Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity

Abstract
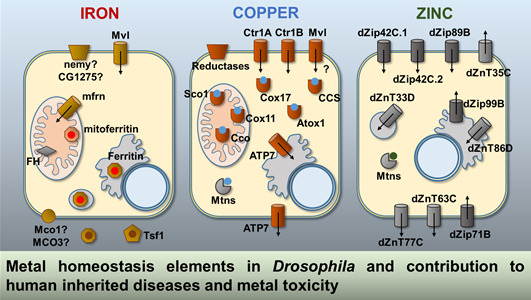
:
1. Introduction
2. Drosophila Genes Involved in Metal Homeostasis
2.1. Iron
2.2. Copper
2.3. Zinc
3. Drosophila as a Model of Human Inherited Diseases Related to Metal Homeostasis
3.1. Friedreich’s Ataxia
3.2. Menkes and Wilson Syndromes
3.3. Spondylocheirodysplasia-Ehlers-Danlos Syndrome-Like
3.4. Huntington’s Disease
3.5. Parkinson’s Disease
3.6. Alzheimer’s Disease
4. Drosophila as Model for Testing Metal Toxicity
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACO | Aconitase |
| AD | Alzheimer’s disease |
| Aβ | β-amyloid peptide |
| BBB | Blood brain barrier |
| CCS | Copper chaperones for superoxide dismutase |
| CP | Ceruloplasmin |
| CTR1 | Copper Transporter-1 |
| Cu | Copper |
| DAergic | Dopaminergic |
| DMT1 | Divalent metal transporter-1 |
| EDS | Ehlers-Danlos syndrome |
| ER | Endoplasmic reticulum |
| Fe | Iron |
| FRDA | Friedreich’s ataxia |
| FXN | Frataxin |
| HD | Huntington’s disease |
| Htt | Huntingtin |
| IRE | Iron-responsive element |
| IRP | Iron regulatory protein |
| ISC | Iron-sulfur cluster |
| LH1 | Lysyl hydroxylase |
| MCO | Multicopper oxidase |
| Mef-2 | Myocyte enhancer factor-2 |
| MRE | Metal response elements |
| MTF-1 | Metal-responsive transcription factor-1 |
| PD | Parkinson’s disease |
| Pdk1 | Protein kinase-1 |
| polyQ | Polyglutamine |
| QTL | Quantitative trait locus |
| RNAi | RNA interference |
| ROS | Reactive oxygen species |
| SCD-EDS | Spondylocheirodysplasia form of Ehlers-Danlos syndrome |
| SNpc | Substantia nigra pars compacta |
| SOD | Superoxide dismutase |
| TF | Transferrin |
| Zn | Zinc |
References
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nichols, C.D. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed]
- Hosamani, R. Muralidhara Acute exposure of Drosophila melanogaster to paraquat causes oxidative stress and mitochondrial dysfunction. Arch. Insect Biochem. Physiol. 2013, 83, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Ramirez, L.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Acute and chronic metal exposure impairs locomotion activity in Drosophila melanogaster: A model to study Parkinsonism. Biometals 2011, 24, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Paula, M.T.; Zemolin, A.P.; Vargas, A.P.; Golombieski, R.M.; Loreto, E.L.S.; Saidelles, A.P.; Picoloto, R.S.; Flores, E.M.M.; Pereira, A.B.; Rocha, J.B.T.; et al. Effects of Hg(II) exposure on MAPK phosphorylation and antioxidant system in D. melanogaster. Environ. Toxicol. 2014, 29, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.; Cheah, P.Y.; Ray, K.; Chia, W. Malvolio, the Drosophila homologue of mouse NRAMP-1 (Bcg), is expressed in macrophages and in the nervous system and is required for normal taste behaviour. EMBO J. 1995, 14, 3007–3020. [Google Scholar] [PubMed]
- Folwell, J.L.; Barton, C.H.; Shepherd, D. Immunolocalisation of the D. melanogaster Nramp homologue Malvolio to gut and Malpighian tubules provides evidence that Malvolio and Nramp2 are orthologous. J. Exp. Biol. 2006, 209, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Nichol, H.; Law, J.H.; Winzerling, J.J. Iron metabolism in insects. Annu. Rev. Entomol. 2002, 47, 535–559. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, B. Ferritin is the key to dietary iron absorption and tissue iron detoxification in Drosophila melanogaster. FASEB J. 2013, 27, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Dunkov, B.; Georgieva, T. Insect iron binding proteins: Insights from the genomes. Insect Biochem. Mol. Biol. 2006, 36, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, B. Iron homeostasis in insects: Insights from Drosophila studies. IUBMB Life 2013, 65, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gunkel, N.; Frishman, D.; Cyrklaff, A.; Tomancak, P.; Hentze, M.W. Iron-regulatory protein-1 (IRP-1) is highly conserved in two invertebrate species—Characterization of IRP-1 homologues in Drosophila melanogaster and Caenorhabditis elegans. Eur. J. Biochem. 1998, 254, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.I.; Missirlis, F.; Melefors, O.; Uhrigshardt, H.; Kirby, K.; Phillips, J.P.; Söderhäll, K.; Rouault, T.A. Of two cytosolic aconitases expressed in Drosophila, only one functions as an iron-regulatory protein. J. Biol. Chem. 2006, 281, 18707–18714. [Google Scholar] [CrossRef] [PubMed]
- Metzendorf, C.; Wu, W.; Lind, M.I. Overexpression of Drosophila mitoferrin in l(2)mbn cells results in dysregulation of Fer1HCH expression. Biochem. J. 2009, 421, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Cañizares, J.; Blanca, J.M.; Navarro, J.A.; Monrós, E.; Palau, F.; Moltó, M.D. dfh is a Drosophila homolog of the Friedreich’s ataxia disease gene. Gene 2000, 256, 35–42. [Google Scholar] [CrossRef]
- Verelst, W.; Asard, H. A phylogenetic study of cytochrome b561 proteins. Genome Biol. 2003, 4, R38. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Braun, C.L.; Kanost, M.R.; Gorman, M.J. Multicopper oxidase-1 is a ferroxidase essential for iron homeostasis in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2012, 109, 13337–13342. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Dittmer, N.T.; Lang, M.; Brummett, L.M.; Braun, C.L.; Davis, L.C.; Kanost, M.R.; Gorman, M.J. Multicopper oxidase-1 orthologs from diverse insect species have ascorbate oxidase activity. Insect Biochem. Mol. Biol. 2015, 59, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Isaya, G.; O’Neill, H.A.; Santambrogio, P.; Cozzi, A.; Arosio, P.; Levi, S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004, 13, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.-L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [PubMed]
- Bettedi, L.; Aslam, M.F.; Szular, J.; Mandilaras, K.; Missirlis, F. Iron depletion in the intestines of Malvolio mutant flies does not occur in the absence of a multicopper oxidase. J. Exp. Biol. 2011, 214, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Mandilaras, K.; Pathmanathan, T.; Missirlis, F. Iron absorption in Drosophila melanogaster. Nutrients 2013, 5, 1622–1647. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, P.V.E.; Klomp, L.W.J. New developments in the regulation of intestinal copper absorption. Nutr. Rev. 2009, 67, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Petris, M.J. The SLC31 (Ctr) copper transporter family. Pflug. Arch. 2004, 447, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, A.; Le Blanc, S.; Olivares, M.; Pizarro, F.; Ruz, M.; Arredondo, M. Iron, copper, and zinc transport: Inhibition of divalent metal transporter 1 (DMT1) and human copper transporter 1 (hCTR1) by shRNA. Biol. Trace Elem. Res. 2012, 146, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Illing, A.C.; Shawki, A.; Cunningham, C.L.; Mackenzie, B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol. Chem. 2012, 287, 30485–30496. [Google Scholar] [CrossRef] [PubMed]
- Guedes, S.M.; Vitorino, R.; Tomer, K.; Domingues, M.R.M.; Correia, A.J.F.; Amado, F.; Domingues, P. Drosophila melanogaster larval hemolymph protein mapping. Biochem. Biophys. Res. Commun. 2003, 312, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Vierstraete, E.; Cerstiaens, A.; Baggerman, G.; Van den Bergh, G.; De Loof, A.; Schoofs, L. Proteomics in Drosophila melanogaster: First 2D database of larval hemolymph proteins. Biochem. Biophys. Res. Commun. 2003, 304, 831–838. [Google Scholar] [CrossRef]
- Southon, A.; Burke, R.; Camakaris, J. What can flies tell us about copper homeostasis? Metallomics 2013, 5, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R.M.; Postlethwait, J.H.; Lefevre, H.W. MeV-ion microprobe analyses of whole Drosophila suggest that zinc and copper accumulation is regulated storage not deposit excretion. J. Exp. Biol. 1997, 200, 3235–3243. [Google Scholar] [PubMed]
- O’Donnell, M.J.; Maddrell, S.H. Fluid reabsorption and ion transport by the lower Malpighian tubules of adult female Drosophila. J. Exp. Biol. 1995, 198, 1647–1653. [Google Scholar] [PubMed]
- Zhou, H.; Cadigan, K.M.; Thiele, D.J. A copper-regulated transporter required for copper acquisition, pigmentation, and specific stages of development in Drosophila melanogaster. J. Biol. Chem. 2003, 278, 48210–48218. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Thiele, D.J. Drosophila Ctr1A functions as a copper transporter essential for development. J. Biol. Chem. 2007, 282, 24017–24026. [Google Scholar] [CrossRef] [PubMed]
- Southon, A.; Farlow, A.; Norgate, M.; Burke, R.; Camakaris, J. Malvolio is a copper transporter in Drosophila melanogaster. J. Exp. Biol. 2008, 211, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Kunst, C.; Culotta, V.C. Copper activation of superoxide dismutase 1 (SOD1) in vivo. Role for protein-protein interactions with the copper chaperone for SOD1. J. Biol. Chem. 2000, 275, 33771–33776. [Google Scholar] [CrossRef] [PubMed]
- Kirby, K.; Jensen, L.T.; Binnington, J.; Hilliker, A.J.; Ulloa, J.; Culotta, V.C.; Phillips, J.P. Instability of superoxide dismutase 1 of Drosophila in mutants deficient for its cognate copper chaperone. J. Biol. Chem. 2008, 283, 35393–35401. [Google Scholar] [CrossRef] [PubMed]
- Horng, Y.-C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 2004, 279, 35334–353340. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, D.; Oliva, M.; Duchi, S.; Latorre, D.; Cavaliere, V.; Barsanti, P.; Villani, G.; Gargiulo, G.; Caggese, C. Genetic, functional and evolutionary characterization of scox, the Drosophila melanogaster ortholog of the human SCO1 gene. Mitochondrion 2010, 10, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Günther, V.; Georgiev, O.; Schaffner, W. Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. Biometals 2011, 24, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R.; Gybina, A.A. Intracellular copper transport in mammals. J. Nutr. 2004, 134, 1003–1006. [Google Scholar] [PubMed]
- Harris, E.D. Basic and clinical aspects of copper. Crit. Rev. Clin. Lab. Sci. 2003, 40, 547–586. [Google Scholar] [CrossRef] [PubMed]
- Southon, A.; Palstra, N.; Veldhuis, N.; Gaeth, A.; Robin, C.; Burke, R.; Camakaris, J. Conservation of copper-transporting P(IB)-type ATPase function. Biometals 2010, 23, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Norgate, M.; Lee, E.; Southon, A.; Farlow, A.; Batterham, P.; Camakaris, J.; Burke, R. Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Mol. Biol. Cell 2006, 17, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Binks, T.; Lye, J.C.; Camakaris, J.; Burke, R. Tissue-specific interplay between copper uptake and efflux in Drosophila. J. Biol. Inorg. Chem. 2010, 15, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sellami, A.; Wegener, C.; Veenstra, J.A. Functional significance of the copper transporter ATP7 in peptidergic neurons and endocrine cells in Drosophila melanogaster. FEBS Lett. 2012, 586, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.; Commons, E.; Camakaris, J. Expression and localisation of the essential copper transporter DmATP7 in Drosophila neuronal and intestinal tissues. Int. J. Biochem. Cell. Biol. 2008, 40, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Wijmenga, C.; Klomp, L.W.J. Molecular regulation of copper excretion in the liver. Proc. Nutr. Soc. 2004, 63, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Uriu-Adams, J.Y.; Keen, C.L. Copper, oxidative stress, and human health. Mol. Asp. Med. 2005, 26, 268–298. [Google Scholar] [CrossRef] [PubMed]
- Bonneton, F.; Théodore, L.; Silar, P.; Maroni, G.; Wegnez, M. Response of Drosophila metallothionein promoters to metallic, heat shock and oxidative stresses. FEBS Lett. 1996, 380, 33–38. [Google Scholar] [CrossRef]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Egli, D.; Selvaraj, A.; Yepiskoposyan, H.; Zhang, B.; Hafen, E.; Georgiev, O.; Schaffner, W. Knockout of “metal-responsive transcription factor” MTF-1 in Drosophila by homologous recombination reveals its central role in heavy metal homeostasis. EMBO J. 2003, 22, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. The elusive function of metallothioneins. Proc. Natl. Acad. Sci. USA 1998, 95, 8428–8430. [Google Scholar] [CrossRef] [PubMed]
- Egli, D.; Domènech, J.; Selvaraj, A.; Balamurugan, K.; Hua, H.; Capdevila, M.; Georgiev, O.; Schaffner, W.; Atrian, S. The four members of the Drosophila metallothionein family exhibit distinct yet overlapping roles in heavy metal homeostasis and detoxification. Genes Cells 2006, 11, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Atanesyan, L.; Günther, V.; Celniker, S.E.; Georgiev, O.; Schaffner, W. Characterization of MtnE, the fifth metallothionein member in Drosophila. J. Biol. Inorg. Chem. 2011, 16, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Lichtlen, P.; Schaffner, W. Putting its fingers on stressful situations: The heavy metal-regulatory transcription factor MTF-1. Bioessays 2001, 23, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Egli, D.; Georgiev, O.; Schaffner, W. The Drosophila homolog of mammalian zinc finger factor MTF-1 activates transcription in response to heavy metals. Mol. Cell. Biol. 2001, 21, 4505–4514. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Egli, D.; Selvaraj, A.; Zhang, B.; Georgiev, O.; Schaffner, W. Metal-responsive transcription factor (MTF-1) and heavy metal stress response in Drosophila and mammalian cells: A functional comparison. Biol. Chem. 2004, 385, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.; Balamurugan, K.; Yepiskoposyan, H.; Zhou, H.; Egli, D.; Georgiev, O.; Thiele, D.J.; Schaffner, W. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Genes Dev. 2005, 19, 891–896. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.S. The role of zinc in growth and cell proliferation. J. Nutr. 2000, 130, 1500S–1508S. [Google Scholar] [PubMed]
- Srivastava, R.C.; Farookh, A.; Ahmad, N.; Misra, M.; Hasan, S.K.; Husain, M.M. Reduction of cis-platinum induced nephrotoxicity by zinc histidine complex: The possible implication of nitric oxide. Biochem. Mol. Biol. Int. 1995, 36, 855–862. [Google Scholar] [PubMed]
- Hennig, B.; Wang, Y.; Ramasamy, S.; McClain, C.J. Zinc protects against tumor necrosis factor-induced disruption of porcine endothelial cell monolayer integrity. J. Nutr. 1993, 123, 1003–1009. [Google Scholar] [PubMed]
- Rodriguez, P.; Darmon, N.; Chappuis, P.; Candalh, C.; Blaton, M.A.; Bouchaud, C.; Heyman, M. Intestinal paracellular permeability during malnutrition in guinea pigs: Effect of high dietary zinc. Gut 1996, 39, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [PubMed]
- Kitamura, H.; Morikawa, H.; Kamon, H.; Iguchi, M.; Hojyo, S.; Fukada, T.; Yamashita, S.; Kaisho, T.; Akira, S.; Murakami, M.; et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat. Immunol. 2006, 7, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Chimienti, F.; Giblin, L.J.; Gross, G.W.; Korichneva, I.; Li, Y.; Libert, S.; Maret, W.; Parviz, M.; Frederickson, C.J.; et al. Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. 2010, 235, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.G. Zinc transport in mammalian cells. Am. J. Physiol. 1996, 270, C401–C410. [Google Scholar] [PubMed]
- Kambe, T.; Yamaguchi-Iwai, Y.; Sasaki, R.; Nagao, M. Overview of mammalian zinc transporters. Cell. Mol. Life Sci. 2004, 61, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Redenti, S.; Chappell, R.L. Localization of zinc transporter-3 (ZnT-3) in mouse retina. Vis. Res. 2004, 44, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Cousins, R.J. Mammalian zinc transporters. Annu. Rev. Nutr. 2004, 24, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Findley, S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995, 14, 639–649. [Google Scholar] [PubMed]
- Bosomworth, H.J.; Thornton, J.K.; Coneyworth, L.J.; Ford, D.; Valentine, R.A. Efflux function, tissue-specific expression and intracellular trafficking of the Zn transporter ZnT10 indicate roles in adult Zn homeostasis. Metallomics 2012, 4, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Illades, D.; Chen, P.; Zogzas, C.E.; Hutchens, S.; Mercado, J.M.; Swaim, C.D.; Morrisett, R.A.; Bowman, A.B.; Aschner, M.; Mukhopadhyay, S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 2014, 34, 14079–14095. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Pérez, J.M.; Dell’Angelica, E.C. Zinc transporter 2 (SLC30A2) can suppress the vesicular zinc defect of adaptor protein 3-depleted fibroblasts by promoting zinc accumulation in lysosomes. Exp. Cell. Res. 2007, 313, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Itsumura, N.; Inamo, Y.; Okazaki, F.; Teranishi, F.; Narita, H.; Kambe, T.; Kodama, H. Compound heterozygous mutations in SLC30A2/ZnT2 results in low milk zinc concentrations: A novel mechanism for zinc deficiency in a breast-fed infant. PLoS ONE 2013, 8, e64045. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Findley, S.D. ZnT-2, a mammalian protein that confers resistance to zinc by facilitating vesicular sequestration. EMBO J. 1996, 15, 1784–1791. [Google Scholar] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, F.; Devergnas, S.; Favier, A.; Seve, M. Identification and cloning of a β-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes 2004, 53, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, F.; Devergnas, S.; Pattou, F.; Schuit, F.; Garcia-Cuenca, R.; Vandewalle, B.; Kerr-Conte, J.; van Lommel, L.; Grunwald, D.; Favier, A.; et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J. Cell Sci. 2006, 119, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Kukic, I.; Lee, J.K.; Coblentz, J.; Kelleher, S.L.; Kiselyov, K. Zinc-dependent lysosomal enlargement in TRPML1-deficient cells involves MTF-1 transcription factor and ZnT4 (Slc30a4) transporter. Biochem. J. 2013, 451, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Fukunaka, A.; Kurokawa, Y.; Teranishi, F.; Sekler, I.; Oda, K.; Ackland, M.L.; Faundez, V.; Hiromura, M.; Masuda, S.; Nagao, M.; et al. Tissue nonspecific alkaline phosphatase is activated via a two-step mechanism by zinc transport complexes in the early secretory pathway. J. Biol. Chem. 2011, 286, 16363–16373. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Yamazaki, T.; Ishida, Y.; Suzuki, T.; Oda, K.; Nagao, M.; Yamaguchi-Iwai, Y.; Kambe, T. Zinc transport complexes contribute to the homeostatic maintenance of secretory pathway function in vertebrate cells. J. Biol. Chem. 2006, 281, 17743–17750. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Kim, J.H.; Stallcup, M.R. GAC63, a GRIP1-dependent nuclear receptor coactivator. Mol. Cell. Biol. 2005, 25, 5965–5972. [Google Scholar] [CrossRef] [PubMed]
- Dufner-Beattie, J.; Langmade, S.J.; Wang, F.; Eide, D.; Andrews, G.K. Structure, function, and regulation of a subfamily of mouse zinc transporter genes. J. Biol. Chem. 2003, 278, 50142–50150. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Segawa, S.; Matsuo, T.; Nakamura, S.; Kikkawa, Y.; Nishida, K.; Nagasawa, K. Microglial zinc uptake via zinc transporters induces ATP release and the activation of microglia. Glia 2011, 59, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Gaither, L.A.; Eide, D.J. Functional expression of the human hZIP2 zinc transporter. J. Biol. Chem. 2000, 275, 5560–5564. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Hasegawa, S.; Ban, S.; Yamada, T.; Date, Y.; Mizutani, H.; Nakata, S.; Tanaka, M.; Hirashima, N. ZIP2 protein, a zinc transporter, is associated with keratinocyte differentiation. J. Biol. Chem. 2014, 289, 21451–21462. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, S.L.; Lönnerdal, B. Zn transporter levels and localization change throughout lactation in rat mammary gland and are regulated by Zn in mammary cells. J. Nutr. 2003, 133, 3378–3385. [Google Scholar] [PubMed]
- Kelleher, S.L.; Lopez, V.; Lönnerdal, B.; Dufner-Beattie, J.; Andrews, G.K. Zip3 (Slc39a3) functions in zinc reuptake from the alveolar lumen in lactating mammary gland. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R194–R201. [Google Scholar] [CrossRef] [PubMed]
- Dufner-Beattie, J.; Kuo, Y.-M.; Gitschier, J.; Andrews, G.K. The adaptive response to dietary zinc in mice involves the differential cellular localization and zinc regulation of the zinc transporters ZIP4 and ZIP5. J. Biol. Chem. 2004, 279, 49082–49090. [Google Scholar] [CrossRef] [PubMed]
- Geiser, J.; De Lisle, R.C.; Andrews, G.K. The zinc transporter Zip5 (Slc39a5) regulates intestinal zinc excretion and protects the pancreas against zinc toxicity. PLoS ONE 2013, 8, e82149. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.Y.; Duncan, F.E.; Que, E.L.; Kim, A.M.; O’Halloran, T.V.; Woodruff, T.K. Maternally-derived zinc transporters ZIP6 and ZIP10 drive the mammalian oocyte-to-egg transition. Mol. Hum. Reprod. 2014, 20, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Xie, F.; Shen, H.; Liu, Q.; Zheng, T.; Kou, X.; Wang, D.; Yang, J. Negative correlation of LIV-1 and E-cadherin expression in hepatocellular carcinoma cells. PLoS ONE 2013, 8, e56542. [Google Scholar] [CrossRef] [PubMed]
- Lichten, L.A.; Ryu, M.-S.; Guo, L.; Embury, J.; Cousins, R.J. MTF-1-mediated repression of the zinc transporter Zip10 is alleviated by zinc restriction. PLoS ONE 2011, 6, e21526. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Miyai, T.; Fujishiro, H.; Kawamura, M.; Yasuda, T.; Hijikata, A.; Bin, B.-H.; Irié, T.; Tanaka, J.; Atsumi, T.; et al. Zinc transporter SLC39A10/ZIP10 controls humoral immunity by modulating B-cell receptor signal strength. Proc. Natl. Acad. Sci. USA 2014, 111, 11786–11791. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kirschke, C.P.; Zhang, Y.; Yu, Y.Y. The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. J. Biol. Chem. 2005, 280, 15456–15463. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Nield, A.; Chew, G.-S.; Myers, M.A. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS ONE 2013, 8, e79316. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, W.; Yamazaki, T.; Yamaguchi-Iwai, Y.; Masuda, S.; Nagao, M.; Andrews, G.K.; Kambe, T. SLC39A9 (ZIP9) regulates zinc homeostasis in the secretory pathway: Characterization of the ZIP subfamily I protein in vertebrate cells. Biosci. Biotechnol. Biochem. 2009, 73, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Fukunaka, A.; Hagihara, M.; Watanabe, K.; Kamino, S.; Kambe, T.; Enomoto, S.; Hiromura, M. Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS ONE 2013, 8, e58022. [Google Scholar] [CrossRef] [PubMed]
- Gaither, L.A.; Eide, D.J. Eukaryotic zinc transporters and their regulation. Biometals 2001, 14, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Wan, Z.; Fan, Q.; Tang, X.; Zhou, B. The metal transporter ZIP13 supplies iron into the secretory pathway in Drosophila melanogaster. Elife 2014, 3, e03191. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Stathakis, D.G.; Burton, D.Y.; McIvor, W.E.; Krishnakumar, S.; Wright, T.R.; O’Donnell, J.M. The catecholamines up (Catsup) protein of Drosophila melanogaster functions as a negative regulator of tyrosine hydroxylase activity. Genetics 1999, 153, 361–382. [Google Scholar] [PubMed]
- Wang, Z.; Ferdousy, F.; Lawal, H.; Huang, Z.; Daigle, J.G.; Izevbaye, I.; Doherty, O.; Thomas, J.; Stathakis, D.G.; O’Donnell, J.M. Catecholamines up integrates dopamine synthesis and synaptic trafficking. J. Neurochem. 2011, 119, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Sasamura, T.; Khanna, M.R.; Whitley, M.; Fortini, M.E. Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 zinc transporter Catsup. Development 2013, 140, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Mathews, W.R.; Ong, D.; Milutinovich, A.B.; Van Doren, M. Zinc transport activity of Fear of Intimacy is essential for proper gonad morphogenesis and DE-cadherin expression. Development 2006, 133, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, M.; Mathews, W.R.; Samuels, M.; Moore, L.A.; Broihier, H.T.; Lehmann, R. Fear of intimacy encodes a novel transmembrane protein required for gonad morphogenesis in Drosophila. Development 2003, 130, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Pielage, J.; Kippert, A.; Zhu, M.; Klämbt, C. The Drosophila transmembrane protein Fear-of-intimacy controls glial cell migration. Dev. Biol. 2004, 275, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Rando, M.; Atienza-Manuel, A.; Martín, P.; Burke, R.; Ruiz-Gómez, M. Fear-of-intimacy-mediated zinc transport controls the function of zinc-finger transcription factors involved in myogenesis. Development 2016, 143, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, X.; Zhou, B. Functional studies of Drosophila zinc transporters reveal the mechanism for dietary zinc absorption and regulation. BMC Biol. 2013, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.D.; Burke, R. Local and systemic effects of targeted zinc redistribution in Drosophila neuronal and gastrointestinal tissues. Biometals 2015, 28, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Y.; Zhou, B. Dietary zinc absorption is mediated by ZnT1 in Drosophila melanogaster. FASEB J. 2009, 23, 2650–2661. [Google Scholar] [CrossRef] [PubMed]
- Dechen, K.; Richards, C.D.; Lye, J.C.; Hwang, J.E.C.; Burke, R. Compartmentalized zinc deficiency and toxicities caused by ZnT and Zip gene over expression result in specific phenotypes in Drosophila. Int. J. Biochem. Cell. Biol. 2015, 60, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lye, J.C.; Richards, C.D.; Dechen, K.; Paterson, D.; de Jonge, M.D.; Howard, D.L.; Warr, C.G.; Burke, R. Systematic functional characterization of putative zinc transport genes and identification of zinc toxicosis phenotypes in Drosophila melanogaster. J. Exp. Biol. 2012, 215, 3254–3265. [Google Scholar] [CrossRef] [PubMed]
- Yepiskoposyan, H.; Egli, D.; Fergestad, T.; Selvaraj, A.; Treiber, C.; Multhaup, G.; Georgiev, O.; Schaffner, W. Transcriptome response to heavy metal stress in Drosophila reveals a new zinc transporter that confers resistance to zinc. Nucleic Acids Res. 2006, 34, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Qin, Q.; Zhou, B. Functional studies of Drosophila zinc transporters reveal the mechanism for zinc excretion in Malpighian tubules. BMC Biol. 2017, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome-An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Palau, F.; Espinós, C. Autosomal recessive cerebellar ataxias. Orphanet J. Rare Dis. 2006, 1, 47. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Yandim, C.; Natisvili, T.; Festenstein, R. Gene regulation and epigenetics in Friedreich’s ataxia. J. Neurochem. 2013, 126, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Puccio, H. Multicellular models of Friedreich ataxia. J. Neurol. 2009, 256, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Puccio, H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum. Mol. Genet. 2010, 19, R103–R110. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Cazzalini, O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.R.; Kirby, K.; Hilliker, A.J.; Phillips, J.P. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum. Mol. Genet. 2005, 14, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.V.; Navarro, J.A.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.-L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Kovacevic, Z.; Sahni, S.; Lane, D.J.R.; Merlot, A.M.; Kalinowski, D.S.; Huang, M.L.-H.; Richardson, D.R. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clin. Sci. 2016, 130, 853–870. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Calap-Quintana, P.; Llorens, J.V.; Al-Ramahi, I.; Gutiérrez, L.; Martínez-Sebastián, M.J.; Botas, J.; Moltó, M.D. Metal Homeostasis Regulators Suppress FRDA Phenotypes in a Drosophila Model of the Disease. PLoS ONE 2016, 11, e0159209. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.-Y.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife 2016, 5, e16043. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ho, T.S.-Y.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 2016, 5, e20732. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; Le Quan Sang, K.H.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.-C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Tümer, Z.; Møller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Tümer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mutat. 2013, 34, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Sato, E.; Gu, Y.-H.; Shiga, K.; Fujisawa, C.; Kozuma, T. Effect of copper and diethyldithiocarbamate combination therapy on the macular mouse, an animal model of Menkes disease. J. Inherit. Metab. Dis. 2005, 28, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Johnson, P.; Jansen, L.A.; Kaler, S.G. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am. J. Med. Genet. A 2010, 152A, 2529–2534. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Yi, L.; Zerfas, P.M.; Brinster, L.R.; Sullivan, P.; Goldstein, D.S.; Prohaska, J.; Centeno, J.A.; Rushing, E.; Kaler, S.G. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol. Ther. 2011, 19, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Castro, K.I.; Hevia-Urrutia, F.J.; Sturniolo, G.C. Wilson’s disease: A review of what we have learned. World J. Hepatol. 2015, 7, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Bahadorani, S.; Bahadorani, P.; Marcon, E.; Walker, D.W.; Hilliker, A.J. A Drosophila model of Menkes disease reveals a role for DmATP7 in copper absorption and neurodevelopment. Dis. Model. Mech. 2010, 3, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.B.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Bucossi, S.; Polimanti, R.; Mariani, S.; Ventriglia, M.; Bonvicini, C.; Migliore, S.; Manfellotto, D.; Salustri, C.; Vernieri, F.; Rossini, P.M.; et al. Association of K832R and R952K SNPs of Wilson’s disease gene with Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 913–919. [Google Scholar] [PubMed]
- Squitti, R.; Polimanti, R.; Bucossi, S.; Ventriglia, M.; Mariani, S.; Manfellotto, D.; Vernieri, F.; Cassetta, E.; Ursini, F.; Rossini, P.M. Linkage disequilibrium and haplotype analysis of the ATP7B gene in Alzheimer’s disease. Rejuvenation Res. 2013, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.W.; Wang, J.; Burke, R. In Vivo Modeling of the Pathogenic Effect of Copper Transporter Mutations That Cause Menkes and Wilson Diseases, Motor Neuropathy, and Susceptibility to Alzheimer’s Disease. J. Biol. Chem. 2017, 292, 4113–4122. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P. The Ehlers-Danlos syndromes. In McKusick’s Heritable Disorders of Connective Tissue, 5th ed.; Beighton, P., Ed.; Mosyb: Maryland Heights, MO, USA, 1993; pp. 189–251. [Google Scholar]
- Myllyharju, J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 2008, 40, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Yeowell, H.N.; Walker, L.C. Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI. Mol. Genet. Metab. 2000, 71, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Walker, J.M.; Wang, F.; Park, J.G.; Palmer, A.E.; Giunta, C.; Rohrbach, M.; Steinmann, B.; Eide, D.J. Promotion of vesicular zinc efflux by ZIP13 and its implications for spondylocheiro dysplastic Ehlers-Danlos syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, E3530–E3538. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.S.; Strong, M.; Quarrell, O.W. The global prevalence of Huntington’s disease: A systematic review and discussion. Neurodegener. Dis. Manag. 2016, 6, 331–343. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.; Ambrose, C.; Duyao, M.; Myers, R.; Lin, C. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef] [PubMed]
- Hands, S.L.; Mason, R.; Sajjad, M.U.; Giorgini, F.; Wyttenbach, A. Metallothioneins and copper metabolism are candidate therapeutic targets in Huntington’s disease. Biochem. Soc. Trans. 2010, 38, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114 Pt 4, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; Flores, A.; Santamaría, A.; Ríos, C.; Galván-Arzate, S. Changes in transition metal contents in rat brain regions after in vivo quinolinate intrastriatal administration. Arch. Med. Res. 1996, 27, 449–452. [Google Scholar] [PubMed]
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc. Natl. Acad. Sci. USA 2013, 110, 14995–15000. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of Parkinson’s disease. Curr. Top. Behav. Neurosci. 2015, 22, 205–220. [Google Scholar] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [PubMed]
- Zhang, Y.; Gao, J.; Chung, K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Andrews, L.A.; Parker, T.J.; Pallanck, L.J. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum. Mol. Genet. 2005, 14, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Pesah, Y.; Pham, T.; Burgess, H.; Middlebrooks, B.; Verstreken, P.; Zhou, Y.; Harding, M.; Bellen, H.; Mardon, G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 2004, 131, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Oelhafen, S.; Hua, H.; Georgiev, O.; Schaffner, W.; Büeler, H. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol. Dis. 2010, 40, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Schaffner, W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 2010, 391, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bray, T.M.; Bettger, W.J. The physiological role of zinc as an antioxidant. Free Radic. Biol. Med. 1990, 8, 281–291. [Google Scholar] [CrossRef]
- Südmeyer, M.; Saleh, A.; Wojtecki, L.; Cohnen, M.; Gross, J.; Ploner, M.; Hefter, H.; Timmermann, L.; Schnitzler, A. Wilson’s disease tremor is associated with magnetic resonance imaging lesions in basal ganglia structures. Mov. Disord. 2006, 21, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P.; Olanow, C.W. The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol. 2007, 66, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Sengstock, G.J.; Olanow, C.W.; Dunn, A.J.; Barone, S.; Arendash, G.W. Progressive changes in striatal dopaminergic markers, nigral volume, and rotational behavior following iron infusion into the rat substantia nigra. Exp. Neurol. 1994, 130, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Morello, M.; Zatta, P.; Zambenedetti, P.; Martorana, A.; D’Angelo, V.; Melchiorri, G.; Bernardi, G.; Sancesario, G. Manganese intoxication decreases the expression of manganoproteins in the rat basal ganglia: An immunohistochemical study. Brain Res. Bull. 2007, 74, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Cass, W.A.; Grondin, R.; Andersen, A.H.; Zhang, Z.; Hardy, P.A.; Hussey-Andersen, L.K.; Rayens, W.S.; Gerhardt, G.A.; Gash, D.M. Iron accumulation in the striatum predicts aging-related decline in motor function in rhesus monkeys. Neurobiol. Aging 2007, 28, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.C.; Guilarte, T.R. Manganese neurotoxicity: Lessons learned from longitudinal studies in nonhuman primates. Environ. Health Perspect. 2009, 117, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova-Golts, N.; Petrucelli, L.; Hardy, J.; Lee, J.M.; Farer, M.; Wolozin, B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 2000, 20, 6048–6054. [Google Scholar] [PubMed]
- Davies, P.; Moualla, D.; Brown, D.R. Α-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Zhu, Z.-J.; Wu, K.-C.; Yung, W.-H.; Qian, Z.-M.; Ke, Y. Differential interaction between iron and mutant α-synuclein causes distinctive Parkinsonian phenotypes in Drosophila. Biochim. Biophys. Acta 2016, 1862, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar]
- Overk, C.R.; Masliah, E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 2014, 88, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Yoshiike, Y.; Kayed, R.; Milton, S.C.; Takashima, A.; Glabe, C.G. Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromol. Med. 2007, 9, 270–275. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Kudo, W.; Lee, H.-P.; Zou, W.-Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 2012, 21, 1138–1344. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Men, L.; Wang, J.; Zhang, Y.; Chickenyen, S.; Wang, Y.; Zhou, F. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathological [correction of neuropathalogical] relevance. Biochemistry 2007, 46, 9270–9282. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1–42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef] [PubMed]
- Rival, T.; Page, R.M.; Chandraratna, D.S.; Sendall, T.J.; Ryder, E.; Liu, B.; Lewis, H.; Rosahl, T.; Hider, R.; Camargo, L.M.; et al. Fenton chemistry and oxidative stress mediate the toxicity of the β-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur. J. Neurosci. 2009, 29, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.; Dziadulewicz, N.; Crowther, D.C. Iron is a specific cofactor for distinct oxidation-and aggregation-dependent Aβ toxicity mechanisms in a Drosophila model. Dis. Model. Mech. 2015, 8, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Zhang, Y.; Xu, H.; Thinakaran, G. Pathological and physiological functions of presenilins. Mol. Neurodegener. 2006, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Volitakis, I.; Li, Q.-X.; Laughton, K.; Evin, G.; Ho, M.; Dalziel, A.H.; Camakaris, J.; Bush, A.I. Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J. Biol. Chem. 2011, 286, 9776–9786. [Google Scholar] [CrossRef] [PubMed]
- Southon, A.; Greenough, M.A.; Ganio, G.; Bush, A.I.; Burke, R.; Camakaris, J. Presenilin promotes dietary copper uptake. PLoS ONE 2013, 8, e62811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Haapasalo, A.; Kim, D.Y.; Ingano, L.A.M.; Pettingell, W.H.; Kovacs, D.M. Presenilin/γ-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006, 20, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, P.; Wang, P.; Boissy, R.E.; Zheng, H. Regulation of tyrosinase trafficking and processing by presenilins: Partial loss of function by familial Alzheimer’s disease mutation. Proc. Natl. Acad. Sci. USA 2006, 103, 353–358. [Google Scholar] [CrossRef] [PubMed]
- He, Z.L.; Yang, X.E.; Stoffella, P.J. Trace elements in agroecosystems and impacts on the environment. J. Trace Elem. Med. Biol. 2005, 19, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy Metal Toxicity and the Environment; Springer: Basel, Switzerland, 2012; pp. 133–164. [Google Scholar]
- Tamás, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Finley, E.J.; Chakraborty, S.; Fretham, S.J.B.; Aschner, M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics 2012, 4, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kijak, E.; Rosato, E.; Knapczyk, K.; Pyza, E. Drosophila melanogaster as a model system of aluminum toxicity and aging. Insect Sci. 2014, 21, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Du, Y.; Xue, H.; Wu, Y.; Zhou, B. Aluminum induces neurodegeneration and its toxicity arises from increased iron accumulation and reactive oxygen species (ROS) production. Neurobiol. Aging 2012, 33, 199.e1–199.e12. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Yokel, R.A.; Nieboer, E.; Borchelt, D.; Cohen, J.; Harry, J.; Kacew, S.; Lindsay, J.; Mahfouz, A.M.; Rondeau, V. Human health risk assessment for aluminium, aluminium oxide, and aluminium hydroxide. J. Toxicol. Environ. Health. B Crit. Rev. 2007, 10, 1–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Yan, Y.; Xu, Y.; Jin, Y.; Lei, J.; Zou, X.; Ran, D.; Zhang, H.; Luan, S.; Gu, H. Alumina nanoparticles alter rhythmic activities of local interneurons in the antennal lobe of Drosophila. Nanotoxicology 2013, 7, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.H.; Lingas, E.O.; Rahman, M. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bull. World Health Organ. 2000, 78, 1093–1103. [Google Scholar] [PubMed]
- Rizki, M.; Kossatz, E.; Velázquez, A.; Creus, A.; Farina, M.; Fortaner, S.; Sabbioni, E.; Marcos, R. Metabolism of arsenic in Drosophila melanogaster and the genotoxicity of dimethylarsinic acid in the Drosophila wing spot test. Environ. Mol. Mutagen. 2006, 47, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.G.M.; Opoka, R.; Kane, D.; Cartwright, I.L. Investigating arsenic susceptibility from a genetic perspective in Drosophila reveals a key role for glutathione synthetase. Toxicol. Sci. 2009, 107, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Muñiz Ortiz, J.G.; Shang, J.; Catron, B.; Landero, J.; Caruso, J.A.; Cartwright, I.L. A transgenic Drosophila model for arsenic methylation suggests a metabolic rationale for differential dose-dependent toxicity endpoints. Toxicol. Sci. 2011, 121, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Niehoff, A.-C.; Schulz, J.; Soltwisch, J.; Meyer, S.; Kettling, H.; Sperling, M.; Jeibmann, A.; Dreisewerd, K.; Francesconi, K.A.; Schwerdtle, T.; et al. Imaging by Elemental and Molecular Mass Spectrometry Reveals the Uptake of an Arsenolipid in the Brain of Drosophila melanogaster. Anal. Chem. 2016, 88, 5258–5263. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Schulz, J.; Jeibmann, A.; Taleshi, M.S.; Ebert, F.; Francesconi, K.A.; Schwerdtle, T. Arsenic-containing hydrocarbons are toxic in the in vivo model Drosophila melanogaster. Metallomics 2014, 6, 2010–2014. [Google Scholar] [CrossRef] [PubMed]
- Griswold, W.; Martin, S. Human Health Effects of Heavy Metals. Environ. Sci. Technol. 2009, 15, 1–6. [Google Scholar]
- Guan, D.; Mo, F.; Han, Y.; Gu, W.; Zhang, M. Digital gene expression profiling (DGE) of cadmium-treated Drosophila melanogaster. Environ. Toxicol. Pharmacol. 2015, 39, 300–306. [Google Scholar] [CrossRef]
- Bernard, A. Cadmium & its adverse effects on human health. Indian J. Med. Res. 2008, 128, 557–564. [Google Scholar] [PubMed]
- Rizki, M.; Kossatz, E.; Creus, A.; Marcos, R. Genotoxicity modulation by cadmium treatment: Studies in the Drosophila wing spot test. Environ. Mol. Mutagen. 2004, 43, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Pragya, P.; Shukla, A.K.; Murthy, R.C.; Abdin, M.Z.; Kar Chowdhuri, D. Characterization of the effect of Cr(VI) on humoral innate immunity using Drosophila melanogaster. Environ. Toxicol. 2015, 30, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Sharma, A.; Negi, M.P.S.; Dwivedi, U.N.; Chowdhuri, D.K. Tracing the tracks of genotoxicity by trivalent and hexavalent chromium in Drosophila melanogaster. Mutat. Res. 2011, 722, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Brochin, R.; Leone, S.; Phillips, D.; Shepard, N.; Zisa, D.; Angerio, A. The Cellular Effect of Lead Poisoning and Its Clinical Picture. Georg. Undergrad. J. Health Sci. 2008, 5, 1–8. [Google Scholar]
- Hirsch, H.V.B.; Lnenicka, G.; Possidente, D.; Possidente, B.; Garfinkel, M.D.; Wang, L.; Lu, X.; Ruden, D.M. Drosophila melanogaster as a model for lead neurotoxicology and toxicogenomics research. Front. Genet. 2012, 3, 68. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Hirsch, H.V.B.; Ruden, D.M.; Lnenicka, G.A. Chronic lead exposure alters presynaptic calcium regulation and synaptic facilitation in Drosophila larvae. Neurotoxicology 2009, 30, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.V.B.; Possidente, D.; Averill, S.; Despain, T.P.; Buytkins, J.; Thomas, V.; Goebel, W.P.; Shipp-Hilts, A.; Wilson, D.; Hollocher, K.; et al. Variations at a quantitative trait locus (QTL) affect development of behavior in lead-exposed Drosophila melanogaster. Neurotoxicology 2009, 30, 305–311. [Google Scholar] [CrossRef]
- Ruden, D.M.; Chen, L.; Possidente, D.; Possidente, B.; Rasouli, P.; Wang, L.; Lu, X.; Garfinkel, M.D.; Hirsch, H.V.B.; Page, G.P. Genetical toxicogenomics in Drosophila identifies master-modulatory loci that are regulated by developmental exposure to lead. Neurotoxicology 2009, 30, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Carmona, E.R.; Creus, A.; Marcos, R. Genotoxicity testing of two lead-compounds in somatic cells of Drosophila melanogaster. Mutat. Res. 2011, 724, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.V.B.; Possidente, D.; Possidente, B. Pb2+: An endocrine disruptor in Drosophila? Physiol. Behav. 2010, 99, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.K.; Wilson, D.T.; Possidente, B.; McDaniel, P.; Morley, E.J.; Possidente, D.; Hollocher, K.T.; Ruden, D.M.; Hirsch, H.V.B. Accumulation, elimination, sequestration, and genetic variation of lead (Pb2+) loads within and between generations of Drosophila melanogaster. Chemosphere 2017, 181, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Horning, K.J.; Caito, S.W.; Tipps, K.G.; Bowman, A.B.; Aschner, M. Manganese Is Essential for Neuronal Health. Annu. Rev. Nutr. 2015, 35, 71–108. [Google Scholar] [CrossRef]
- Herrero Hernandez, E.; Discalzi, G.; Valentini, C.; Venturi, F.; Chiò, A.; Carmellino, C.; Rossi, L.; Sacchetti, A.; Pira, E. Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNa2EDTA. Neurotoxicology 2006, 27, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Ternes, A.P.L.; Zemolin, A.P.; da Cruz, L.C.; da Silva, G.F.; Saidelles, A.P.F.; de Paula, M.T.; Wagner, C.; Golombieski, R.M.; Flores, É.M.M.; Picoloto, R.S.; et al. Drosophila melanogaster—An embryonic model for studying behavioral and biochemical effects of manganese exposure. EXCLI J. 2014, 13, 1239–1253. [Google Scholar] [PubMed]
- Adedara, I.A.; Abolaji, A.O.; Rocha, J.B.T.; Farombi, E.O. Diphenyl Diselenide Protects Against Mortality, Locomotor Deficits and Oxidative Stress in Drosophila melanogaster Model of Manganese-Induced Neurotoxicity. Neurochem. Res. 2016, 41, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Contreras, R.; Medina-Leendertz, S.; Mora, M.; Villalobos, V.; Bravo, Y. Minocycline increases the life span and motor activity and decreases lipid peroxidation in manganese treated Drosophila melanogaster. Toxicology 2012, 294, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, M.C.; Gularte, C.O.A.; Escoto, D.F.; Pereira, G.; Gayer, M.C.; Roehrs, R.; Soares, F.A.A.; Puntel, R.L. Peumus boldus (Boldo) Aqueous Extract Present Better Protective Effect than Boldine Against Manganese-Induced Toxicity in D. melanogaster. Neurochem. Res. 2016, 41, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Chen, C.-F.; Dong, C.-D. Distribution and Accumulation of Mercury in Sediments of Kaohsiung River Mouth, Taiwan. APCBEE Procedia 2012, 1, 153–158. [Google Scholar] [CrossRef]
- Trasande, L.; Landrigan, P.J.; Schechter, C. Public health and economic consequences of methyl mercury toxicity to the developing brain. Environ. Health Perspect. 2005, 113, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Morais, S.; Costa, F.G.; Lourdes Pereir, M. De Heavy Metals and Human Health. In Environmental Health—Emerging Issues and Practice; InTech: Rijeka, Croatia, 2012. [Google Scholar]


{kind=link}
{kind=link}
| Human Gene | Primary Metals | Metal-Related Function | Drosophila Orthologue | References |
|---|---|---|---|---|
| Divalent Metal Transporter 1 (DMT1, SLC11A2) | Fe | Divalent metals transport | Malvolio (Mvl) | [10,11] |
| Iron absorption | ||||
| Ferritin Heavy Chain 1 (FTH1) | Fe | A component of ferritin | Ferritin 1 heavy chain homologue (Fer1HCH) | [12,15] |
| Iron storage | ||||
| Ferritin Light Chain (FTL) | Fe | A component of ferritin | Ferritin 2 light chain homologue (Fer2LCH) | [12,15] |
| Iron storage | ||||
| Ferritin Mitochondrial (FTMT) | Fe | Iron storage | Ferritin 3 heavy chain homologue (Fer3HCH) | [19] |
| Oxidative stress protection | ||||
| Transferrin (TF) | Fe | Serum iron binding transport protein | Transferrin 1 (Tsf1) | [13,14] |
| Aconitase 1 (ACO1) | Fe | Iron sensor | Iron regulatory protein 1A (Irp-1A) | [16,18] |
| Iron regulatory protein 1B (Irp-1B) | ||||
| Mitoferrin 1 (SLC25A37) | Fe | Mitochondrial iron importer | Mitoferrin (mfrn) | [19] |
| Frataxin (FXN) | Fe | Mitochondrial iron chaperone | Frataxin (fh) | [20] |
| Duodenal cytochrome b (DCYTB, CYBRD1) | Fe | Ferric-chelate reductase that reduces Fe3+ to Fe2+ | No extended memory (nemy) | [21] |
| CG1275 | ||||
| Hephaestin (HEPH) | Fe | Ferroxidase activity oxidizing Fe2+ to Fe3+ | Multicopper oxidase-1 (Mco1) | [22] |
| Ceruloplasmin (CP) | Fe | Ferroxidase activity oxidizing Fe2+ to Fe3+ | Multicopper oxidase-3 (MCO3) | [23] |
| Human Gene | Primary Metals | Metal-Related Function | Drosophila Orthologue | References |
|---|---|---|---|---|
| Solute carrier family 31 member 1 (SLC31A1, CTR1) | Cu | Copper uptake | Copper transporter 1A (Ctr1A) Copper transporter 1B (Ctr1B) Copper transporter 1C (Ctr1C) | [33,41,42] |
| Copper chaperone for superoxide dismutase (CCS) | Cu | Chaperone; copper donor to SOD1 | Copper chaperone for superoxide dismutase (Ccs) | [44,45] |
| Cytochrome c oxidase copper chaperone (COX17) | Cu | Chaperone; copper donor to COX11 and SCO1 | CG9065 | [46] |
| Cytochrome c oxidase copper chaperone (COX11) | Cu | Chaperone; copper transfer to cytochrome c oxidase | CG31648 | [46] |
| Cytochrome c oxidase assembly protein (SCO1) | Cu | Chaperone; copper transfer to cytochrome c oxidase | Synthesis of cytochrome c oxidase (Scox) | [46,47] |
| Antioxidant 1 copper chaperone (ATOX1) | Cu | Chaperone; copper donor to ATP7A and ATP7B | Antioxidant 1 copper chaperone (Atox1) | [48] |
| ATPase copper transporting α (ATP7A) | Cu | Copper delivery to proteins in the secretory pathway; copper efflux | ATP7 (ATP7) | [49,50,51,52,53,54,55] |
| ATPase copper transporting β (ATP7B) | Cu | Copper delivery to proteins in the secretory pathway; copper efflux | ATP7 (ATP7) | [49,50,51,52,53,54,55,56] |
| Human Gene | Primary Metals | Metal-Related Function | Drosophila Orthologue | References |
|---|---|---|---|---|
| SLC30A1 (hZnT1) | Zn | Exporting cytosolic zinc into the extracellular space | dZnT63C dZnT77C | [80] |
| SLC30A10 (hZnT10) | Mn, Zn | Zinc transporter localized to early/recycling endosomes or Golgi | [81,82] | |
| SLC30A2 (hZnT2) | Zn | Transporting zinc into the lumen of vesicular compartments | dZnT33D dZnT35C | [83,84,85] |
| SLC30A3 (hZnT3) | [86] | |||
| SLC30A8 (hZnT8) | [87,88] | |||
| SLC30A4 (hZnT4) | Zn | Maintenance of cytosolic zinc; homeostasis by controlling zinc; translocation to the lysosomes | dZnT41F | [89] |
| SLC30A7 (hZnT7) | Zn | Transports zinc into early secretory pathway and contributing to its homeostatic control | dZnT86D | [90,91] |
| SLC30A9 (hZnT9) | Zn | No zinc transport functions; acts as nuclear receptor coactivator | dZnT49B | [92] |
| SLC39A1 (hZIP1) | Zn | Imports zinc from the extracellular space | dZip42C.1 dZip42C.2 dZip89B dZip88E | [93,94] |
| SLC39A2 (hZIP2) | [95,96] | |||
| SLC39A3 (hZIP3) | [97,98] | |||
| SLC39A5 (hZIP5) | Zn | Zinc importer | dZip71B | [99,100] |
| SLC39A6 (hZIP6) | Zn | Zinc importer that can be a growth factor-elicited signaling molecule | fear of intimacy (foi) | [101,102] |
| SLC39A10 (hZIP10) | [103,104] | |||
| SLC39A7 (hZIP7) | Zn | Zinc importer from endoplasmic reticulum and Golgi apparatus; implicated in the glycemic control in skeletal muscle | Catecholamines up (catsup) | [105,106] |
| SLC39A9 (hZIP9) | Zn | Zinc importer localized to the Golgi apparatus and the cell surface; plays a crucial role in B-cell receptor | dZip102B | [107,108] |
| SLC39A11 (hZIP11) | Zn | Not well defined | dZip48C | [109] |
| SLC39A13 (hZIP13) | Fe | Mobilizes zinc from the lumen of Golgi apparatus and cytoplasmic vesicles to cytosol and plays a pivotal role in cellular signaling | dZip99C | [110] |
| Zn | [111] |
| Metal | Natural and Human Sources | Main Human Exposure | Symptoms | Drosophila Findings | Reference |
|---|---|---|---|---|---|
| Aluminum | Water treatment agents, aerosol, cosmetics, food additives, beverage cans, cookware, fireworks, explosives, rubber manufacturing | Drinking water, food, inhalation, dermal contact, pharmaceuticals | Mouth ulcers, skin lesions, bone, lung and brain damage, neurodegeneration, loss of memory, problems with balance and loss of coordination | Neurological injury, neurodegeneration, developmental alterations, behavior impairment, lifespan reduction and daily rhythm alterations, increase iron accumulation and ROS production | [215,216,217,218,219,220] |
| Arsenic | Arsenic minerals, sedimentary bed rocks, mining, melting, pesticides, fertilizers, drugs, soaps | Drinking contaminated water | Abnormal heart beat, damage in blood vessels, skin lesions, cancer, neurological problems, high rate of mortality | Genotoxicity of methylated metabolites, susceptibility related with genes of the biosynthesis of glutathione, brain injury, developmental alterations | [215,221,222,223,224,225,226] |
| Cadmium | Batteries, plastics, pigments, weathering, volcanic eruptions, river transport, fertilizers, pesticides, smelting, mining | Contaminated food and drinking water, inhalation, occupational exposure | Renal dysfunction, bone and lung damage and kidney disease | Changes in transferase enzymatic activity, stress response, cell cycle alterations, interference in DNA repair mechanism | [215,227,228,229,230] |
| Chromium | Burning of petroleum, coil and oil, pigment oxidants, fertilizers, metal planting tanneries, sewage, metallurgy, paper production | Water, occupational exposure | Ulcers, fever, renal failure, liver damage and hemorrhagic diathesis | DNA damage, alterations in pre- and post-replication mechanism implicated in repair DNA, changes in humoral innate immune response | [215,227,231,232] |
| Lead | Pipes, paints, gasoline, cosmetics, bullets, pesticides, fertilizers, mining, fossil fuel burning | Occupational exposure, food, smoking and water | Arthritis, renal dysfunction, vertigo, hallucinations, birth defects, mental retardation, psychosis, hyperactivity, autism, brain damage | Alterations in presynaptic calcium regulation, identification of QTL associated with behavioral lead-dependent changes, weak mutagenic effect, endocrine disruption | [215,227,233,234,235,236,237,238,239,240] |
| Manganese | Steel industry, mining, soil erosion, fungicides, fertilizers, dry-cell batteries, fireworks, ceramics, paint, cosmetics | Occupational exposure, water and food | Manganism, tremors, psychosis, fatigue, irritability | Reduced cell viability, induction of ROS, decrease in lifespan and locomotor activity | [6,216,241,242,243,244,245,246] |
| Mercury | Agriculture, mining, wastewater discharges, batteries | Contaminated water and marine food | Brain damage, memory problems, depression, hair loss, fatigue, tremors, changes in vision and hearing | Morphometric changes, interference in cellular signaling pathways and enzymatic mechanisms, inhibition of Notch cleavage by γ-secretase | [6,215,243,244,245,246,247,248,249] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calap-Quintana, P.; González-Fernández, J.; Sebastiá-Ortega, N.; Llorens, J.V.; Moltó, M.D. Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity. Int. J. Mol. Sci. 2017, 18, 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071456
Calap-Quintana P, González-Fernández J, Sebastiá-Ortega N, Llorens JV, Moltó MD. Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity. International Journal of Molecular Sciences. 2017; 18(7):1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071456
Chicago/Turabian StyleCalap-Quintana, Pablo, Javier González-Fernández, Noelia Sebastiá-Ortega, José Vicente Llorens, and María Dolores Moltó. 2017. "Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity" International Journal of Molecular Sciences 18, no. 7: 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071456