Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution

Abstract
:1. Introduction
2. Results
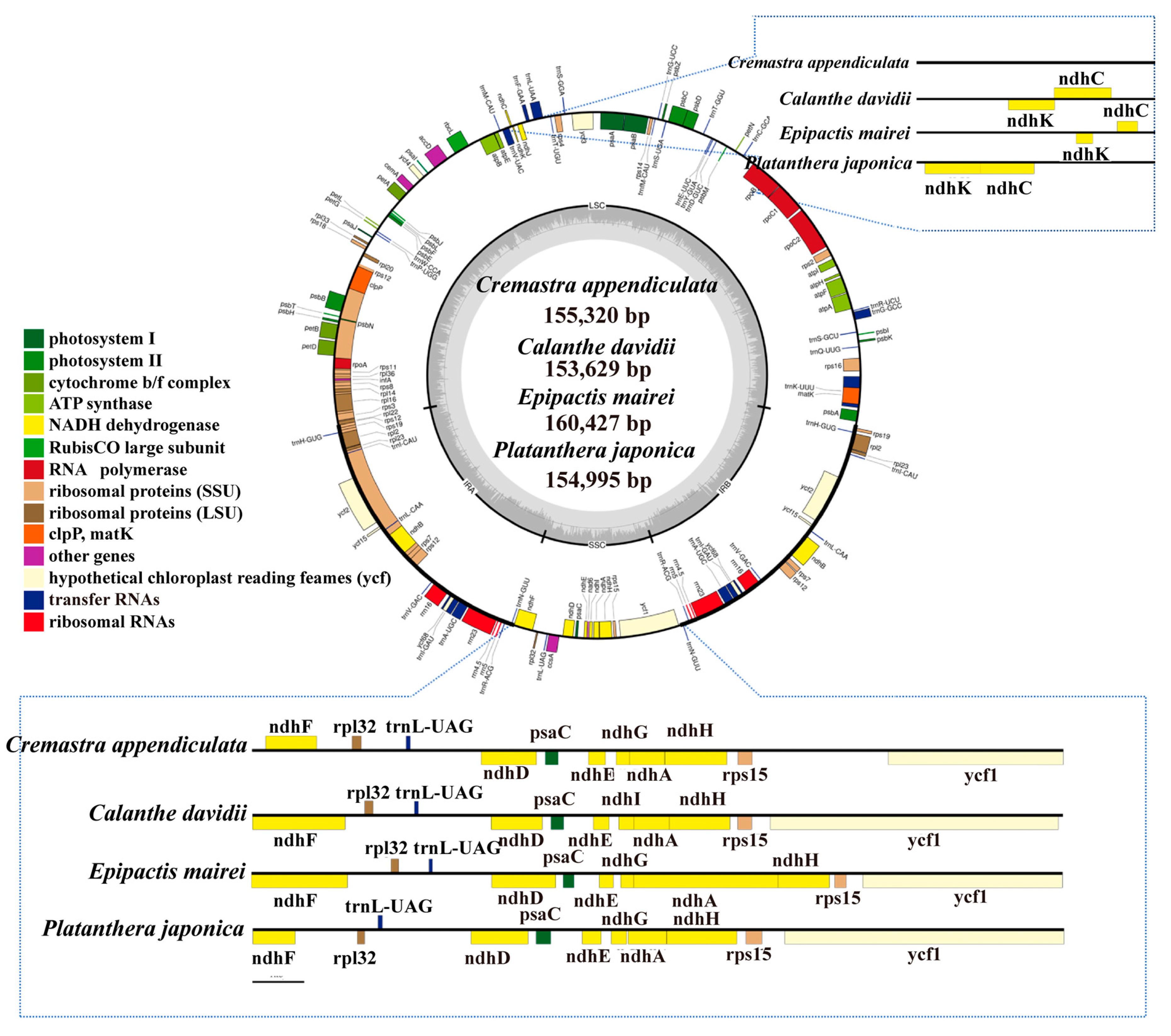
2.1. The Chloroplast Genome Structures
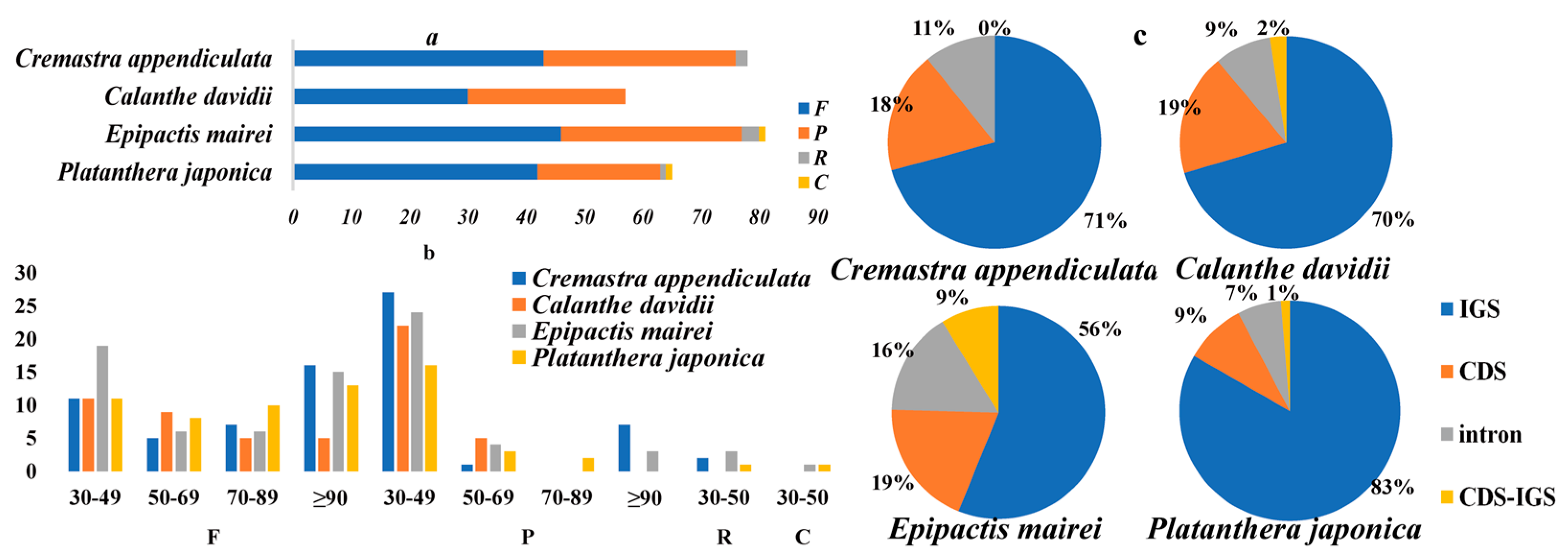
2.2. Repeat Structure and Simple Sequence Repeats
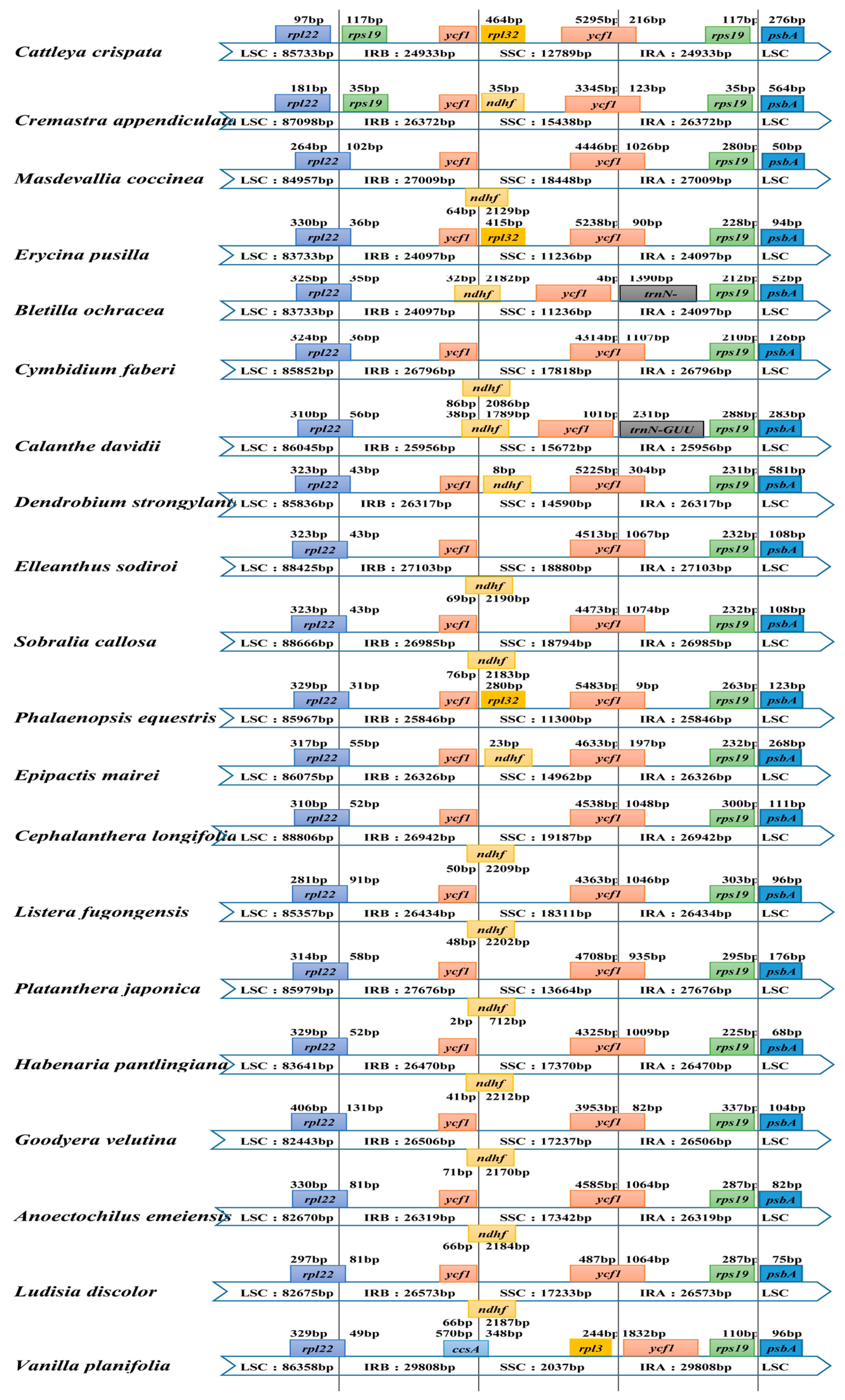
2.3. IR Contraction and Expansion
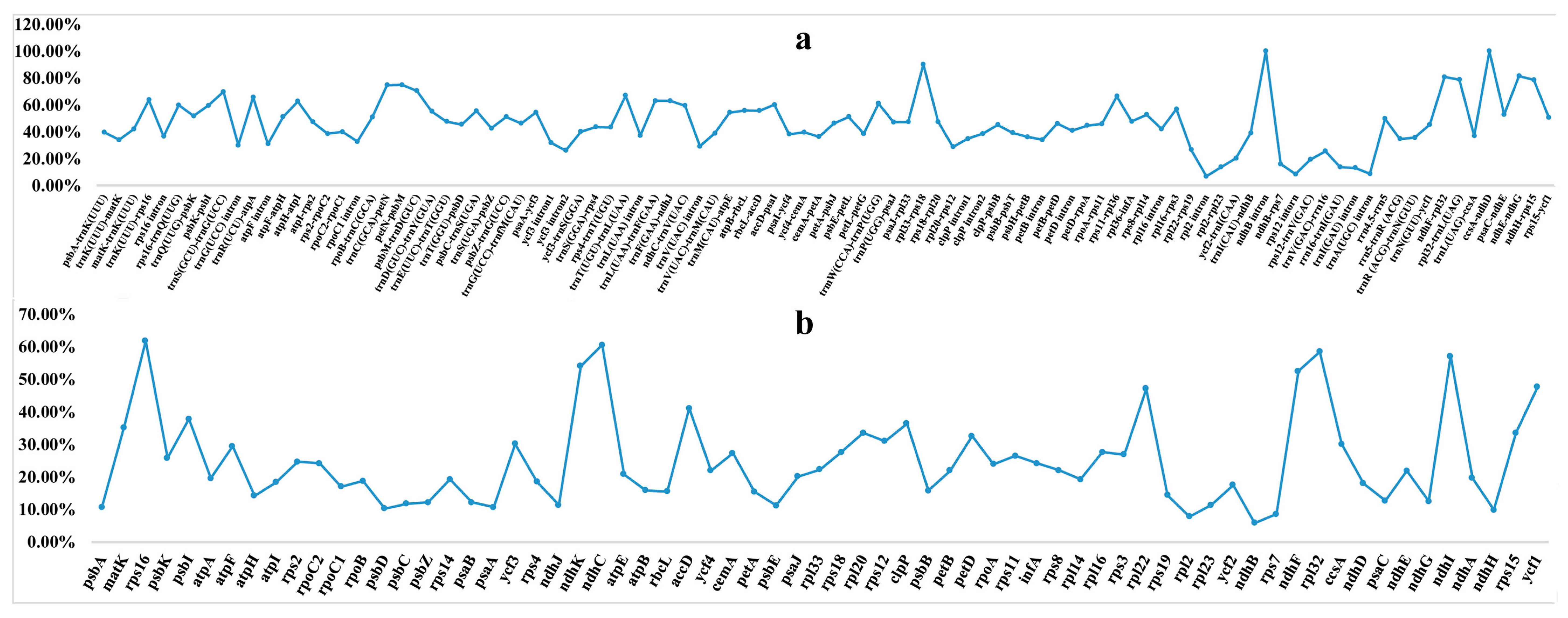
2.4. Sequence Divergence and Mutational Hotspot
2.5. Gene Selective Analysis
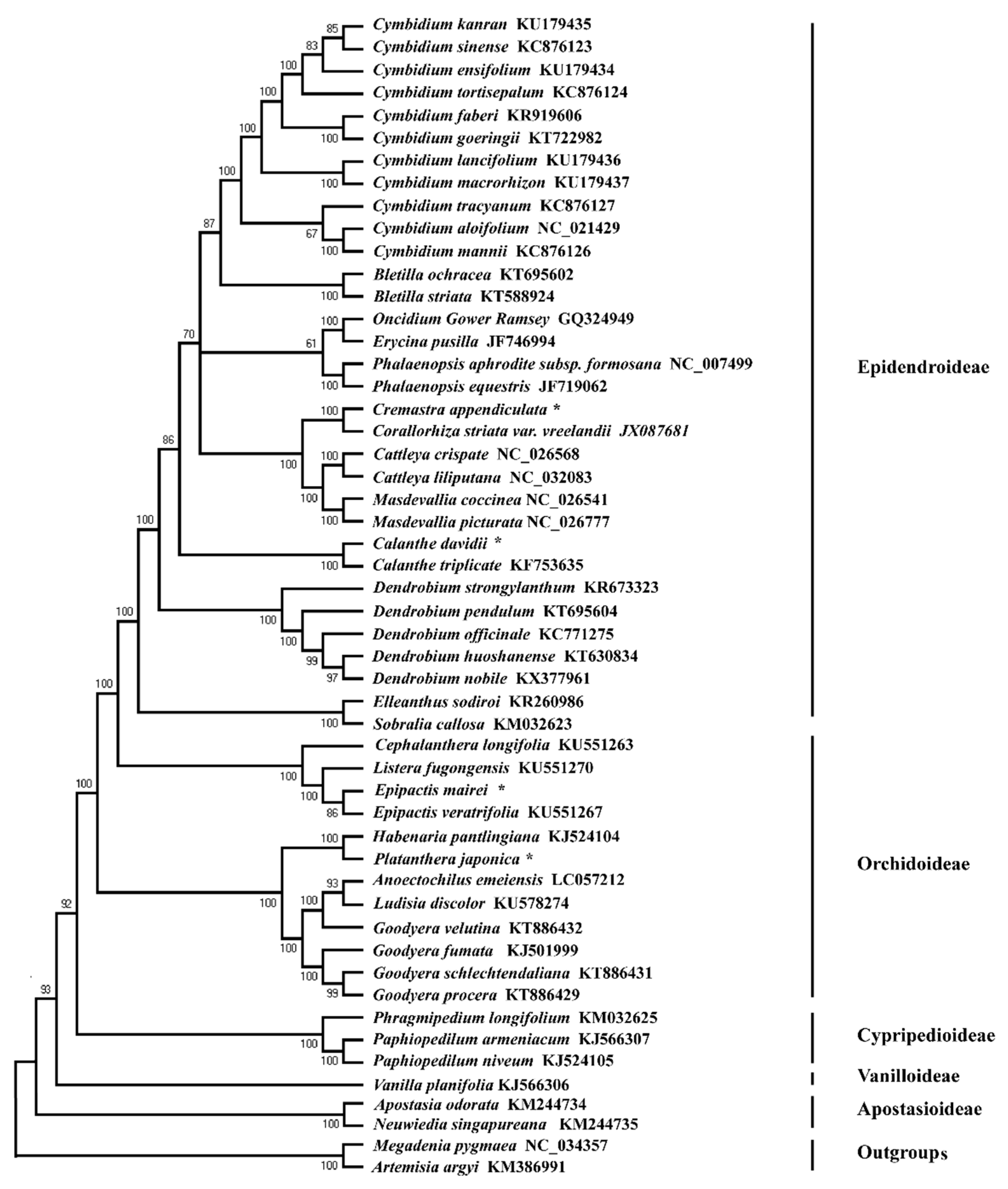
2.6. Phylogenetic Relationship
3. Discussion
3.1. Sequence Variation
3.2. Adaptive Evolution
3.3. Phylogenetic Relationship
4. Materials and Methods
4.1. Plant Material, DNA Extraction, Library Construction, and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Repeat Sequence Analyses
4.4. Genome Structure and Mutational Hotspot
4.5. Gene Selective Pressure Analysis
4.6. Phylogenetic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chase, M.W.; Cameron, K.M.; Barrett, R.L.; Freudenstein, J.V. DNA data and Orchidaceae systematics: A new phylogenetic classification. In Orchid Conservation; Dixon, K.W., Kell, S.P., Barrett, R.L., Cribb, P.J., Eds.; Natural History Publications: Kota Kinabalu, Malaysia, 2003; pp. 69–89. [Google Scholar]
- Dressler, R.L. The Orchids: Natural History and Classification; Harvard University Press: Cambridge, MA, USA, 1990. [Google Scholar]
- Chase, M.W. Classification of Orchidaceae in the age of DNA data. Curtis’s Bot. Mag. 2005, 22, 2–7. [Google Scholar] [CrossRef]
- Luo, J.; Hou, B.W.; Niu, Z.T.; Liu, W.; Xue, Q.Y.; Ding, X.Y. Comparative chloroplast genomes of photosynthetic orchids: Insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS ONE 2014, 9, e99016. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CAB International: Wallingford, UK, 2005; pp. 45–68. [Google Scholar]
- Van den Berg, C.; Goldman, D.H.; Freudenstein, J.V.; Pridgeon, A.M.; Cameron, K.M.; Chase, M.W. An overview of the phylogenetic relationships within Epidendroideae inferred from multiple DNA regions and recircumscription of Epidendreae and Arethuseae (Orchidaceae). Am. J. Bot. 2005, 92, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, M.P.; Lins, L.V. Revisao das Listas das Especies da Flora eda Fauna Ameaçadas de Extincao do Estado de Minas Gerais; Fundacao Biodiversitas: BeloHorizonte, Brazil, 2007. [Google Scholar]
- Cameron, K.M.; Chase, M.W.; Whitten, W.M.; Kores, P.J.; Jarrell, D.C.; Albert, V.A.; Yukawa, T.; Hills, H.G.; Goldman, D.H. A phylogenetic analysis of the Orchidaceae: Evidence from rbcL nucleotide. Am. J. Bot. 1999, 86, 8–24. [Google Scholar] [CrossRef]
- Van den Berg, C.; Higgins, W.E.; Dressler, R.L.; Whitten, W.M.; Soto-Arenas, M.; Chase, M.W. A phylogenetic study of Laeliinae (Orchidaceae) based on combined nuclear and plastid DNA sequences. Ann. Bot. 2009, 104, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Verlynde, S.; D’Haese, C.A.; Plunkett, G.M.; Simo-Droissart, M.; Edwards, M.; Droissart, V.; Stévart, T. Molecular phylogeny of the genus Bolusiella (Orchidaceae, Angraecinae). Plant Syst. Evol. 2017, 304, 269–279. [Google Scholar] [CrossRef]
- Niu, Z.T.; Xue, Q.Y.; Zhu, S.Y.; Sun, J.; Liu, W.; Ding, X.Y. The complete plastome sequences of four orchid species: Insights into the evolution of the Orchidaceae and the utility of plastomic mutational hotspots. Front. Plant. Sci. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Rudall, P.J. Clarified relationship between Dactylorhiza viridis and Dactylorhiza iberica renders obsolete the former genus Coeloglossum (Orchidaceae: Orchidinae). Kew Bull. 2018, 73, 1–17. [Google Scholar] [CrossRef]
- Wilson, M.; Frank, G.S.; Lou, J.; Pridgeon, A.M.; Vieira-Uribe, S.; Karremans, A.P. Phylogenetic analysis of Andinia (Pleurothallidinae; Orchidaceae) and a systematic re-circumscription of the genus. Phytotaxa 2017, 295, 101–131. [Google Scholar] [CrossRef]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Rodríguezezpeleta, N.; Brinkmann, H.; Burey, S.C.; Roure, B.; Burger, G.; Löffelhardt, W.; Bohnert, H.J.; Philippe, H.; Lang, B.F. Monophyly of primary photosynthetic eukaryotes: Green plants, red algae, and glaucophytes. Curr. Biol. 2005, 15, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.Y.; Rohner, T.; Greenfield, A.; Van Der Merwe, M.; McPherson, H.; Glenn, W.; Kornfeld, G.; Marendy, E.; Pan, A.Y.; Wilton, A.; et al. Complete chloroplast genome of the Wollemi pine (Wollemia nobilis): Structure and evolution. PLoS ONE 2015, 106, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, L.; Yan, T.L.; Liu, Q. Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene 2014, 549, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Dong, W.P.; Li, W.Q.; Lu, Y.Z.; Xie, X.M.; Jin, X.B.; Shi, J.; He, K.; Suo, Z. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 2017, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jer, J.D. Plastid chromosomes: Structure and evolution. In Cell Culture and Somatic Cell Genetics in Plants, the Molecular Biology of Plastids 7A; Vasil, I.K., Bogorad, L., Eds.; Academic Press: San Diego, CA, USA, 1991; pp. 5–53. [Google Scholar]
- Bendich, A.J. Circular chloroplast chromosomes: The grand illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2015, 395, 348–384. [Google Scholar]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two new world bamboos-Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)-extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Civan, P.; Foster, P.G.; Embley, M.T.; Séneca, A.; Cox, C.J. Analyses of charophyte chloroplast genomes help characterize the ancestral chloroplast genome of land plants. Genome Biol. Evol. 2014, 6, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Grewe, F.; Cobo-Clark, A.; Fan, W.; Duan, Z.; Adams, R.P.; Schwarzbach, A.E.; Mower, J.P. Predominant and substoichiometric isomers of the plastid genome coexist within Juniperus plants and have shifted multiple times during cupressophyte evolution. Genome Biol. Evol. 2014, 6, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relation-ships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 4302–4315. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.F.; Zanis, M.J.; Emery, N.C. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 2014, 101, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, D.J.; Bendich, A.J. The linear plastid chromosomes of maize: Terminal sequences, structures, and implications for DNA replication. Curr. Genet. 2015, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Doose, D.; Grand, C.; Lesire, C. MAUVE Runtime: A Component-Based Middleware to Reconfigure Software Architectures in Real-Time. In Proceedings of the IEEE International Conference on Robotic Computing (IRC), Taichung, Taiwan, 10–12 April 2017; pp. 208–211. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H., Jr.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, E.; Fujii, S.; Colas des Francs-Small, C.; Brundrett, M.S. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 2011, 28, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Logacheva, M.D.; Schelkunov, M.I.; Penin, A.A. Sequencing and analysis of plastid genome in mycoheterotrophic orchid Neottia nidus-avis. Genome Biol. Evol. 2011, 3, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F.; Davis, J.I. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am. J. Bot. 2012, 99, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.H.; Chan, M.T.; Liao, D.C.; Hsu, C.T.; Lee, Y.W.; Daniell, H.; Duvall, M.R.; Lin, C.S. Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 2010, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associatedinstability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar]
- Ni, L.; Zhao, Z.; Xu, H.; Chen, S.; Dorje, G. The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the Sino-Himalayan subregion. Gene 2016, 577, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhao, Z.; Xu, H.; Chen, S.; Dorje, G. Chloroplast genome structures in Gentiana (Gentianaceae), based on three medicinal alpine plants used in Tibetan herbal medicine. Curr. Genet. 2017, 63, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.B.; Yu, H.; Wang, J.H.; Lei, W.J.; Gao, J.H.; Qiu, X.P.; Wang, J.S. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed]
- Kanga, J.Y.; Lua, J.J.; Qiua, S.; Chen, Z.; Liu, J.J.; Wang, H.Z. Dendrobium SSR markers play a good role in genetic diversity and phylogenetic analysis of Orchidaceae species. Sci. Hortic. 2015, 183, 160–166. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 2017, 7, 3427–3434. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Drew, B.T.; Yang, J.B.; Gao, L.M.; Li, D.Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, B.; Zhou, Y.; Su, Y.; Wang, T. Higher substitution rates and lower dN/dS for the plastid genes in Gnetales than other gymnosperms. Biochem. Syst. Ecol. 2015, 59, 278–287. [Google Scholar] [CrossRef]
- Givnish, T.J.; Spalink, D.; Ames, M.; Lyon, S.P.; Hunter, S.J.; Zuluaga, A.; Iles, W.J.; Clements, M.A.; Arroyo, M.T.; Leebens-Mack, J.; et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. Biol. Sci. B 2015, 282, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Hakamada, K.; Suama, Y.; Nagano, Y.; Furusawa, I.; Matsuno, R. Chloroplast encoded protein as a subunit of acetyl-COA carboxylase in pea plant. J. Biol. Chem. 1993, 268, 25118–25123. [Google Scholar] [PubMed]
- Konishi, T.; Shinohara, K.; Yamada, K.; Sasaki, Y. Acetyl-CoA carboxylase in higher plants: Most plants other than Gramineae have both the prokaryotic and the eukaryotic forms of this enzyme. Plant Cell Physiol. 1996, 37, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Nakkaew, A.; Chotigeat, W.; Eksomtramage, T.; Phongdara, A. Cloning and expression of a plastid-encoded subunit, beta-carboxyltransferase gene (accD) and a nuclear-encoded subunit, biotin carboxylase of acetyl-CoA carboxylase from oil palm (Elaeis guineensis Jacq.). Plant Sci. 2008, 175, 497–504. [Google Scholar] [CrossRef]
- Drescher, A.; Ruf, S.; Calsa, T.J.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Tsudzuki, T.; Takahashi, S.; Shimada, H.; Kadowaki, K. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: A comparative analysis of four monocot chloroplast genomes. DNA Res. 2004, 11, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Wang, X.; Herrmann, R.G.; Rauwolf, U.; Mayer, K.; Haberer, G.; Meurer, J. The complete nucleotide sequences of the 5 genetically distinct plastid genomes of Oenothera, subsection Oenothera: II. A microevolutionary view using bioinformatics and formal genetic data. Mol. Biol. Evol. 2008, 25, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sablok, G.; Wang, B.; Qu, D.; Barbaro, E.; Viola, R.; Li, M.; Varotto, C. Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Allahverdiyeva, Y.; Mamedov, F.; Mäenpää, P.; Vass, I.; Aro, E.M. Modulation of photosynthetic electron transport in the absence of terminal electron acceptors: Characterization of the rbcL deletion mutant of tobacco. Biochim. Biophys. Acta Bioenerg. 2005, 1709, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Piot, A.; Hackel, J.; Christin, P.A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, M.V.; Filatov, D.A. Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol. Biol. 2007, 7, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, Z.; Sablok, G.; Daskalova, E.; Zahmanova, G.; Apostolova, E.; Yahubyan, G.; Baev, V. Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yang, L.F.; Yang, Z.Y.; Ji, Y.H. The complete chloroplast genome of Pleione bulbocodioides (Orchidaceae). Conserv. Genet. Resour. 2017, 1–5. [Google Scholar] [CrossRef]
- Górniak, M.; Paun, O.; Chase, M.W. Phylogenetic relationships with Orchidaceae based on a low-copy nuclear-coding gene, Xdh: Congruence with organellar and nuclear ribosomal DNA results. Mol. Phylogenet. Evol. 2010, 56, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, F.N. The families of the monocotyledones—Structure, evolution and taxonomy. In Orchids; Dahlgren, R., Cliford, H.T., Yeo, P.F., Eds.; Springer: Berlin/Heidelberg, Germany, 1985; pp. 249–274. [Google Scholar]
- Szlachetko, D.L. Systema orchidalium. Fragm. Florist. Geobot. Pol. 1995, 3, 1–152. [Google Scholar]
- Doyle, J.J. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.; Wetter, T.; Suhai, S. Using the mira EST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-abaiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Steven, S.H.; Matthew, C.; Shane, S.; Simon, B.; Alex, C.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 12, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar Genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats incomplete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Cremastra appendiculata | Calanthe davidii | Epipactis mairei | Platanthera japonica |
|---|---|---|---|---|
| Accession number | MG925366 | MG925365 | MG925367 | MG925368 |
| Genome size (bp) | 155,320 | 153,629 | 160,427 | 154,995 |
| LSC length (bp) | 87,098 | 86,045 | 88,328 | 85,979 |
| SSC length (bp) | 15,478 | 15,672 | 18,513 | 13,664 |
| IR length (bp) | 26,372 | 25,956 | 26,790 | 27,676 |
| Coding (bp) | 100,018 | 104,531 | 113,915 | 107,028 |
| Non-coding (bp) | 55,302 | 49,098 | 46,512 | 47,967 |
| Number of genes | 130 (0) | 132 (19) | 131 (19) | 128 (17) |
| Number of protein-coding genes | 83 (7) | 86 (7) | 85 (7) | 85 (7) |
| Number of tRNA genes | 38 (8) | 38 (8) | 38 (8) | 38 (8) |
| Number of rRNA genes | 8 (4) | 8 (4) | 8 (4) | 8 (4) |
| GC content (%) | 37.2 | 36.9 | 37.2 | 37 |
| GC content in LSC (%) | 34.5 | 34.5 | 34.9 | 34.2 |
| GC content in SSC (%) | 30.4 | 30.2 | 31.0 | 29 |
| GC content in IR (%) | 43.5 | 43.1 | 43.1 | 43.2 |
| Mapped read number | 551,680 | 324,741 | 230,968 | 322,259 |
| Chloroplast coverage | 544.9 | 217.4 | 216 | 313.6 |
| Category of Genes | Group of Gene | Name of Gene | Name of Gene | Name of Gene | Name of Gene | Name of Gene |
|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn16 (×2) | rrn2 (×2) | rrn4.5 (×2) | rrn5 (×2) | |
| Transfer RNA genes | trnA-UGC *,(×2) | trnC-GCA | trnD-GUC | trnE-UUC | trnF-GAA | |
| trnfM-CAU | trnG-GCC * | trnG-UCC | trnH-GUG (×2) | trnI-CAU (×2) | ||
| trnI-GAU *,(×2) | trnK-UUU * | trnL-CAA (×2) | trnL-UAA * | trnL-UAG | ||
| trnM-CAU | trnN-GUU (×2) | trnP-UGG | trnQ-UUG | trnR-ACG (×2) | ||
| trnR-UCU | trnS-GCU | trnS-GGA | trnS-UGA | trnT-GGU | ||
| trnT-UGU | trnV-GAC (×2) | trnV-UAC (×2) | trnW-CCA | trnY-GUA | ||
| Small subunit of ribosome | rps2 | rps3 | rps4 | rps7 (×2) | rps8 | |
| rps11 | rps12 **,(×2) | rps14 | rps15 | rps16 * | ||
| rps18 | rps19 (×2) | |||||
| Large subunit of ribosome | rpl2 *,(×2) | rpl14 | rpl16 * | rpl20 | rpl22 | |
| rpl23 (×2) | rpl32 | rpl33 | rpl36 | |||
| DNA-dependent RNA polymerase | rpoA | rpoB | rpoC1 * | rpoC2 | ||
| Translational initiation factor | infA | |||||
| Genes for photosynthesis | Subunits of NADH-dehydrogenase | ndhA * | ndhB *,(×2) | ndhC a | ndhD | ndhE |
| ndhF | ndhG | ndhH | ndhI a,c,d | ndhJ | ||
| ndhK a | ||||||
| Subunits of photosystem I | psaA | psaB | psaC | psaI | psaJ | |
| ycf3 ** | ycf4 | |||||
| Subunits of photosystem II | psbA | psbB | psbC | psbD | psbE | |
| psbF | psbH | psbI | psbJ | psbK | ||
| psbL | psbM | psbN | psbT | psZ | ||
| Subunits of cytochrome b/f complex | petA | petB* | petD * | petG | petL | |
| petN | ||||||
| Subunits of ATP synthase | atp A | atp B | atp E | atp F * | atp H | |
| atpI | ||||||
| Subunits of rubisco | rbcL | |||||
| Other genes | Maturase | matK | ||||
| Protease | clpP ** | |||||
| Envelope membrane protein | cemA | |||||
| Subunit of acetyl-CoA carboxylase | accD | |||||
| C-type cytochrome synthesis gene | ccsA | |||||
| Genes of unknown function | Conserved open reading frames | ycf1 | ycf2 (×2) |
| Subfamily | Species | Accession Number |
|---|---|---|
| Orchidaceae subfamily. Epidendroideae | Cattleya crispata | KP168671 |
| Cremastra appendiculata | MG925366 | |
| Masdevallia coccinea | KP205432 | |
| Erycina pusilla | JF746994 | |
| Phalaenopsis equestris | JF719062 | |
| Bletilla ochracea | KT695602 | |
| Cymbidium faberi | KR919606 | |
| Calanthe davidii | MG925365 | |
| Dendrobium strongylanthum | KR673323 | |
| Elleanthus sodiroi | KR260986 | |
| Sobralia callosa | KM032623 | |
| Orchidaceae subfamily. Orchidoideae | Epipactis mairei | MG925367 |
| Cephalanthera longifolia | KU551263 | |
| Listera fugongensis | KU551270 | |
| Platanthera japonica | MG925368 | |
| Habenaria pantlingiana | KJ524104 | |
| Goodyera velutina | KT886432 | |
| Anoectochilus emeiensis | LC057212 | |
| Ludisia discolor | KU578274 | |
| Orchidaceae subfamily. Vanilloideae | Vanilla planifolia | KJ566306 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030716
Dong W-L, Wang R-N, Zhang N-Y, Fan W-B, Fang M-F, Li Z-H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. International Journal of Molecular Sciences. 2018; 19(3):716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030716
Chicago/Turabian StyleDong, Wan-Lin, Ruo-Nan Wang, Na-Yao Zhang, Wei-Bing Fan, Min-Feng Fang, and Zhong-Hu Li. 2018. "Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution" International Journal of Molecular Sciences 19, no. 3: 716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030716