The Complete Chloroplast Genome of Catha edulis: A Comparative Analysis of Genome Features with Related Species


Abstract
:1. Introduction
2. Results and Discussion
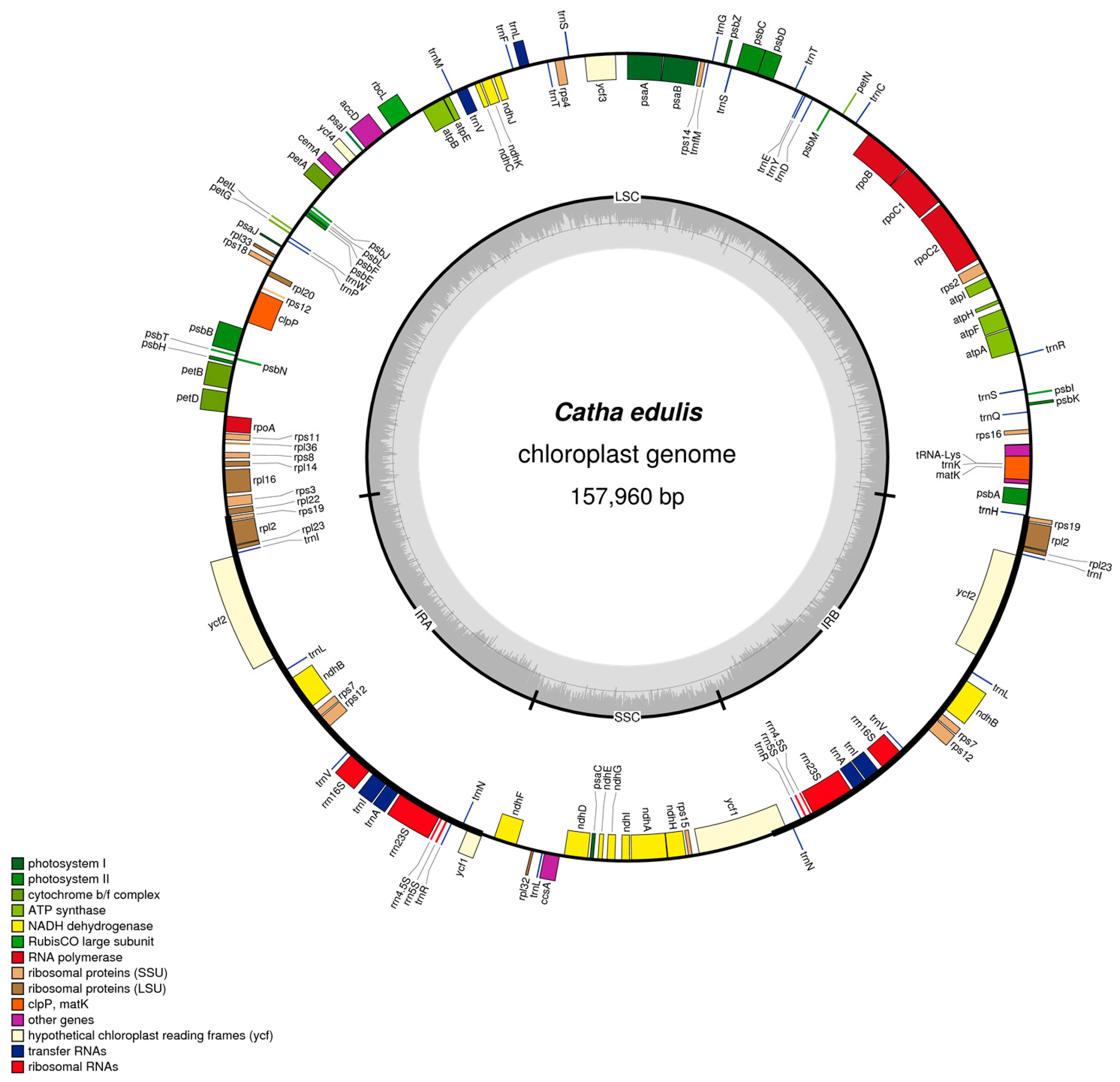
2.1. Chloroplast Assembly and Genome Features
2.2. Gene Content and Structure
2.3. Comparison of the cp Genomes
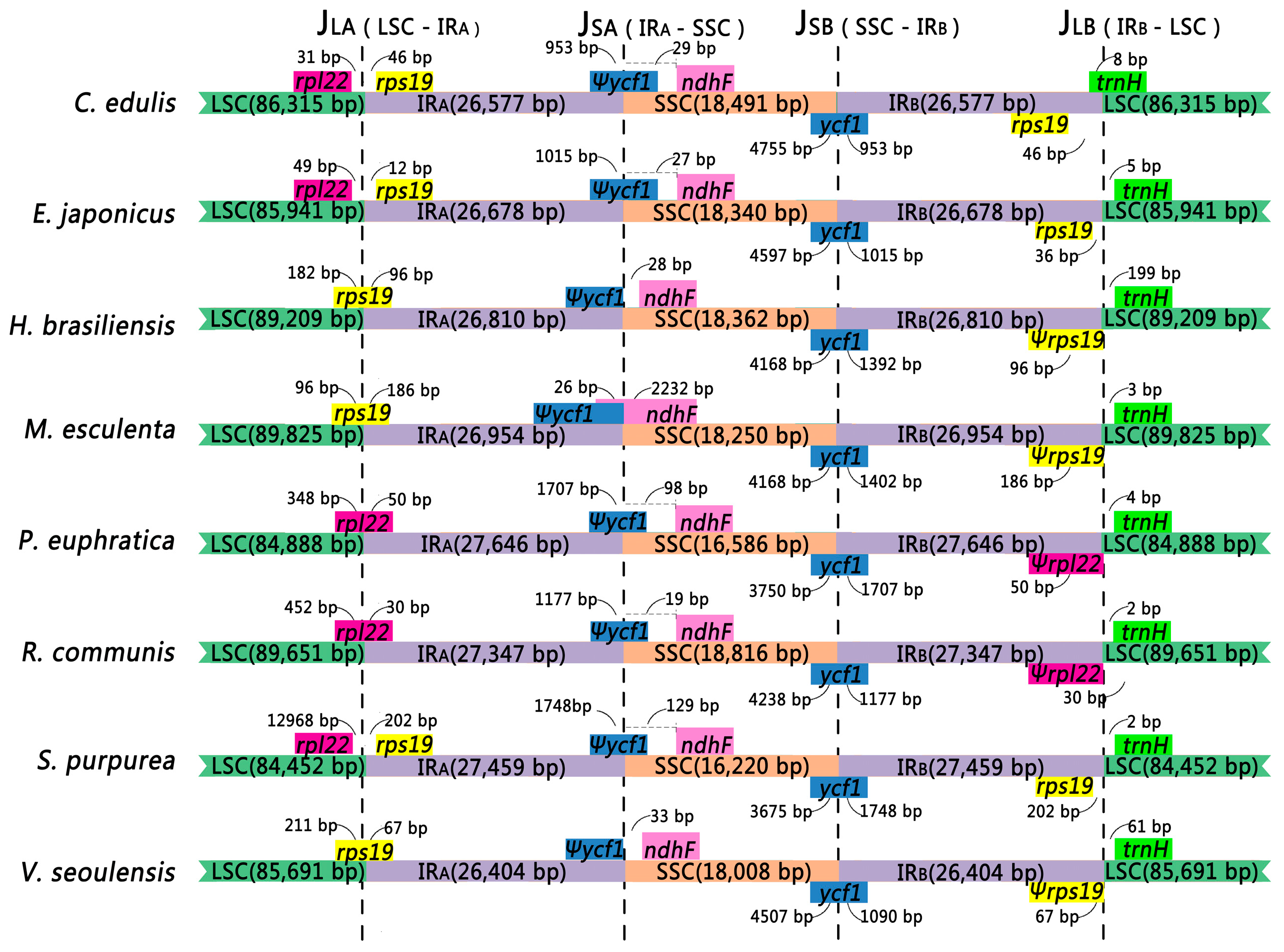
2.4. Contraction and Expansion in the Four Junction Regions
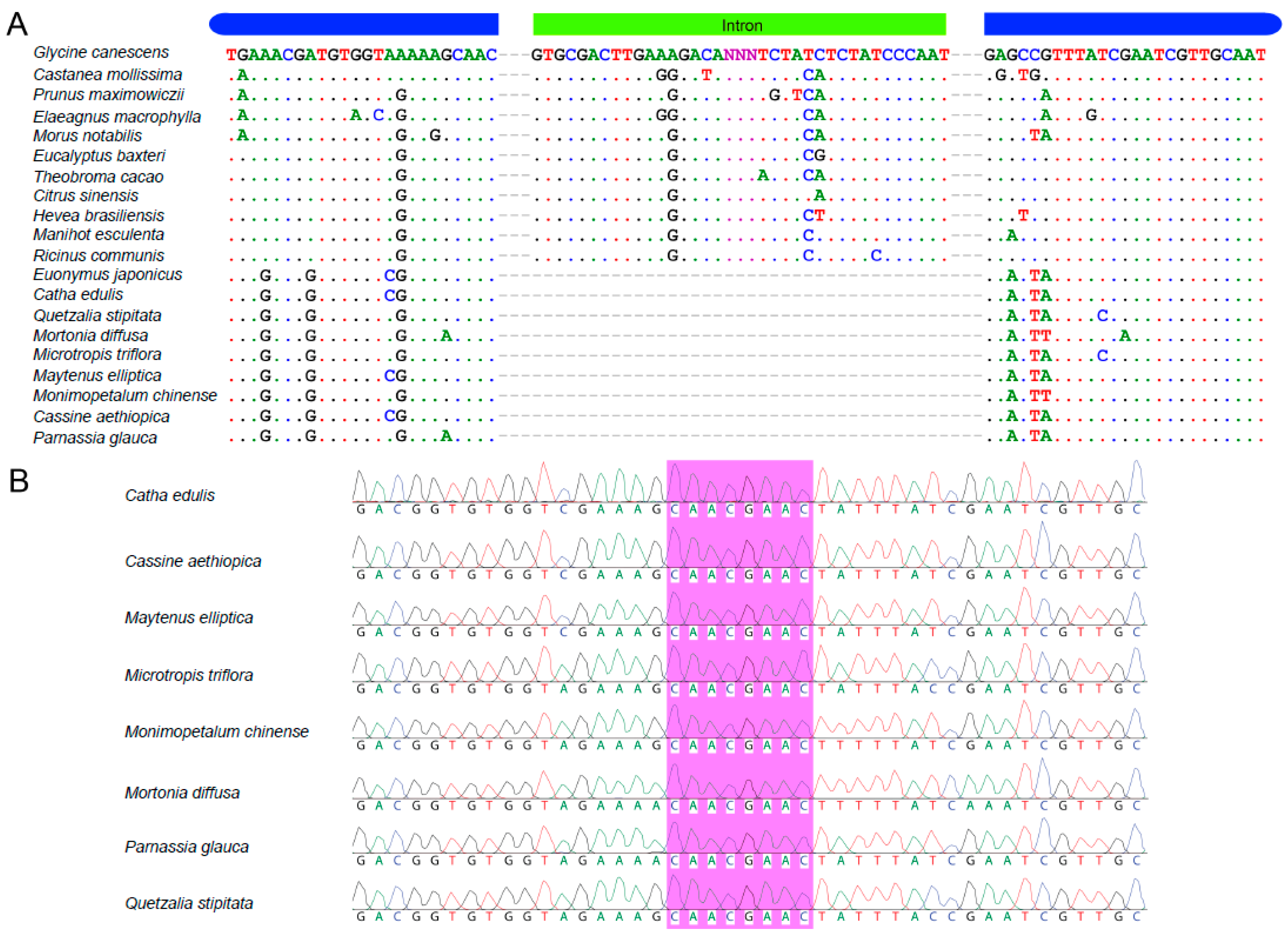
2.5. Verification of the rps16 Intron Loss from Catha and Seven Other Celastraceae Species
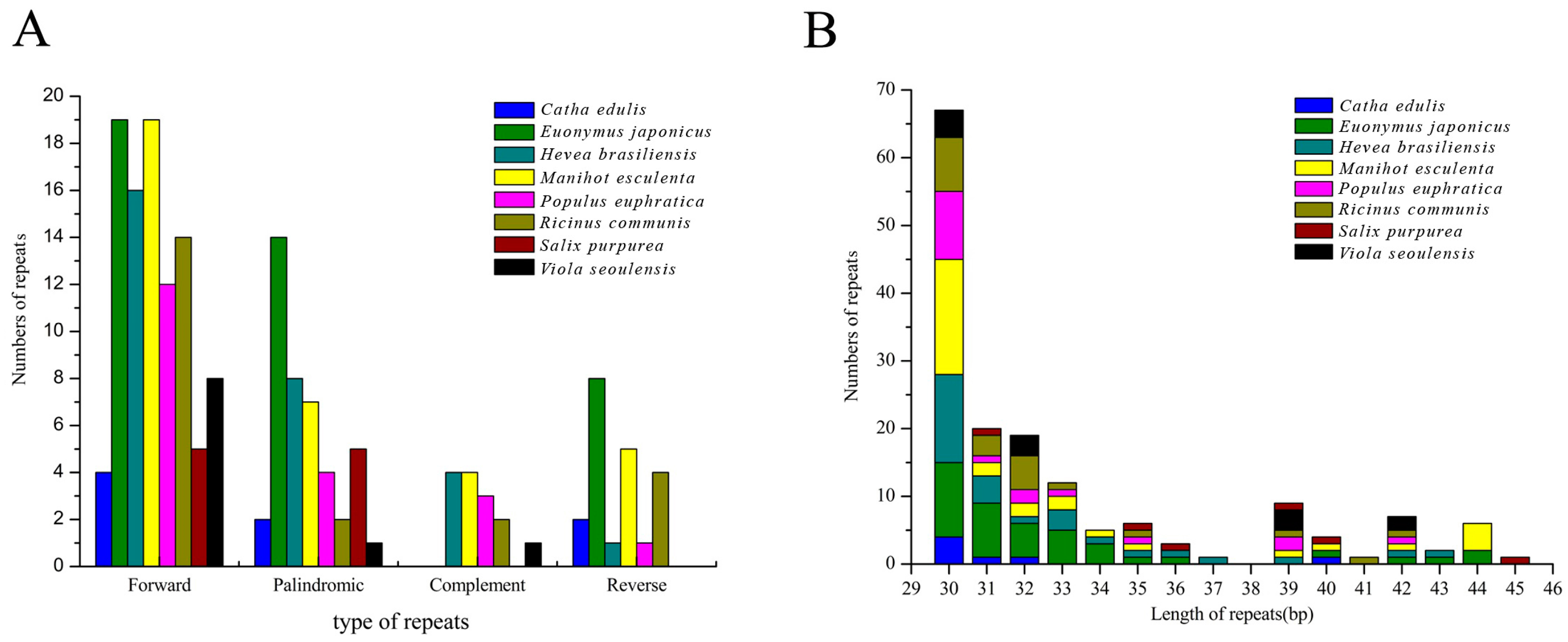
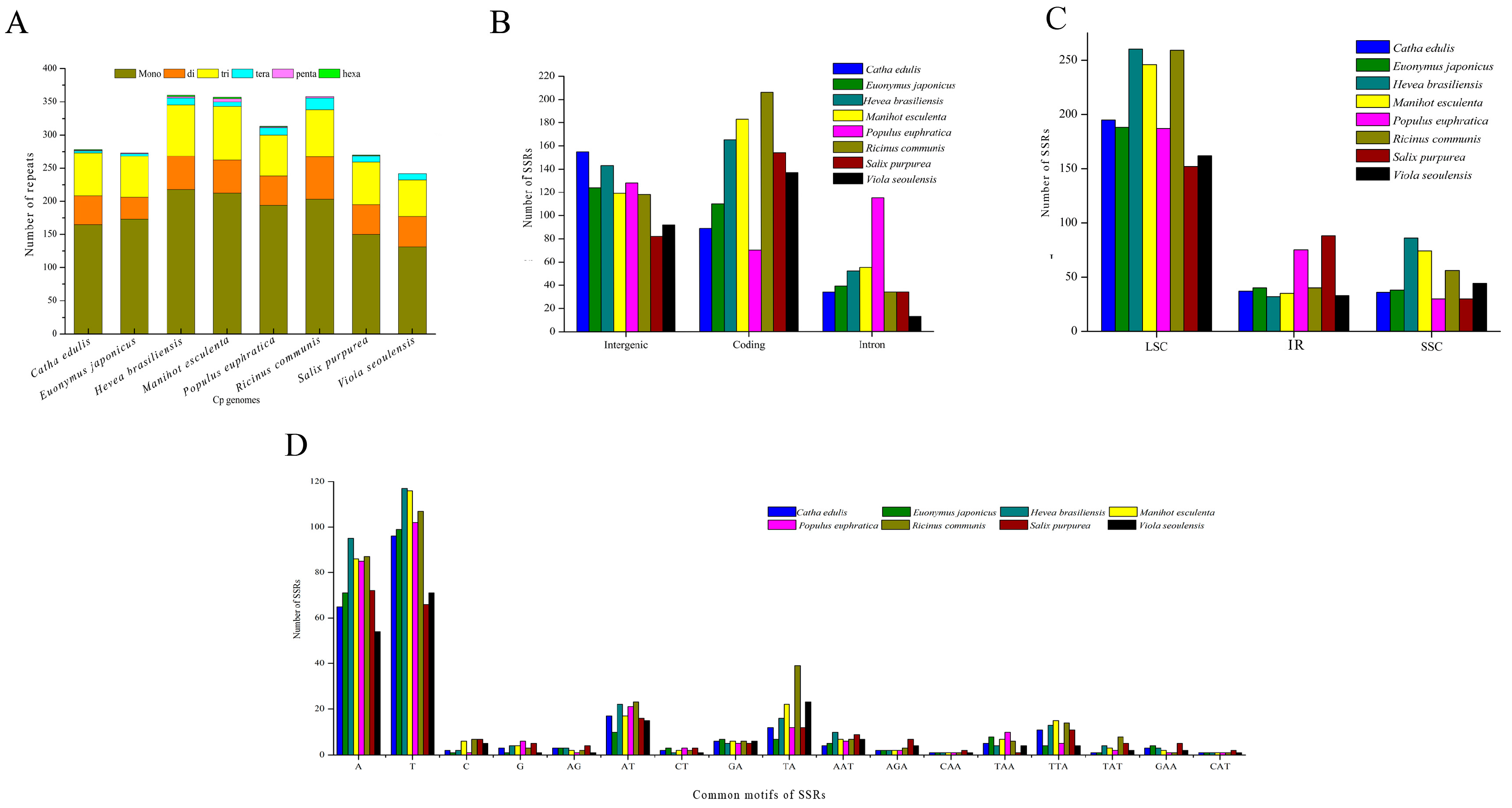
2.6. Identification of Long Repetitive Sequences
2.7. Chloroplast Genome Simple Sequence Repeats (SSRs)
2.8. Highly Informative Coding Genes and Markers for Phylogenomic Analysis
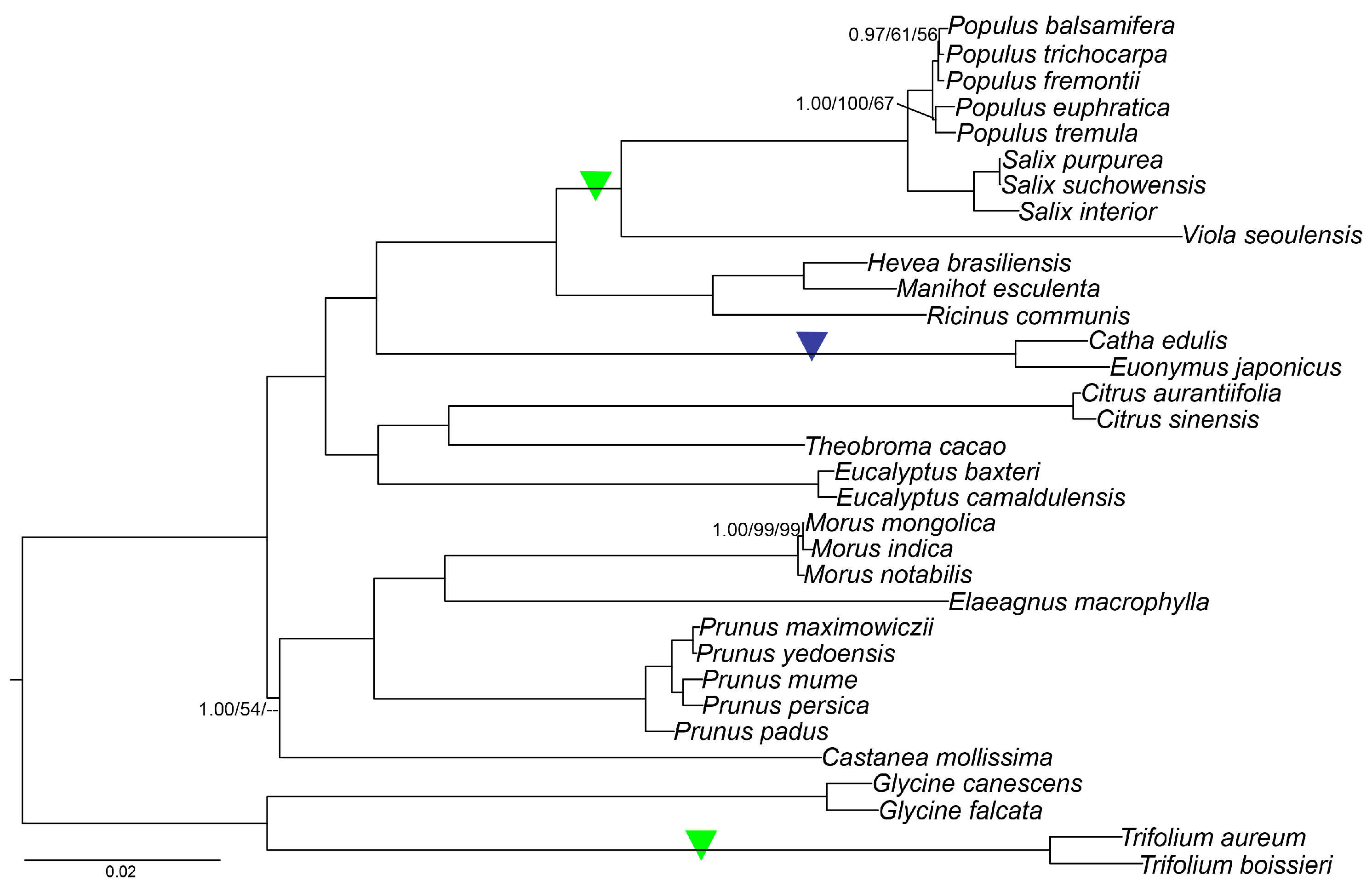
2.9. Phylogenetic Analysis
3. Materials and Methods
3.1. DNA Extraction and Sequencing
3.2. Chloroplast Genome Assembly and Sequence Analysis
3.3. Chloroplast Genomes Comparison
3.3.1. IR Expansion and Contraction
3.3.2. Repeat Analysis
3.3.3. Dot-Plot Analysis
3.3.4. Verification of the rps16 Intron Loss from Catha and Seven Other Celastraceae Genera
3.3.5. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Al-Motarreb, A.; Baker, K.; Broadly, K.J. Khat: Pharmacological and medical aspects and its social use in Yemen. Phytother. Res. 2002, 16, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Beckerleg, S.; Hailu, D.; Klein, A. The Khat Controversy: Stimulating the Debate on Drugs; Berg: Oxford, UK, 2007. [Google Scholar]
- Carrier, N.C.M. The Social Life of a Stimulant; Brill: Leiden, The Netherlands, 2007. [Google Scholar]
- Kennedy, J.G. The flower of paradise: The Institutional Use of the Drug Qat in North Yemen. Q. Rev. Biol. 1988, 63, 364–365. [Google Scholar]
- World Bank. Yemen: Towards Qat Demand Reduction; World Bank Document Report 39738-YE; World Bank: Washington, DC, USA, 2007. [Google Scholar]
- Gebissa, E. Leaf of Allah: Khat & Agricultural Transformation in Harerge, Ethiopia; James Currey Ltd.: Oxford, UK, 2004. [Google Scholar]
- Curto, M.A.; Tembrock, L.R.; Puppo, P.; Nogueira, M.; Simmons, M.P.; Meimberg, H. Evaluation of microsatellites of Catha edulis (qat; Celastraceae) identified using pyrosequencing. Biochem. Syst. Ecol. 2013, 49, 1–9. [Google Scholar] [CrossRef]
- Hagel, J.M.; Krezevski, K.; Sitrit, Y.; Marsolais, F.; Facchini, J.P.; Krizevski, R.; Lewinsohn, E. Expressed sequence tag analysis of khat (Catha edulis) provides a putative molecular biochemical basis for the biosynthesis of phenylpropylamino alkaloids. Genet. Mol. Biol. 2011, 34, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Tembrock, L.R.; Broeckling, C.D.; Heuberger, A.L.; Simmons, M.P.; Stermitz, F.R.; Uvarov, J.M. Employing two-stage derivatisation and GC–MS to assay for cathine and related stimulant alkaloids across the Celastraceae. Phytochem. Anal. 2017, 28, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.P.; Cappa, J.J.; Archer, R.H.; Ford, A.J.; Eichstedt, D.; Clevinger, C.C. Phylogeny of the Celastreae (Celastraceae) and the relationships of Catha edulis (qat) inferred from morphological characters and nuclear and plastid genes. Mol. Phylogenet. Evol. 2008, 48, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Tembrock, L.R.; Simmons, M.P.; Richards, C.M.; Reeves, P.A.; Reilley, A.; Curto, M.A.; Al-Thobhani, M.; Varisco, D.M.; Simpson, S.; Ngugi, G.; et al. Phylogeography of the wild and cultivated stimulant plant qat (Catha edulis, Celastraceae) in areas of historical cultivation. Am. J. Bot. 2017, 104, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Gu, C.H.; Tembrock, L.R.; Johnson, N.G.; Simmons, M.P.; Wu, Z.Q. The complete plastid genome of Lagerstroemia fauriei and loss of rpl2 intron from Lagerstroemia (Lythraceae). PLoS ONE 2016, 11, e0150752. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; DePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Gitzendanner, M.; Stull, G.; Chester, M.; Chanderbali, A.; Jordon-Thaden, I.; Soltis, P.S.; Schnable, P.S.; Barbazuk, W.B. The potential of genomics in plant systematics. Taxon 2013, 62, 886–898. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Tembrock, L.R.; Ge, S. Are Differences in Genomic Data Sets due to True Biological Variants or Errors in Genome Assembly: An Example from Two Chloroplast Genomes. PLoS ONE 2015, 10, e0118019. [Google Scholar] [CrossRef] [PubMed]
- CBOL. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar]
- Day, A.; Goldschmidt-Clermont, M. The chloroplast transformation toolbox: Selectable markers and marker removal. Plant Biotechnol. J. 2011, 9, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Ge, S. The phylogeny of the BEP clade in grasses revisited: Evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 2012, 62, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, H.; Johnson, A.M.; Boynton, J.E.; Gillham, N.W. Inhibition of chloroplast DNA recombination and repair by dominant negative mutants of Escherichia coli RecA. Mol. Cell. Biol. 1995, 15, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Maliga, P. Plastid transformation in higher plants. Annu. Rev. Plant Biol. 2004, 55, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Maliga, P.; Staub, J.; Carrer, H.; Kanevski, I.; Svab, Z. Homologous Recombination and Integration of Foreign DNA in Plastids of Higher Plants; Paszkowski, J., Ed.; Kluwer Academic: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Svab, Z.; Maliga, P. High-frequency plastid transformation in tobacco by selection for a chimeric aadA gene. Proc. Natl. Acad. Sci. USA 1993, 90, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.J.; Stanyon, R. Phylogenomics. Ancestral primate viewed. Nature 1999, 402, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Hogenhout, S.A.; Al-sadi, A.M.; Kuo, C. Complete chloroplast genome sequence of Omani Lime (Citrus aurantiifolia) and comparative analysis within the Rosids. PLoS ONE 2014, 9, e113049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Uthaipaisanwong, P.; Sangsrakru, D.; Chanprasert, J.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 2011, 475, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax ginseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Ryzhova, N.N.; Kholda, O.A.; Kochieva, E.Z. Structure characteristics of the chloroplast rps16 intron in Allium sativum and related Allium species. Mol. Biol. 2009, 43, 766–775. [Google Scholar] [CrossRef]
- Schwarz, E.N.; Ruhlman, T.A.; Sabir, J.S.; Hajrah, N.H.; Alharbi, N.S.; Al-Malki, A.L.; Bailey, C.D.; Jansen, R.K. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 2015, 53, 458–468. [Google Scholar] [CrossRef]
- Downie, S.R.; Olmstead, R.G.; Zurawski, G.; Soltis, D.E.; Soltis, S.; Watson, J.C.; Palmer, J.D. Six independent losses of the Chloroplast DNA rpl2 intron in Dicotyledons: Molecular and Phylogenetic Implications. Evolution 1991, 45, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Tank, D.C.; Eastman, J.M.; Pennell, M.W.; Soltis, P.S.; Soltis, D.E.; Hinchliff, C.E.; Brown, J.W.; Sessa, E.B.; Harmon, L.J. Nested radiations and the pulse of angiosperm diversification: Increased diversification rates often follow whole genome duplications. New Phytol. 2015, 207, 454–467. [Google Scholar] [CrossRef] [PubMed]
- CavalierSmith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, 62–64. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Amos, W.; Leggo, J.; Goodburn, S.; Jain, S.; Li, S.H.; Margolis, R.L.; Ross, C.A.; Ferguson-Smith, M.A. Microsatellite evolution—Evidence for directionality and variation in rate between species. Nat. Genet. 1995, 10, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Cho, J.; Boeynaems, S.; Verstrepen, K.J. Beyond junk-variable tandem repeats as facilitators of rapid evolution of regulatory and coding sequences. Genes 2012, 3, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Voronova, A.; Belevich, V.; Jansons, A.; Rungis, D. Stress-induced transcriptional activation of retrotransposon-like sequences in the Scots pine (Pinus sylvestris L.) genome. Tree Genet. Genomes 2014, 10, 937–951. [Google Scholar] [CrossRef]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Labra, M.; Scienza, A.; Imazio, S. Chloroplast SSR markers to assess DNA diversity in wild and cultivated grapevines. Vitis 2002, 41, 157–158. [Google Scholar]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Erickson, D.L. A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2014, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hilu, K.W.; Black, C.; Diouf, D.; Burleigh, J.G. Phylogenetic signal in matK vs. trnK: A case study in early diverging eudicots (angiosperms). Mol. Phylogenet. Evol. 2008, 48, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Jansen, R.K. ndhF sequence evolution and the major clades in the sunflower family. Proc. Natl. Acad. Sci. USA 1995, 92, 10379–10383. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Phylogeny of Catalpa (Bignoniaceae) inferred from sequences of chloroplast ndhF and nuclear ribosomal DNA. J. Syst. Evol. 2008, 46, 341–348. [Google Scholar]
- Yan, H.F.; Liu, Y.J.; Xie, X.F.; Zhang, C.Y.; Hu, C.M.; Hao, G.; Ge, X.J. DNA barcoding evaluation and its taxonomic implications in the species-rich genus Primula L. in China. PLoS ONE 2015, 10, e0122903. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Su, Y.J.; Wang, T. Plastid genome sequencing, comparative genomics, and phylogenomics: Current status and prospects. J. Syst. Evol. 2010, 48, 77–93. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Park, S. The complete chloroplast genome sequence of Euonymus japonicus (Celastraceae). Mitochondrial DNA 2015, 1736, 1–2. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2016, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wan, J.M. SSRHunter: Development of local searching software for SSR sites. Yi Chuan 2005, 27, 808–810. [Google Scholar] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dang, Y.; Li, Q.; Lu, J.J.; Li, X.W.; Wang, Y.T. Complete Chloroplast genome sequence of poisonous and medicinal plant Datura stramonium: Organizations and implications for genetic engineering. PLoS ONE 2014, 9, e110656. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.P. Independence of alignment and tree search. Mol. Phylogenet. Evol. 2004, 31, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. Paup*: Phylogenetic Analysis Using Parsimony (and other methods). Mccarthy 1993, 1–142. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M. New alogrithms and methods to estimate maximum-likelihoods phylogenies: Assessing the performance of PhyML 30. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Features | C. edulis | E. japonicus | H. brasiliensis | M. esculenta | P. euphratica | R. communis | S. purpurea | V. seoulensis |
|---|---|---|---|---|---|---|---|---|---|
| LSC | Length (bp) | 86,315 | 85,941 | 89,209 | 89,295 | 84,888 | 89,651 | 84,452 | 85,691 |
| GC Content (%) | 35.1 | 35.1 | 33.2 | 33.3 | 34.5 | 33.3 | 34.4 | 33.8 | |
| Length Percentage (%) | 54.6 | 54.5 | 55.3 | 55.3 | 54.1 | 54.9 | 54.3 | 54.8 | |
| SSC | Length (bp) | 18,491 | 18,340 | 18,362 | 18,250 | 16,586 | 18,816 | 16,220 | 18,008 |
| GC Content (%) | 31.8 | 31.8 | 29.5 | 29.6 | 30.6 | 29.5 | 31 | 29.6 | |
| Length Percentage (%) | 11.7 | 11.6 | 11.4 | 11.3 | 10.6 | 11.5 | 10.4 | 11.5 | |
| IR | Length (bp) | 26,577 | 26,678 | 26,810 | 26,954 | 27,646 | 27,347 | 27,459 | 26,404 |
| GC Content (%) | 42.7 | 42.7 | 42.2 | 42.3 | 41.9 | 41.9 | 41.9 | 42.6 | |
| Length Percentage (%) | 16.8 | 16.9 | 16.6 | 16.7 | 17.6 | 16.8 | 17.6 | 16.9 | |
| Total | Length (bp) | 157,960 | 157,637 | 161,191 | 161,453 | 156,766 | 163,161 | 155,590 | 156,507 |
| GC Content (%) | 37.3 | 37.3 | 35.7 | 35.9 | 36.7 | 35.7 | 36.7 | 36.3 |
| Region | Species | C. edulis | E. japonicus | H. brasiliensis | M. esculenta | P. euphratica | R. communis | S. purpurea | V. seoulensis |
|---|---|---|---|---|---|---|---|---|---|
| Protein coding | length (bp) | 78,471 | 77,331 | 78,852 | 79,089 | 78,728 | 78,119 | 77,898 | 78,310 |
| Length Percentage (%) | 49.7 | 49.1 | 48.9 | 49.0 | 50.2 | 47.9 | 50.1 | 50.0 | |
| GC Content (%) | 38 | 38.2 | 37.1 | 37.2 | 37.6 | 37.5 | 37.6 | 37.2 | |
| tRNA | length (bp) | 2806 | 2806 | 2798 | 2742 | 2796 | 2802 | 2792 | 2810 |
| Length Percentage (%) | 1.8 | 1.8 | 1.7 | 1.7 | 1.8 | 1.7 | 1.8 | 1.8 | |
| GC Content (%) | 52.6 | 53.3 | 53.2 | 53.3 | 53 | 53.2 | 52.9 | 53 | |
| rRNA | length (bp) | 9,050 | 9050 | 9050 | 9050 | 9050 | 9050 | 9,050 | 9050 |
| Length Percentage (%) | 5.7 | 5.7 | 5.6 | 5.6 | 5.8 | 5.5 | 5.8 | 5.8 | |
| GC Content (%) | 55.2 | 55.4 | 55.4 | 55.5 | 55.5 | 55.5 | 55.4 | 55.4 | |
| Intron | length (bp) | 18,474 | 19,287 | 18,538 | 18,479 | 18,210 | 18,278 | 17,321 | 18,348 |
| Length Percentage (%) | 11.7 | 12.2 | 11.5 | 11.4 | 11.6 | 11.2 | 11.1 | 11.7 | |
| GC Content (%) | 37.1 | 36.6 | 36.6 | 36.9 | 36.9 | 37.1 | 37.3 | 36.7 | |
| Intergenic | length (bp) | 49,159 | 49,163 | 51,953 | 52,093 | 47,982 | 54,912 | 48,529 | 47,989 |
| Length Percentage (%) | 31.1 | 31.2 | 32.2 | 32.3 | 30.6 | 33.7 | 31.2 | 30.7 | |
| GC Content (%) | 31.9 | 31.7 | 29 | 29 | 31 | 28.7 | 30.7 | 30.1 |
| Gene Category | Groups of Genes | Name of Genes |
|---|---|---|
| Self-replication | Transfer RNA genes | trnAUGC a,b trnCGCA trnDGUC trnEUUC trnFGAA trnfMCAU trnGUCC trnGGCC trnHGUG trnICAU b trnIGAU a,b trnKUUU a trnLCAA b trnLUAA a trnLUAG trnMCAU trnNGUU b trnPUGG trnQUUG trnRACG b trnRUCU trnSGCU trnSGGA trnSUGA trnTGGU trnTUGU trnVGAC b trnVUAC a trnWCCA trnYGUA |
| Small subunit of ribosome | rps2 rps3 rps4 rps7b rps8 rps11 rps12 a,b rps14 rps15 rps16 rps18 rps19 | |
| Ribosomal RNA genes | rrn16 b rrn23 b rrn4.5 b rrn5 b | |
| Large subunit of ribosome | rpl2 b rpl14 rpl16 a rpl20 rpl22 rpl23 b rpl32 rpl33 rpl36 | |
| DNA dependent RNA polymerase | rpoA rpoB rpoC1 a rpoC2 | |
| Photosynthesis | Subunits of photosystem I | psaA psaB psaC psaI psaJ |
| Subunits of photosystem II | psbA psbB psbC psbD psbE psbF psbH psbI psbJ psbK psbL psbM psbN psbT psbZ | |
| Subunits of cytochrome | petA petB a petD a petG petL petN | |
| Subunits of ATP synthase | atpA atpB atpE atpF a atpH atpI | |
| ATP-dependent protease subunit p gene | clpP a | |
| Large subunit of Rubisco | rbcL | |
| Subunits of NADH dehydrogenase | ndhA a ndhB a,b ndhC ndhD ndhE ndhF ndhG ndhH ndhI ndhJ ndhK | |
| Other genes | Maturase | matK |
| Envelop membrane protein | cemA | |
| Subunit of acetyl-CoA-carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Genes of unknown function | Conserved open reading frames | ycf1 ycf2 b ycf3 a ycf4 |
| Gene Name | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| rpoC1 | LSC | 1632 | 817 | 441 | ||
| atpF | LSC | 396 | 699 | 159 | ||
| petB | LSC | 6 | 773 | 642 | ||
| petD | LSC | 8 | 784 | 475 | ||
| ndhB | IR | 756 | 687 | 777 | ||
| ndhA | SSC | 540 | 1178 | 573 | ||
| rpl16 | LSC | 399 | 1119 | 9 | ||
| rpl2 | IR | 471 | 648 | 393 | ||
| rps12 | LSC | 114 | 27 | 546 | 231 | |
| ycf3 | LSC | 153 | 727 | 228 | 731 | 126 |
| clpP | LSC | 231 | 676 | 291 | 849 | 69 |
| trnK-UUU | LSC | 29 | 2495 | 37 | ||
| trnL-UAA | LSC | 37 | 540 | 50 | ||
| trnV-UAC | LSC | 37 | 663 | 39 | ||
| trnI-GAU | IR | 42 | 939 | 35 | ||
| trnA-UGC | IR | 38 | 801 | 35 | ||
| trnG-UCC | LSC | 23 | 761 | 48 |
| No. | Region | Length (bp) 1 | Aligned Length (bp) 2 | Conserved Sites | Parsimony Informative 3 | Parsimony Informative % 4 | CI. 5 | RI 6 | SI 7 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | matK | 1518 | 1575 | 1028 | 265 | 16.83 | 0.82 | 0.7 | 0.9 |
| 2 | ycf1 | 5640 | 6327 | 3970 | 1063 | 16.80 | 0.82 | 0.6 | 0.8 |
| 3 | ccsA | 969 | 987 | 689 | 160 | 16.21 | 0.84 | 0.7 | 0.9 |
| 4 | accD | 1509 | 1401 | 242 | 227 | 16.20 | 0.83 | 0.7 | 0.8 |
| 5 | rps3 | 648 | 663 | 467 | 107 | 16.14 | 0.82 | 0.7 | 0.9 |
| 6 | ndhF | 2232 | 2331 | 1606 | 368 | 15.79 | 0.81 | 0.6 | 0.8 |
| 7 | rps8 | 405 | 411 | 294 | 64 | 15.57 | 0.8 | 0.7 | 0.9 |
| 8 | rpl22 | 399 | 551 | 345 | 82 | 14.88 | 0.83 | 0.6 | 0.7 |
| 9 | petL | 96 | 96 | 70 | 14 | 14.58 | 0.9 | 0.8 | 0.9 |
| 10 | ndhD | 1503 | 1527 | 1116 | 207 | 13.56 | 0.82 | 0.7 | 0.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, C.; Tembrock, L.R.; Zheng, S.; Wu, Z. The Complete Chloroplast Genome of Catha edulis: A Comparative Analysis of Genome Features with Related Species. Int. J. Mol. Sci. 2018, 19, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020525
Gu C, Tembrock LR, Zheng S, Wu Z. The Complete Chloroplast Genome of Catha edulis: A Comparative Analysis of Genome Features with Related Species. International Journal of Molecular Sciences. 2018; 19(2):525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020525
Chicago/Turabian StyleGu, Cuihua, Luke R. Tembrock, Shaoyu Zheng, and Zhiqiang Wu. 2018. "The Complete Chloroplast Genome of Catha edulis: A Comparative Analysis of Genome Features with Related Species" International Journal of Molecular Sciences 19, no. 2: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020525