclonal antibody BIIB033 after acute optic ne uritis. In Proceedings of the 67th Annual Meeting of the American Academy of Neurology, Wasgington, DC, USA, 18–25 April 2015.
- Phan, H.; DeReese, A.; Day, A.J.; Carvalho, M. The dual role of domperidone in gastroparesis and lactation. Int. J. Pharm. Compd. 2014, 18, 203–207. [Google Scholar] [PubMed]
- Bartels, C.; Spate, K.; Krampe, H.; Ehrenreich, H. Recombinant human erythropoietin: Novel strategies for neuroprotective/neuroregenerative treatment of multiple sclerosis. Ther. Adv. Neurol. Disord. 2008, 1, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Fischer, B.; Norra, C.; Schellenberger, F.; Stender, N.; Stiefel, M.; Sirén, A.L.; Paulus, W.; Nave, K.A.; Gold, R.; et al. Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 2007, 130, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Maeda, Y.; Yuan, R.R.; Elkabes, S.; Cook, S.; Dowling, P. Beneficial effect of erythropoietin on experimental allergic encephalomyelitis. Ann. Neurol. 2004, 56, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, M.; Merlini, A.; Greco, R.; Sangalli, F.; Comi, G.; Ciceri, F.; Martino, G. Autologous bone marrow transplantation for the treatment of multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Sadiq, S.A. Stem cell therapy in multiple sclerosis: A future perspective. Neurodegener. Dis. Manag. 2015, 5, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.Q.; Luan, S.L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef]
- Gibson, L.C.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; MacKenzie, F.L.; Nagasawa, M.; Stevens, P.A.; MacKenzie, S.J. The inhibitory profile of Ibudilast against the human phosphodiesteraseenzyme family. Eur. J. Pharmacol. 2006, 538, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Misu, T.; Fujihara, K.; Sakoda, S.; Nakatsuji, Y.; Fukaura, H.; Kikuchi, S.; Tashiro, K.; Suzumura, A.; Ishii, N.; et al. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Mult. Scler. 2004, 10, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Kurotani, T.; Komatsu, Y.; Kawanokuchi, J.; Kato, H.; Mitsuma, N.; Suzumura, A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004, 46, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: Antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Bezzi, P.; Trotti, D.; Volterra, A.; Racagni, G. Inhibitory effect of the neuroprotective agent idebenone on arachidonic acid metabolism in astrocytes. Eur. J. Pharmacol. 1999, 370, 161–167. [Google Scholar] [CrossRef]
- Fiebiger, S.M.; Bros, H.; Grobosch, T.; Janssen, A.; Chanvillard, C.; Paul, F.; Dörr, J.; Millward, J.M.; Infante-Duarte, C. The antioxidant idebenone fails to prevent or attenuate chronic experimental autoimmune encephalomyelitis in the mouse. J. Neuroimmunol. 2013, 262, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Salinthone, S.; Yadav, V.; Bourdette, D.N.; Carr, D.W. Lipoic acid: A novel therapeutic approach for multiple sclerosis and other chronic inflammatory diseases of the CNS. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Marracci, G.H.; Jones, R.E.; McKeon, G.P.; Bourdette, D.N. Alpha lipoic acid inhibits T cell migration into the spinal cord and suppresses and treats experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 131, 104–114. [Google Scholar] [CrossRef]
- Yadav, V.; Marracci, G.; Lovera, J.; Woodward, W.; Bogardus, K.; Marquardt, W.; Shinto, L.; Morris, C.; Bourdette, D. Lipoic acid in multiple sclerosis: A pilot study. Mult. Scler. 2005, 11, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3(GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- De Sarno, P.; Axtell, R.C.; Raman, C.; Roth, K.A.; Alessi, D.R.; Jope, R.S. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Benrabah, R.; Schmidt, N.; Zéphir, H.; Clavelou, P.; Vongsouthi, C.; Dubreuil, P.; Moussy, A.; Hermine, O. Masitinib treatment in patients with progressive multiple sclerosis: A randomized pilot study. BMC Neurol 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theoharides, T.C.; Kempuraj, D.; Kourelis, T.; Manola, A. Human mast cells stimulate activated T cells: Implications for multiple sclerosis. Ann. N. Y. Acad. Sci. 2008, 1144, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The crucial role of mast cells in blood–brain barrier alterations. Exp. Cell Res. 2015, 338, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Girvan, R.C.; Knight, D.A.; O’Loughlin, C.J.; Hayman, C.M.; Hermans, I.F.; Webster, G.A. MIS416, a non-toxic microparticle adjuvant derived from Propionibacterium acnes comprising immunostimulatorymuramyl dipeptide and bacterial DNA promotes cross-priming and Th1 immunity. Vaccine 2011, 29, 545–557. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Webster, G.; O’Sullivan, D.; Stone, S.; la Flamme, A.C. Targeting innate receptors with MIS416 reshapes Th responses and suppresses CNS disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, e87712. [Google Scholar] [CrossRef] [PubMed]
- Stuve, O.; Cravens, P.D.; Eagar, T.N. DNA-based vaccines: The future of multiple sclerosis therapy? Expert Rev. Neurother. 2008, 8, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Vandenbark, A.A. TCR peptide vaccination in multiple sclerosis: Boosting a deficient natural regulatory network that may involve TCR-specific CD4+CD25+ Treg cells. Curr. Drug Targets Inflamm. Allergy 2005, 4, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, R.E.; Kapoor, R. Neuroprotection for acute optic neuritis can it work? Mult. Scler. Relat. Disord. 2013, 2, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Liu, S.; Carrithers, M.; Carrithers, L.M.; Waxman, S.G. Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Ann. Neurol. 2007, 62, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Morsali, D.; Bechtold, D.; Lee, W.; Chauhdry, S.; Palchaudhuri, U.; Hassoon, P.; Snell, D.M.; Malpass, K.; Piers, T.; Pocock, J.; et al. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain 2013, 136, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Raftopoulos, R.; Hickman, S. Phenytoin is neuroprotective in acute optic neuritis: Results of a phase 2 randomized controlled trial. In Proceedings of the 67th Annual Meeting of the American Academy of Neurology, Wasgington, DC, USA, 18–25 April 2015.
- Van der Most, P.J.; Dolga, A.M.; Nijholt, I.M.; Luiten, P.G.; Eisel, U.L. Statins: Mechanisms of neuroprotection. Prog. Neurobiol. 2009, 88, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Youssef, S.; Dunn, S.E.; Prod’homme, T.; Neuhaus, O.; Stuve, O.; Greenwood, J.; Steinman, L.; Zamvil, S.S. Statins in the treatment of central nervous system autoimmune disease. J. Neuroimmunol. 2006, 178, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Stuve, O.; Zamvil, S.S.; Hartung, H.P. Are statins a treatment option for multiple sclerosis? Lancet Neurol. 2004, 3, 369–371. [Google Scholar] [CrossRef]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.; Clegg, S.; Nielsen, C.; et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 2014, 383, 2213–2222. [Google Scholar] [CrossRef]
- Gonzalez-Cabrera, P.J.; Brown, S.; Studer, S.M.; Rosen, H. S1P signaling: New therapies and opportunities. F1000Prime Rep. 2014, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Novgorodov, A.S.; El-Alwani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 2007, 21, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Brana, C.; Frossard, M.J.; Pescini Gobert, R.; Martinier, N.; Boschert, U.; Seabrook, T.J. Immunohistochemical detection of sphingosine-1-phosphate receptor 1 and 5 in human multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2014, 40, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bar-Or, A.; Cree, B.; Fox, R.; Giovannoni, G.; Gold, R.; Vermersch, P.; Lam, E.; Pohlmann, H.; Zhang-Auberson, L.; et al. Siponimod (BAF312) for the Treatment of Secondary Progressive Multiple Sclerosis: Design of the Phase 3 EXPAND Trial. In Proceedings of the 65th Annual Meeting of the American Academy of Neurology, San Diego, CA, USA, 16–23 March 2013.
- Lutterotti, A.; Martin, R. Getting specific: Monoclonal antibodies in multiple sclerosis. Lancet Neurol. 2008, 7, 538–547. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, E.; Patti, F.; Zanghì, A.; Zappia, M. A Personalized Approach in Progressive Multiple Sclerosis: The Current Status of Disease Modifying Therapies (DMTs) and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101725
D’Amico E, Patti F, Zanghì A, Zappia M. A Personalized Approach in Progressive Multiple Sclerosis: The Current Status of Disease Modifying Therapies (DMTs) and Future Perspectives. International Journal of Molecular Sciences. 2016; 17(10):1725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101725
Chicago/Turabian StyleD’Amico, Emanuele, Francesco Patti, Aurora Zanghì, and Mario Zappia. 2016. "A Personalized Approach in Progressive Multiple Sclerosis: The Current Status of Disease Modifying Therapies (DMTs) and Future Perspectives" International Journal of Molecular Sciences 17, no. 10: 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101725