Multifaceted Role of Pneumolysin in the Pathogenesis of Myocardial Injury in Community-Acquired Pneumonia


Abstract
:
1. Introduction
2. Community-Acquired Pneumonia (CAP)
2.1. Incidence of CAP
2.2. Role of Streptococcus Pneumoniae in CAP
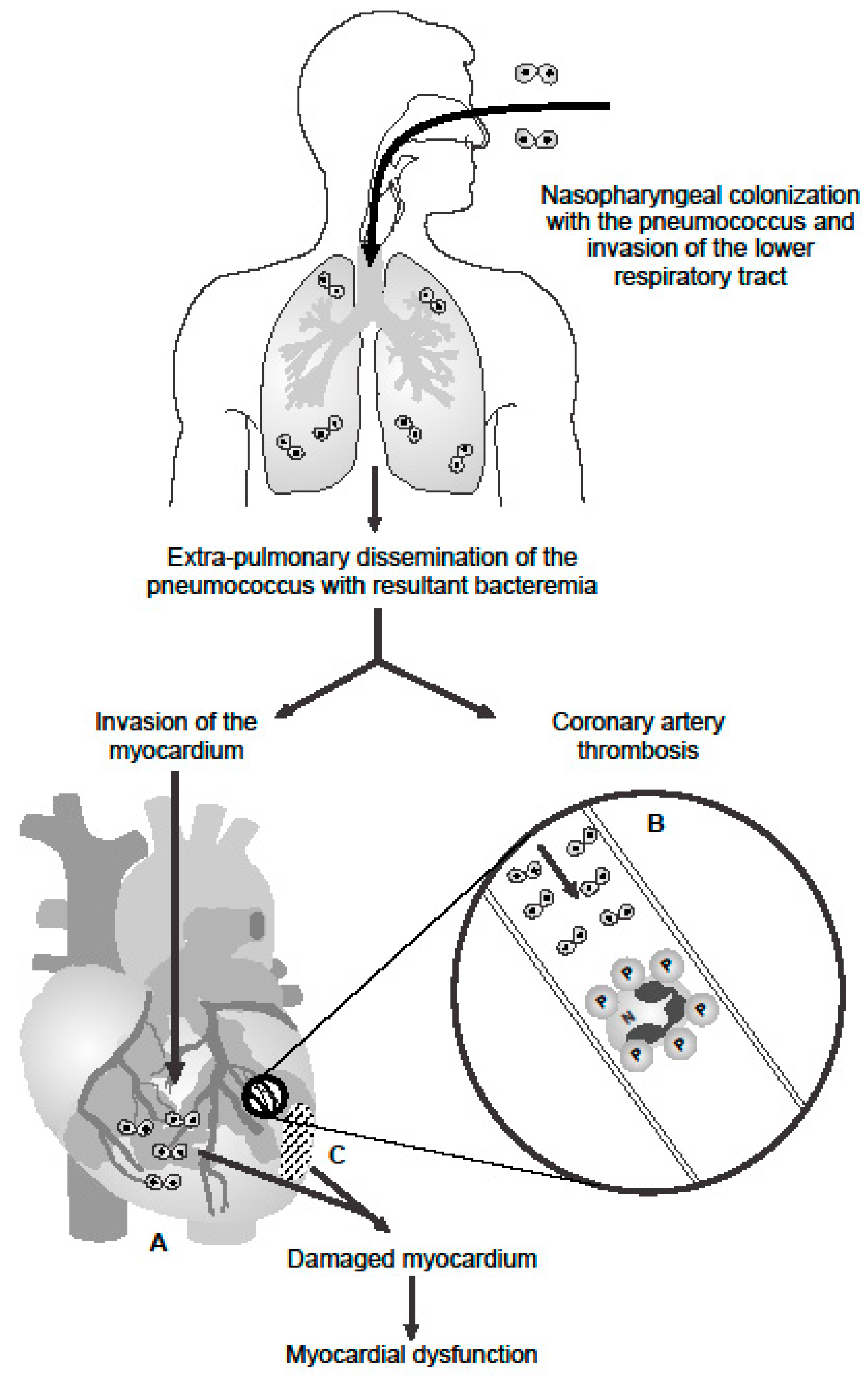
3. Cardiac Complications of CAP
4. Structure and Biological Activities of Pneumolysin
- potentiation of the generation of reactive oxygen species (ROS) and release of elastase by chemoattractant-activated human neutrophils, as well as production of prostaglandin E2, leukotriene B4 and interleukin (IL)-8, and release of matrix metalloproteinase-9 by both unstimulated and chemoattractant-activated cells [54,57,58,59].
- induction of the formation of neutrophil extracellular traps (NETs) via a process of vital NETosis [62]. NET formation involves the extrusion of decondensed nucleosomes and granule-derived proteins into the extracellular milieu by neutrophils, a process which contributes to the intravascular host innate immune response via entrapment of blood-borne pathogens. However, if excessive and poorly controlled, the process of NETosis poses the potential hazard of generation of coronary thrombi by mechanisms described below
- activation of human platelets detected according to up-regulation of the expression of the adhesion molecule, CD62P (P-selectin), which is stored in platelet α-granules [56]
- activation of the formation of pro-inflammatory/prothrombotic neutrophil:platelet heterotypic aggregates, an event which is critically dependent on up-regulated expression of CD62P on platelets and interaction with its counter-ligand, P-selectin glycoprotein ligand 1 (PSGL-1), on neutrophils [63].
5. Direct Cardiotoxic Activity of Pneumolysin
- myocardial invasion by the pneumococcus in the murine model of IPD is associated with strain-specific, PLY-mediated necroptosis (programmed necrotic cell death) of resident and infiltrating cardiac macrophages, which may contribute to the observed paucity of immune and inflammatory cells in microlesions [65]. In this context, it is noteworthy, as reported by others, that bacterial pore-forming toxins, including PLY, induce necroptosis of alveolar macrophages in a murine model of experimental pneumonia [66], which appears to be strain-specific [67]
- although CbpA and phosphorylcholine appear to be determinants of myocardial invasion via interaction of these adhesins with vascular endothelium, invasion of cardiomyocytes by the pneumococcus in vitro results from an alternative mechanism involving clathrin-mediated endocytosis of the pathogen [68]. In this setting, intracellular pneumococci were found to reside in the cytoplasm and cytoplasmic vacuoles, with resultant killing of infected cardiomyocytes mediated by exposure to PLY acting in concert with pneumococcus-derived hydrogen peroxide [68]
- infection of cardiomyocytes and/or macrophages or fibroblasts by the pneumococcus during experimental IPD results in intracellular proliferation of the pneumococcus and transition of the pathogen from a planktonic to a biofilm-forming phenotype, which is associated with formation of cardiac microlesions [69]. Acquisition of the biofilm-forming phenotype, in turn, is associated with an altered transcriptional profile of the pneumococcus, which results in high-level production of PLY and resultant elimination of cardiac macrophages [69]
- extrapolation of these findings to a scenario more representative of severe pneumococcal disease in humans was achieved by using a non-human primate (adult baboon, species Papio cynocephalus) model of IPD in which the TIGR4 strain of the pneumococcus is inoculated directly into the right middle lobe of the lung [70]. Using this model, pneumococcal bacteremia was found to result in myocardial invasion by the pathogen, cardiomyocyte death by both apoptotic and necroptotic mechanisms, cardiac dysfunction, and cardiac scarring in convalescent, antibiotic-treated animals [71].
6. Indirect Pro-Inflammatory/Pro-Thrombotic Potential of Pneumolysin in the Pathogenesis of Cardiac Damage and Dysfunction
7. Update on Pneumolysin-Targeted Therapeutic Strategies
7.1. Macrolide Antibiotics
7.1.1. Macrolides and Inhibition of the Synthesis of Pneumolysin
7.1.2. Anti-inflammatory Activities of Macrolides
7.2. Low Molecular Weight Antagonists of the Pore-Forming Action of Pneumolysin
7.2.1. Naturally Occurring Antagonists
- phytosterols: β-sitosterol was initially identified as a cholesterol-mimic, interacting with the conserved cholesterol-binding site located on domain 4 of PLY, with potency comparable with that of cholesterol [99]. A follow-up, mechanistic study, using a molecular dynamics simulation approach, revealed two additional phytosterols which also antagonized the membrane-binding activity of PLY viz. campesterol and brassicasterol, with the former demonstrating activity comparable with that of β-sitosterol and the latter being somewhat less potent [100]
- polyphenols: verbascoside (a caffeoyl polyethanol glycoside) and the catechin, epigallocatechin gallate, are naturally-occurring polyphenol anti-oxidant antagonists of PLY, which have demonstrated almost complete attenuation of the hemolytic activity of the toxin at concentrations of about 2.0 and 2.7 µg/mL respectively [101,102]. Unlike the phytosterols, the polyphenol antagonists appear to interfere with the oligomerization of PLY monomers on the target cell membrane via binding to the cleft between domains 3 and 4 of the toxin molecule [101,102]
- bioflavonoids: three bioflavonoids viz. apigenin (4′,5,7-trihyddroxyflavone) [103], morin [2-(2,4-dihydroxyphenol)-3,5,7-trihydroxychromen-4-one] [104] and amentoflavone {8-[5-(5,7-dihydroxy-4-oxo-chromen-2-yl)-2-hydroxy-phenyl]-5,7-dihydroxy-2-(4-hydroxyphenyl)chromen-4-one} [105] have also been found to attenuate the harmful activities of PLY. All three agents were found to cause inhibition of the hemolytic activity of PLY at concentrations of around 16 µg/mL by apparent interference with the oligomerization of toxin monomers [103,104,105]
- naphthoquinones: two members of this class of chemical agent viz. shikonin {also known as alkanin: 5,8-dihydroxy-2-[(1S)-1-hydroxy-4-methylpent-3-en-1 yl]naphthalene-1,4-dione} [106] and juglone (5-hydroxy-1,4-naphthalenedione) [107] have also been reported to inhibit the pore-forming activity of PLY via interference with toxin oligomerization. Shikonin demonstrated superior activity, causing almost complete inhibition of the hemolytic activity of PLY at concentrations ≥1 µg/mL [106]. Although not addressed in these studies [106,107], it is noteworthy that 1,4-naphthoquinones possess pro-oxidative properties that may promote oxidative inactivation of membrane pore formation mediated by PLY and other CDCs via oxidative inactivation of the essential cysteine residue located in the undecapeptide membrane insertion region of the toxin [51].
7.2.2. Magnesium Chloride
7.2.3. Liposome-Based Strategies
7.2.4. Monoclonal Antibody-Based Strategies
7.2.5. Indirect Strategies to Counter the Harmful Activities of Pneumolysin
8. Pneumolysin as a Candidate Vaccine Antigen
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mitchell, T.J.; Dalziel, C.E. The biology of pneumolysin. In MACP/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer Science + Business Media: Dordrecht, The Netherlands, 2014; Volume 80, pp. 145–160. ISBN 978-94-017-8880-9. [Google Scholar]
- Hirst, R.A.; Kadioglu, A.; O’Callaghan, C.; Andrew, P.W. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin. Exp. Immunol. 2004, 138, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.A.; Wang, Y.; Hamaguchi, S.; Weiser, J.N. Host-to-host transmission of Streptococcus pneumoniae is driven by its inflammatory toxin, pneumolysin. Cell Host Microbe 2017, 21, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Witzenrath, M.; Gutbier, B.; Hocke, A.C.; Schmeck, B.; Hippenstiel, S.; Berger, K.; Mitchell, T.J.; de los Toyos, J.R.; Rosseau, S.; Suttorp, N.; et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit. Care Med. 2006, 34, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Del Mar García-Suárez, M.; Flórez, N.; Astudillo, A.; Vázquez, F.; Villaverde, R.; Fabrizio, K.; Pirofski, L.A.; Méndez, F.J. The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir. Res. 2007, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, C.; Konradsen, H.B.; Samuelsson, S. Clinical presentation and prognostic factors of Streptococcus pneumoniae meningitis according to the focus of infection. BMC Infect. Dis. 2005, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Weisfelt, M.; van de Beek, D.; Spanjaard, L.; Reitsma, J.B.; de Gans, J. Clinical features, complications, and outcome in adults with pneumococcal meningitis: A prospective case series. Lancet Neurol. 2006, 5, 123–129. [Google Scholar] [CrossRef]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, C.J.; Mahdavi, J.; Thornton, J.; Mann, B.; Wooldridge, K.G.; Abouseada, N.; Oldfield, N.J.; Self, T.; Ala’Aldeen, D.A.; Tuomanen, E.I. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Investig. 2009, 119, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Rueda, A.M.; Kaka, A.S.; Mapara, S.M. The association between pneumococcal pneumonia and acute cardiac events. Clin. Infect. Dis. 2007, 45, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Medina, V.F.; Musher, D.M.; Wells, G.A.; Chirinos, J.A.; Chen, L.; Fine, M.J. Cardiac complications in patients with community-acquired pneumonia: Incidence, timing, risk factors, and association with short-term mortality. Circulation 2012, 125, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Rae, N.; Finch, S.; Chalmers, J.D. Cardiovascular disease as a complication of community-acquired pneumonia. Curr. Opin. Pulm. Med. 2016, 22, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Cangemi, R.; Falcone, M.; Taliani, G.; Pieralli, F.; Vannucchi, V.; Nozzoli, C.; Venditti, M.; Chirinos, J.A.; Corrales-Medina, V.F.; for the SIXTUS (Thrombosis-Related Extrapulmonary Outcomes in Pneumonia) Study Group. Cardiovascular complications and short-term mortality risk in community-acquired pneumonia. Clin. Infect Dis. 2017, 64, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Eurich, D.T.; Marrie, T.J.; Minhas-Sandhu, J.K.; majumdar, S.R. Risk of heart failure after community acquired pneumonia: A prospective controlled study with 10 years of follow-up. BMJ 2017, 356, j413. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.O.; Mann, B.; Gao, G.; Hankins, J.S.; Humann, J.; Giardina, J.; Faverio, P.; Restrepo, M.I.; Halade, G.V.; Mortensen, E.M.; et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Neill, D.R.; Abrams, S.T.; Malak, H.A.; Yahya, R.; Barrett-Jolley, R.; Wang, G.; Kadioglu, A.; Toh, C.H. Circulating pneumolysin is a potent inducer of cardiac injury during pneumococcal infection. PLoS Pathog. 2015, 11, e1004836. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Czikora, I.; Sridhar, S.; Zemskov, E.; Gorshkov, B.; Siddaramappa, U.; Oseghale, A.; Lawson, J.; Verin, A.; Rick, F.G.; et al. Mini-review: Novel therapeutic strategies to blunt actions of pneumolysin in the lungs. Toxins 2013, 5, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Feldman, C. Pneumolysin as a potential therapeutic target in severe pneumococcal disease. J. Infect. 2017, 74, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. Community-acquired pneumonia: Pathogenesis of acute cardiac events and potential adjunctive therapies. Chest 2015, 148, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. Prevalence, pathogenesis, therapy, and prevention of cardiovascular events in patients with community-acquired pneumonia. Pneumonia 2016, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Feldman, C. Review manuscript: Mechanisms of platelet activation by the pneumococcus and the role of platelets in community-acquired pneumonia. J. Infect. 2017, 75, 473–485. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. The Top 10 Causes of Death. Updated January 2017. Available online: http:/www.who.int/mediacentre/factsheets/fs310/en/ (accessed on 12 January 2018).
- Kolditz, M.; Ewig, S. Community-acquired pneumonia in adults. Dtsch Arztebl Int. 2017, 114, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.; Campling, J.; Ellsbury, G.; Hawkey, P.M.; Madhava, H.; Slack, M. Community-acquired pneumonia in the United Kingdom: A call to action. Pneumonia 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Kolditz, M.; Tesch, F.; Mocke, L.; Höffken, G.; Ewig, S.; Schmitt, J. Burden and risk factors of ambulatory or hospitalized CAP: A population based cohort study. Respir. Med. 2016, 121, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. CDC EPIC Study Team. Community-acquired pneumonia requiring hospitalization among U.S. adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.A.; Wiemken, T.L.; Peyrani, P.; Arnold, F.W.; Kelley, R.; Mattingly, W.A.; Nakamatsu, R.; Pena, S.; Guinn, B.E.; Furmanek, S.P.; et al. University of Louisville Pneumonia Study Group. Adults hospitalized with pneumonia in the United States: Incidence, epidemiology, and mortality. Clin. Infect. Dis. 2017, 65, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.P.; Fawcett, N.J.; Wrightson, J.M.; Finney, J.; Wyllie, D.; Jeffery, K.; Jones, N.; Shine, B.; Clarke, L.; Crook, D.; et al. Infections in Oxfordshire Research Database (IORD). Increasing burden of community-acquired pneumonia leading to hospitalization, 1998–2014. Thorax 2016, 71, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Abers, M.S.; Bartlett, J.G. Evolving understanding of the causes of pneumonia in adults, with special attention to the role of pneumococcus. Clin. Infect. Dis. 2017, 65, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- McNeil, S.A.; Qizilbash, N.; Ye, J.; Gray, S.; Zanotti, G.; Munson, S.; Dartois, N.; Laferriere, C. A retrospective study of the clinical burden of hospitalized all-cause and pneumococcal pneumonia in Canada. Can. Respir. J. 2016, 2016, 3605834. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.J.; ElSherif, M.; Ye, L.; MacKinnon-Cameron, D.; Li, L.; Ambrose, A.; Hatchette, T.F.; Lang, A.L.; Gillis, H.; Martin, I.; et al. Burden of vaccine-preventable pneumococcal disease in hospitalized adults: A Canadian Immunization Research Network (CIRN) Serious Outcomes Surveillance (SOS) network study. Vaccine 2017, 35, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Earle, K.; Williams, S. Burden of pneumococcal disease in adults aged 65 years and older: An Australian perspective. Pneumonia 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.D.; Campling, J.; Dicker, A.; Woodhead, M.; Madhava, H. A systematic review of the burden of vaccine preventable pneumococcal disease in UK adults. BMC Pulm. Med. 2016, 16, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyhan, M.; Dagan, R.; Sayiner, A.; Chernyshova, L.; Dinleyici, E.C.; Hryniewicz, W.; Kulcsár, A.; Mad’arová, L.; Pazdiora, P.; Sidorenko, S.; et al. Surveillance of pneumococcal disease in central and Eastern Europe. Hum. Vaccin. Immunother. 2016, 12, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Said, M.A.; Johnson, H.L.; Nonyane, B.A.; Deloria-Knoll, M.; O’Brien, K.L.; AGEDD Adult Pneumococcal Burden Study Team. Estimating the burden of pneumococcal pneumonia among adults: A systematic review and meta-analysis of diagnostic techniques. PLoS ONE 2013, 8, e60273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Blasi, F.; Dartois, N.; Akova, M. Which individuals are at increased risk of pneumococcal disease and why? Impact of COPD, asthma, smoking, diabetes, and/or chronic heart disease on community-acquired pneumonia and invasive pneumococcal disease. Thorax 2015, 70, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Curcio, D.; Cané, A.; Isturiz, R. Redefining risk categories for pneumococcal disease in adults: Critical analysis of the evidence. Int. J. Infect. Dis. 2015, 37, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.; Yee, A.; Aukes, L.; Snow, V.; Fireman, B.; Atkinson, B.; Klein, N.P. Risk of underlying chronic medical conditions for invasive pneumococcal disease in adults. Vaccine 2016, 34, 4293–4297. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, O.O.; Norton, N.B.; Gress, T.W.; Stanek, R.J.; Mufson, M.A. Three decades of follow-up of adults after recovery from invasive pneumococcal pneumonia. Am. J. Med. Sci. 2017, 353, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Wagenvoort, G.H.; Sanders, E.A.; de Melker, H.E.; van der Ende, A.; Vlaminckx, B.J.; Knol, M.J. Long-term mortality after IPD and bacteremic versus non-bacteremic pneumococcal pneumonia. Vaccine 2017, 35, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, M.I.; Reyes, L.F.; Anzueto, A. Complication of community-acquired pneumonia (including cardiac complications). Semin. Respir. Crit. Care Med. 2016, 37, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Mullany, D.; Fraser, J.F. Takotsubo cardiomyopathy associated with sepsis due to Streptococcus pneumoniae pneumonia. Crit. Care Resusc. 2008, 10, 231–234. [Google Scholar] [PubMed]
- Marrie, T.J.; Tyrrell, G.J.; Majumdar, S.R.; Eurich, D.T. Invasive pneumococcal disease: Still lots to learn and a need for standardized data collection instruments. Can. Respir. J. 2017, 2017, 2397429. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.; Heywood, A.E.; Mahimbo, A.; Rahman, B.; Newall, A.T.; Macintyre, C.R. Acute myocardial infarction and influenza: A meta-analysis of case-control studies. Heart 2015, 101, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 2006, 19, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.; Calvieri, C.; Falcone, M.; Bucci, T.; Bertazzoni, G.; Scarpellini, M.G.; Barillà, F.; Taliani, G.; Violi, F.; SIXTUS Study Group. Relation of cardiac complications in the early phase of community-acquired pneumonia to long-term mortality and cardiovascular events. Am. J. Cardiol. 2015, 116, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Gámez, G.; Castro, A.; Gómez-Mejia, A.; Gallego, M.; Bedoya, A.; Camargo, M.; Hammerschmidt, S. The variome of pneumococcal virulence factors and regulators. BMC Genom. 2018, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Price, K.E.; Camilli, A. Pneumolysin localizes to the cell wall of Streptococcus pneumoniae. J. Bacteriol. 2009, 191, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Bandara, M.; Skehel, J.M.; Kadioglu, A.; Collinson, I.; Nobbs, A.H.; Blocker, A.J.; Jenkinson, H.F. The accessory Sec sytem (SecY2A2) in Streptococcus pneumoniae is involved in export of pneumolysin toxin, adhesion and biofilm formation. Microbes Infect. 2017, 19, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Hupp, S.; Ribes, S.; Seele, J.; Bischoff, C.; Förtsch, C.; Maier, E.; Benz, R.; Mitchell, T.J.; Nau, R.; Iliev, A.I. Magnesium therapy improves outcome in Streptococcus pneumoniae meningitis by altering pneumolysin pore formation. Br. J. Pharmacol. 2017, 174, 4295–4307. [Google Scholar] [CrossRef] [PubMed]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef] [PubMed]
- Van Pee, K.; Neuhaus, A.; D’Imprima, E.; Mills, D.J.; Kühlbrandt, W.; Yildiz, Ö. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of pneumolysin. Elife 2017, 6, e23644. [Google Scholar] [CrossRef] [PubMed]
- Cockeran, R.; Theron, A.J.; Steel, H.C.; Matlola, N.M.; Mitchell, T.J.; Feldman, C.; Anderson, R. Proinflammatory interactions of pneumolysin with human neutrophils. J. Infect. Dis. 2001, 183, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.; Malak, H.A.; Greene, N.G.; Howell, S.J.; Hise, A.G.; Camilli, A.; Kadioglu, A.; Dubyak, G.R.; Pearlman, E. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J. Immunol. 2015, 194, 1763–1775. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Durandt, C.; Mitchell, T.J.; Feldman, C.; Anderson, R.; Tintinger, G.R. Pneumolysin mediates platelet activation in vitro. Lung 2016, 194, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Cockeran, R.; Steel, H.C.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin potentiates production of prostaglandin E2 and leukotriene B4 by human neutrophils. Infect. Immun. 2001, 69, 3494–3496. [Google Scholar] [CrossRef] [PubMed]
- Cockeran, R.; Durandt, C.; Feldman, C.; Mitchell, T.J.; Anderson, R. Pneumolysin activates the synthesis and release of interleukin-8 by human neutrophils in vitro. J. Infect. Dis. 2002, 186, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Cockeran, R.; Steel, H.C.; Theron, A.J.; Mitchell, T.J.; Feldman, C.; Anderson, R. Characterization of the interaction of the pneumolysoid, Δ6 PLY, with human neutrophils in vitro. Vaccine 2011, 29, 8780–8782. [Google Scholar] [CrossRef] [PubMed]
- McNeela, E.A.; Burke, A.; Neill, D.R.; Baxter, C.; Fernandes, V.E.; Ferreira, D.; Smeaton, S.; El-Rachkidy, R.; McLoughlin, R.M.; Mori, A.; et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- Hassane, M.; Demon, D.; Soulard, D.; Fontaine, J.; Keller, L.E.; Patin, E.C.; Porte, R.; Prinz, I.; Ryffel, B.; Kadioglu, A.; et al. Neutrophilic NLRP3 inflammasome-dependent IL-1β secretion regulates the γδT17 cell response in respiratory bacterial infections. Mucosal Immunol. 2017, 10, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Theron, A.J.; Durandt, C.; Tintinger, G.R.; Pool, R.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin activates neutrophil extracellular trap formation. Clin. Exp. Immunol. 2016, 184, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Durandt, C.; Theron, A.J.; Tintinger, G.R.; Pool, R.; Richards, G.A.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin mediates heterotypic aggregation of neutrophils and platelets in vitro. J. Infect. 2017, 74, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wall, E.C.; Gordon, S.B.; Hussain, S.; Goonetilleke, U.R.; Gritzfeld, J.; Scarborough, M.; Kadioglu, A. Persistence of pneumolysin in the cerebrospinal fluid of patients with pneumococcal meningitis is associated with mortality. Clin. Infect. Dis. 2012, 54, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Gilley, R.P.; González-Juarbe, N.; Shenoy, A.T.; Reyes, L.F.; Dube, P.H.; Restrepo, M.I.; Orihuela, C.J. Infiltrated macrophages die of pneumolysin-mediated necroptosis following pneumococcal myocardial invasion. Infect. Immun. 2016, 84, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.M.; Hughes, C.E.; Paton, A.W.; Trappetti, C.; Tweten, R.K.; Paton, J.C. The impact of pneumolysin on the macrophage response to Streptococcus pneumoniae is strain-dependent. PLoS ONE 2014, 9, e103625. [Google Scholar] [CrossRef] [PubMed]
- Brissac, T.; Shenoy, A.T.; Patterson, L.A.; Orihuela, C.J. Cell invasion and pyruvate oxidase derived H2O2 are critical for Streptococcus pneumoniae mediated cardiomyocyte killing. Infect. Immun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.T.; Brissac, T.; Gilley, R.P.; Kumar, N.; Wang, Y.; González-Juarbe, N.; Hinkle, W.S.; Daugherty, S.C.; Shetty, A.C.; Ott, S.; et al. Streptococcus pneumoniae in the heart subvert the host response through biofilm-mediated resident macrophage killing. PLoS Pathog. 2017, 13, e1006582. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.F.; Restrepo, M.I.; Hinojosa, C.A.; Soni, N.J.; Shenoy, A.T.; Gilley, R.P.; Gonzalez-Juarbe, N.; Noda, J.R.; Winter, V.T.; de la Garza, M.A.; et al. A non-human primate model of severe pneumococcal pneumonia. PLoS ONE 2016, 11, e0166092. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.F.; Restrepo, M.I.; Hinojosa, C.A.; Soni, N.J.; Anzueto, A.; Babu, B.L.; Gonzalez-Juarbe, N.; Rodriguez, A.H.; Jimenez, A.; Chalmers, J.D.; et al. Severe pneumococcal pneumonia causes acute cardiac toxicity and subsequent cardiac remodeling. Am. J. Respir. Crit. Care Med. 2017, 196, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Chen, S.; Yin, J.; Pan, Z.; Liu, K.; Li, L.; Zheng, Y.; Yuan, Y.; Jiang, Y. Effects of suilysin on Streptococcus suis-induced platelet aggregation. Front. Cell Infect. Microbiol. 2016, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenböck, C.K.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, L.; Zhang, Y.; Lin, P.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Tunjungputri, R.N.; de Jonge, M.I.; de Greeff, A.; van Selm, S.; Buys, H.; Harders-Westerveen, J.F.; Stockhofe-Zurwieden, N.; Urbanus, R.T.; de Groot, P.G.; Smith, H.E.; et al. Invasive pneumococcal disease leads to activation and hyperreactivity of platelets. Thromb. Res. 2016, 144, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.; Casciaro, M.; Rossi, E.; Calvieri, C.; Bucci, T.; Calabrese, C.M.; Taliani, G.; Falcone, M.; Phalange, P.; Bertazzoni, G.; et al. Platelet activation is associated with myocardial infarction in patients with pneumonia. J. Am. Coll. Cardiol. 2014, 64, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.M. Macrolides and ketolides: Azithromycin, clarithromycin, telithromycin. Infect. Dis. Clin. N. Am. 2004, 18, 621–649. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Steel, H.C.; Cockeran, R.; von Gottberg, A.; de Gouveia, L.; Klugman, K.P.; Mitchell, T.J.; Feldman, C. Comparison of the effects of macrolides, amoxicillin, ceftriaxone, doxycycline, tobramycin and fluoroquinolones, on the production of pneumolysin by Streptococcus pneumoniae in vitro. J. Antimicrob. Chemother. 2007, 60, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Waterer, G. Empiric antibiotics for community-acquired pneumonia: A macrolide and a beta-lactam please! Respirology 2017. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Bonten, M.J.; Boersma, W.G.; Jonkers, R.E.; Aleva, R.M.; Kullberg, B.J.; Schouten, J.A.; Degener, J.E.; van de Garde, E.M.; Verheij, T.J.; et al. Management of community-acquired pneumonia in adults: 2016 guideline update from the Dutch Working Party on Antibiotic Policy (SWAB) and Dutch Association of Chest Physicians (NVALT). Neth. J. Med. 2018, 76, 4–13. [Google Scholar] [PubMed]
- Gattarello, S.; Borgatta, B.; Solé-Violán, J.; Vallés, J.; Vidaur, L.; Zaragoza, R.; Torres, A.; Rello, J.; Community-Acquired Pneumonia en la Unidad de Cuidados Intensivos II Study Investigators. Decrease in mortality in severe community-acquired pneumococcal pneumonia: Impact of improving antibiotic strategies (2000–2013). Chest 2014, 146, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Okumura, J.; Shindo, Y.; Takahashi, K.; Sano, M.; Sugino, Y.; Yagi, T.; Taniguchi, H.; Saka, H.; Matsui, S.; Hasegawa, Y.; et al. Mortality in patients with community-onset pneumonia at low risk of drug-resistant pathogens: Impact of β-lactam plus macrolide combination therapy. Respirology 2017. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Tintinger, G.; Cockeran, R.; Potjo, M.; Feldman, C. Beneficial and harmful interactions of antibiotics with microbial pathogens and the host innate immune system. Pharmaceuticals 2010, 3, 1694–1710. [Google Scholar] [CrossRef] [PubMed]
- Peyrani, P.; Wiemken, T.L.; Metersky, M.L.; Arnold, F.W.; Mattingly, W.A.; Feldman, C.; Cavallazzi, R.; Fernandez-Botran, R.; Bordon, J.; Ramirez, J.A. The order of administration of macrolides and beta-lactams may impact the outcomes of hospitalized patients with community-acquired pneumonia: Results from the community-acquired pneumonia organization. Infect. Dis. 2018, 50, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Metersky, M.L.; Priya, A.; Mortensen, E.M.; Lindenauer, P.K. Association between the order of macrolide and cephalosporin treatment and outcomes of pneumonia. Open Forum. Infect. Dis. 2017, 4, ofx141. [Google Scholar] [CrossRef] [PubMed]
- Bystrzycka, W.; Manda-Handzlik, A.; Sieczkowska, S.; Moskalik, A.; Demkow, U.; Ciepiela, O. Azithromycin and chloramphenicol diminish neutrophil extracellular traps (NETs) release. Int. J. Mol. Sci. 2017, 18, 2666. [Google Scholar] [CrossRef] [PubMed]
- Manda-Handzlik, A.; Bystrzycka, W.; Sieczkowska, S.; Demkow, U.; Ciepiela, O. Antibiotics modulate the ability of neutrophils to release neutrophil extracellular traps. Adv. Exp. Med. Biol. 2017, 944, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bystrzycka, W.; Moskalik, A.; Sieczkowska, S.; Manda-Handzlik, A.; Demkow, U.; Ciepiela, O. The effect of clindamycin and amoxicillin on neutrophil extracellular trap (NET) release. Cent. Eur. J. Immunol. 2016, 41, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Luke, D.R.; Foulds, G.; Cohen, S.F.; Levy, B. Safety, toleration, and pharmacokinetics of intravenous azithromycin. Antimicrob. Agents Chemother. 1996, 40, 2577–2581. [Google Scholar] [PubMed]
- Konstantinidis, T.; Kambas, K.; Mitsios, A.; Panopoulou, M.; Tsironidou, V.; Dellapporta, E.; Kouklakis, G.; Arampatzioglou, A.; Angelidou, I.; Mitroulis, I.; et al. Immunomodulatory role of clarithromycin in Acinetobacter baumannii infection via formation of neutrophil extracellular traps. Antimicrob. Agents Chemother. 2015, 60, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.; Wang, J.; Dong, Y.; Meng, S.; Li, R.; Niu, X.; Deng, X. β-sitosterol interacts with pneumolysin to prevent Streptococcus pneumoniae infection. Sci. Rep. 2015, 5, 17668. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.; Deng, X.; Wang, J.; Song, M.; Niu, X.; Peng, L. Insights into structure and activity of natural compound inhibitors of pneumolysin. Sci. Rep. 2017, 7, 42015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, H.; Wang, J.; Guo, Y.; Liu, B.; Deng, X.; Niu, X. Verbascoside alleviates pneumococcal pneumonia by reducing pneumolysin oligomers. Mol. Pharmacol. 2016, 89, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Teng, Z.; Li, M.; Niu, X.; Wang, J.; Deng, X. Epigallocatechin gallate inhibits Streptococcus pneumoniae virulence by simultaneously targeting pneumolysin and sortase A. J. Cell. Mol. Med. 2017, 21, 2586–2598. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Li, L.; Li, M.; Cha, Y.; Deng, X.; Wang, J. Apigenin protects mice from pneumococcal pneumonia by inhibiting the cytolytic activity of pneumolysin. Fitoterapia 2016, 115, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhou, Y.; Wang, G.; Shi, D.; Zha, Y.; Yi, P.; Wang, J. Morin moderates the biotoxicity of pneumococcal pneumolysin by weakening the oligomers’ formation. Chem. Pharm. Bull. 2017, 65, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, B.; Liu, S.; Wang, L.; Wang, J. Anticytotoxin effects of amentoflavone to pneumolysin. Biol. Pharm. Bull. 2017, 40, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhou, Y.; Wang, L.; Li, M.; Shi, D.; Li, D.; Wang, J. Shikonin alleviates the biotoxicity produced by pneumococcal pneumolysin. Life Sci. 2017, 177, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Lu, G.; Li, M.; Deng, X.; Wang, J. Juglone alleviates pneumolysin-induced human alveolar epithelial cell injury via inhibiting the haemolytic activity of pneumolysin. Antonie Van Leeuwenhoek 2017, 110, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Mühlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef] [PubMed]
- BioSpace. Combioxin Release: Data Monitoring Committee Unanimously Recommends Continuation of Combioxin First-In-Human Clinical Trial with CAL02 in Patients with Severe Pneumococcal Pneumonia. Available online: https://www.biospace.com/article/releases/combioxin-release-data-monitoring-committee-unanimously-recommends-continuation-of-combioxin-first-in-human-clinical-trial-with-cal02-in-patients-wit/ (accessed on 17 January 2018).
- François, B.; Colin, G.; Dequin, P.F.; Laterre, P.F.; Perez, A. CAL02: A liposomal adjunctive anti-toxin therapy in infections. A new therapeutic approach for severe community-acquired pneumonia. In Proceedings of the 27th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Vienna, Austria, 22–25 April 2017; ESCMID eLibrary, Abstract Number OS0605. Available online: https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=40357 (accessed on 8 January 2018).
- García-Suárez Mdel, M.; Cima-Cabal, M.D.; Flórez, N.; García, P.; Cernuda-Cernuda, R.; Astudillo, A.; Vázquez, F.; De los Toyos, J.R.; Méndez, F.J. Protection against pneumococcal pneumonia in mice by monoclonal antibodies to pneumolysin. Infect. Immun. 2004, 72, 4534–4540. [Google Scholar] [CrossRef] [PubMed]
- Wang-Lin, S.X.; Balthasar, J.P. Pharmacokinetic and pharmacodynamics considerations for the use of monoclonal antibodies in the treatment of bacterial infections. Antibodies 2018, 7, 5. [Google Scholar] [CrossRef]
- Arsanis. Programs & Pipeline. Available online: http://www.arsanis.com/programs-pipelines/ (accessed on 19 January 2018).
- Rouha, H.; Badarau, A.; Visram, Z.C.; Battles, M.B.; Prinz, B.; Magyarics, Z.; Nagy, G.; Mirkina, I.; Stulik, L.; Zerbs, M.; et al. Five birds, one stone: Neutralization of α-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single monoclonal antibody. MAbs 2015, 7, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Badarau, A.; Rouha, H.; Malafa, S.; Battles, M.B.; Walker, L.; Nielson, N.; Dolezilkova, I.; Teubenbacher, A.; Banerjee, S.; Maierhofer, B.; et al. Context matters: The importance of dimerization-induced conformation of the LukGH leucocidin of Staphylococcus aureus for the generation of neutralizing antibodies. MAbs 2016, 8, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Rouha, H.; Maierhofer, B.; Gross, K.; Badarau, A.; Dolezilkova, I.; Weber, S.; Stulik, L.; Nagy, E. Synergistic inhibition of the concerted actions of six Staphylococcus aureus cytotoxins with ASN100, a combination of two human monoclonal antibodies. In Proceedings of the 27th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Vienna, Austria, 22–25 April 2017; Poster Number P0470. Available online: http://www.arsanis.com/wp-content/uploads/2017/04/5237-Rouha-2017-ECCMID-1.pdf (accessed on 8 January 2018).
- David, S.; Ghosh, C.C.; Kümpers, P.; Shushakova, N.; Van Slyke, P.; Khankin, E.V.; Karumanchi, A.; Dumont, D.; Parikh, S.M. Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L851–L862. [Google Scholar] [CrossRef] [PubMed]
- Gutbier, B.; Jiang, X.; Dietert, K.; Ehrler, C.; Lienau, J.; Van Slyke, P.; Kim, H.; Hoang, V.C.; Maynes, J.T.; Dumont, D.J.; et al. Vasculotide reduces pulmonary hyperpermeability in experimental pneumococcal pneumonia. Crit. Care 2017, 21, 274. [Google Scholar] [CrossRef] [PubMed]
- Hazemi, P.; Tzotzos, S.J.; Fischer, B.; Andavan, G.S.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Essential structural features of TNF-α lectin-like domain derived peptides for activation of amiloride-sensitive sodium current in A549 cells. J. Med. Chem. 2010, 53, 8021–8029. [Google Scholar] [CrossRef] [PubMed]
- Czikor, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial sodium channel-α mediates the protective effect of the TNF-derived TIP peptide in pneumolysin-induced endothelial barrier dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef] [PubMed]
- Study in Intensive Care Patients to Investigate the Clinical Effect of Repetitive Orally Inhaled Doses of AP301 on Alveolar Liquid Clearance in Acute Lung Injury. Clinical Trial NCT01627613C01627613. Available online: https://clinicaltrials.gov/ct2/show/NCT01627613 (accessed on 19 January 2018).
- Krenn, K.; Lucas, R.; Croizé, A.; Boehme, S.; Klein, K.U.; Hermann, R.; Markstaller, K.; Ullrich, R. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: A phase IIa randomized placebo-controlled trial. Crit. Care 2017, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. Review: Current and new generation pneumococcal vaccines. J. Infect. 2014, 69, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Sings, H.L. Pneumococcal conjugate vaccine use in adults—Addressing an unmet medical need for non-bacteremic pneumococcal pneumonia. Vaccine 2017, 35, 5406–5417. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hure, A.; Peel, R.; D’Este, C.; Abhayaratna, W.; Tonkin, A.; Hopper, I.; Thrift, A.G.; Levi, C.; Sturm, J.; et al. Rationale and design of a randomized controlled trial of pneumococcal polysaccharide vaccine for prevention of cardiovascular events: The Australian Study for the Prevention through Immunization of Cardiovascular Events (AUSPICE). Am. Heart J. 2016, 177, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Alderson, M.R. Status of research and development of pediatric vaccines for Streptococcus pneumoniae. Vaccine 2016, 34, 2959–2961. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Type | Mechanism | Status | Refs |
|---|---|---|---|
| Macrolide antibiotics | Inhibition of synthesis of PLY | Component of combination antibiotic therapy | [86] |
| Phytosterols (β-sitosterol; campesterol; brassicasterol) | Membrane binding of PLY | Pre-clinical | [99,100] |
| Polyphenols (verbascoside; epigallocatechin gallate) | Oligomerization of PLY monomers | Pre-clinical | [101,102] |
| Bioflavonoids (apigenin; morin; amentoflavone) | Oligomerization of PLY monomers | Pre-clinical | [103,104,105] |
| Naphthoquinones (shikonin; juglone) | Oligomerization of PLY monomers | Pre-clinical | [106,107] |
| Magnesium chloride | Delays pore formation; others | Pre-clinical with respect to severe pneumococcal disease | [50] |
| Liposome-based (CAL02) | Membrane binding of PLY | Phase IIb clinical evaluation | [16,108,109,110] |
| Monoclonal antibody-based | Membrane binding and inhibition of oligomerization depending on target epitope on PLY | Pre-clinical | [111,113] |
| Angiopoietin-1 mimetic (vasculotide) | Protects against PLY-mediated endothelial barrier dysfunction | Pre-clinical with respect to severe pneumococcal disease | [118] |
| TNF-derived TIP peptide | Activates the amiloride-sensitive sodium channel in epithelial and endothelial cells | Phase IIa trial recently completed | [119,120,121,122] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, R.; Nel, J.G.; Feldman, C. Multifaceted Role of Pneumolysin in the Pathogenesis of Myocardial Injury in Community-Acquired Pneumonia. Int. J. Mol. Sci. 2018, 19, 1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041147
Anderson R, Nel JG, Feldman C. Multifaceted Role of Pneumolysin in the Pathogenesis of Myocardial Injury in Community-Acquired Pneumonia. International Journal of Molecular Sciences. 2018; 19(4):1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041147
Chicago/Turabian StyleAnderson, Ronald, Jan G. Nel, and Charles Feldman. 2018. "Multifaceted Role of Pneumolysin in the Pathogenesis of Myocardial Injury in Community-Acquired Pneumonia" International Journal of Molecular Sciences 19, no. 4: 1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041147