Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH)

 ,
, 
Abstract
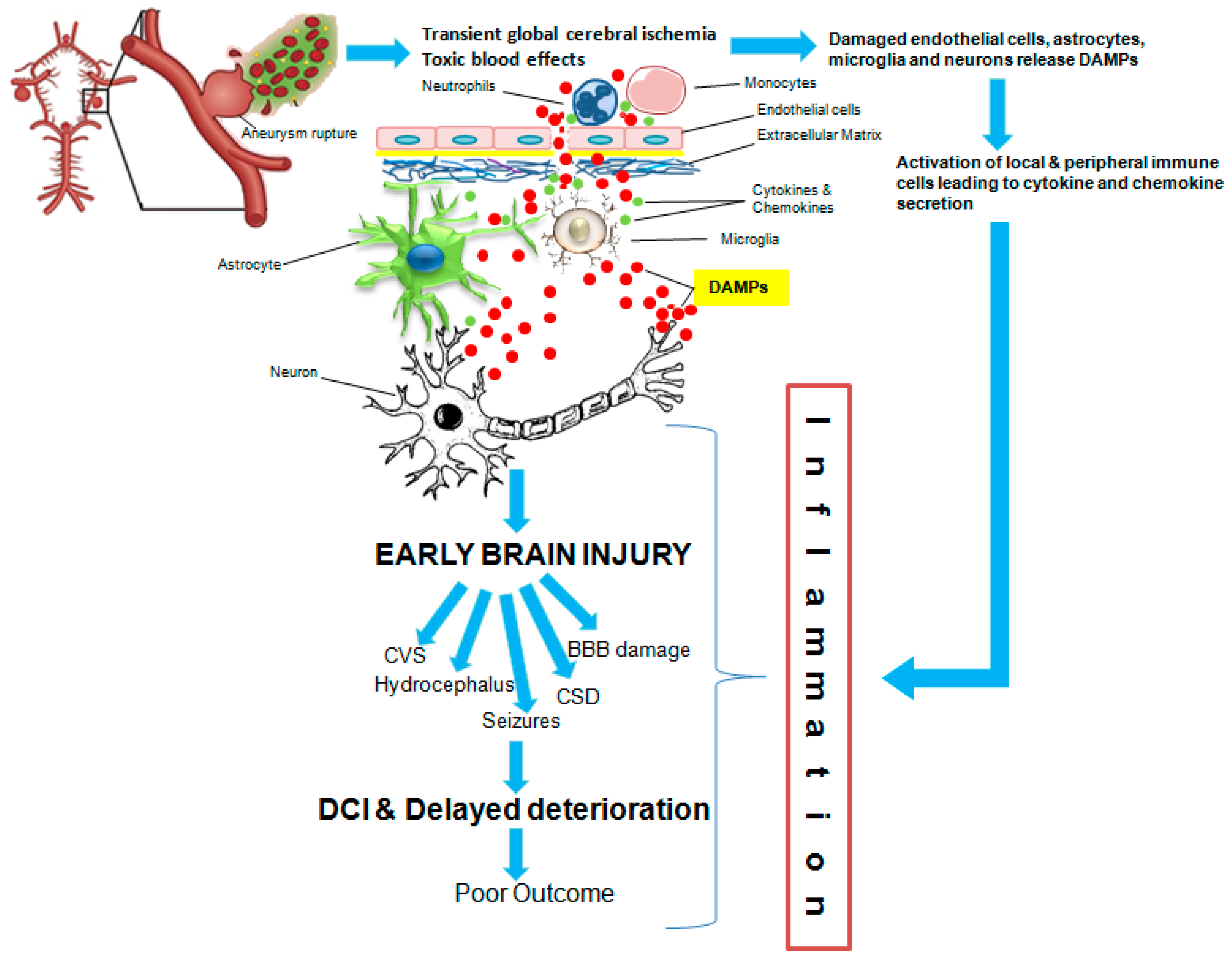
:1. Introduction
2. Methods
3. Results
3.1. High Mobility Group Box 1 (HMGB1) and aSAH
3.2. S100B
3.3. Hemoglobin and Its Derivatives
3.4. Fibrinogen
3.5. IL-1α and IL-33
3.6. Mitochondrial DAMPs
3.7. Extracellular Matrix Derived DAMPs
3.8. Heat Shock Proteins
4. Discussion
Acknowledgments
Conflicts of Interest
Abbreviations
| DAMPs | Damage Associated Molecular Patterns |
| aSAH | Aneurysmal Subarachnoid Hemorrhage |
| IL-1 | Interleukin-1 |
| HMGB1 | High Mobility Group Box-1 |
| mtDNA | Mitochondrial DNA |
| DINDs | Delayed Ischemic Neurological Deficits |
| CVS | Cerebral Vasospasm |
| DCI | Delayed Cerebral Ischemia |
| HSPs | Heat Shock Proteins |
| PRRs | Pattern Recognition Receptors |
| S100B | S100 Calcium Binding Protein Beta |
| ECM | Extracellular Matrix |
| RAGE | Receptor for Advanced Glycation Endproducts |
| CSF | Cerebrospinal Fluid |
| BBB | Blood Brain Barrier |
| TNF-α | Tumor Necrosis Factor-alpha |
| NF-κB | Nuclear Factor-κB |
| TLR-4 | Toll Like Receptor-4 |
| H&H | Hunt and Hess score |
| WFNS | World Federation of Neurological Surgeons score |
| GCS | Glasgow Coma Scale |
| GOS | Glasgow Outcome Scale |
| mRS | Modified Rankin Scale |
| GFAP | Glial Fibrillary Acidic Protein |
| CNS | Central Nervous System |
| EVD | External Ventricular Drain |
| iNOS | Inducible Nitric Oxide Synthase |
| Hb | Hemoglobin |
| PPAR-γ | Peroxisome Proliferator Activated Receptor-γ |
| Hp | Haptoglobin |
| HO-1 | Heme Oxygenase-1 |
| MDA | Malondialdehyde |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| PDGF-AB | Platelet Derived Growth Factor-AB |
| MAPK | Mitogen Activated Protein Kinase |
| LPS | Lipopolysaccharide |
| VSMCs | Vascular Smooth Muscle Cells |
| ODN | Oligodeoxynucleotides |
| Gal-3 | Galectin-3 |
| TFAM | Mitochondrial Transcription Factor A |
| EGF-R | Endothelial Growth Factor-Receptor |
| ST2 | Suppressor of Tumorigenicity-2 |
| MMP-9 | Matrix Metalloproteinase-9 |
| ZO-1 | Zonaocculdens-1 |
| SDC-1 | Syndecan-1 |
| GGA | Geranyl Geranyl Acetone |
| cGAS | cyclic GMP-AMP synthase |
| STING | Stimulator of Interferon Genes |
| NLRP3 | Nucleotide-binding oligomerization domain, Leucine-rich repeat and Pyrin domain containing protein 3 |
| NLRC4 | NLR family CARD domain containing 4 |
| AIM2 | Absent in Melanoma 2 |
References
- Zhao, J.; Lin, H.; Summers, R.; Yang, M.; Cousins, B.G.; Tsui, J. Current treatment strategies for intracranial aneurysms: An overview. Angiology 2018, 69, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Frȍsen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E2–EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Etminan, N.; Rinkel, G.J. Unruptured intracranial aneurysms: Development, rupture and preventive management. Nat. Rev. Neurol. 2016, 12, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Alafaci, C.; Macdonald, R.L. Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives. Surg. Neurol. Int. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Van Gijn, J.; Kerr, R.S.; Rinkel, G.J.E. Subarachnoid haemorrhage. Lancet 2007, 369, 306–318. [Google Scholar] [CrossRef]
- De Rooij, N.K.; Linn, F.H.; van der Plas, J.A.; Algra, A.; Rinkel, G.J. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Korja, M.; Kaprio, J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat. Rev. Neurol. 2016, 12, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Suarez, J.I.; Tarr, R.W.; Selman, W.R. Aneurysmal subarachnoid hemorrhage. N. Engl. J. Med. 2006, 354, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, B.J.; Vergouwen, M.D.I.; Kelfkens, M.M.; Rinkel, G.J.E.; Hol, E.M. Glial cell response after aneurysmal subarachnoid hemorrhage—Functional consequences and clinical implications. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.; Zhang, J.H. Subarachnoid hemorrhage: Is it time for a new direction? Stroke 2009, 40, S86–S87. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Schweizer, T.A. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: A review. Acta Neurochir. Suppl. 2013, 115, 233–238. [Google Scholar] [PubMed]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Manfredi, A.A. Dangers in and out. Science 2009, 323, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit. Care 2009, 11, 362–368. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Laird, M.D.; Sangeetha, S.R.; Youssef, P.; Shakir, B.; Vender, J.R.; Alleyne, C.H.; Dhandapani, K.M. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: An emerging role for neuroproteomics. Neurosurg. Focus 2010, 28, E10. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl. Stroke Res. 2011, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-D.; Chen, J.-S.; Zhou, F.; Liu, Q.-C.; Chen, G.; Zhang, J.-M. Relationship between plasma high mobility group box-1 protein levels and clinical outcomes of aneurysmal subarachnoid hemorrhage. J. Neuroinflamm. 2012, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-Z.; Lin, C.-L.; Wu, S.-C.; Kwan, A.-L. Purpurogallin, a natural phenol, attenuates high-mobility group box 1 in subarachnoid hemorrhage induced vasospasm in a rat model. Int. J. Vasc. Med. 2014, 2014, 254270. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Z.; Wu, S.-C.; Kwan, A.-L.; Lin, C.-L. 4′-O-β-d-glucosyl-5-O-methylvisamminol, an active ingredient of Saposhnikovia divaricata, attenuates high-mobility group box 1 and subarachnoid hemorrhage-induced vasospasm in a rat model. Behav. Brain Funct. 2015, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Z.; Wu, S.C.; Kwan, A.L.; Lin, C.L. Rhinacanthin-C, a fat-soluble extract from Rhinacanthus nasutus, modulates high-mobility group box 1-related neuro-inflammation and subarachnoid hemorrhage-induced brain apoptosis in a rat model. World Neurosurg. 2016, 86, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Sokol, B.; Wozniak, A.; Jankowski, R.; Jurga, S.; Wasik, N.; Shahid, H.; Grzeskowiak, B. HMGB1 level in cerebrospinal fluid as a marker of treatment outcome in patients with acute hydrocephalus following aneurysmal subarachnoid hemorrhage. J. Stroke Cerebrovasc. Dis. 2015, 24, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-C.; Tang, S.-C.; Lee, J.-E.; Li, Y.-I.; Huang, Y.-S.; Yang, W.-S.; Jeng, J.-S.; Arumugam, T.V.; Tu, Y.-K. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2017, 37, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (HMGB1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, F.; Jing, Z.; Wang, X.; Hua, X.; Wan, L. Glycyrrhizic acid exerts anti-inflammatory effect to improve cerebral vasospasm secondary to subarachnoid hemorrhage in a rat model. Neurol. Res. 2017, 39, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francoeur, C.L.; Mayer, S.A. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit. Care 2016, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.D.; Rhind, S.G.; Di Battista, A.P.; Macdonald, R.L.; Baker, A.J. Biomarkers of glycocalyx injury are associated with delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage: A case series supporting a new hypothesis. Neurocrit. Care 2017, 26, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, P.; Foreman, P.M.; Harrigan, M.R.; Fisher, W.S.R.; Vyas, N.A.; Lipsky, R.H.; Lin, M.; Walters, B.C.; Tubbs, R.S.; Shoja, M.M.; et al. Impact of high-mobility group box-1 polymorphism on delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017, 101, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2007, 81, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Bianchi, R.; Riuzzi, F.; Tubaro, C.; Arcuri, C.; Giambanco, I.; Donato, R. S100B protein, a damage-associated molecular pattern protein in the brain and heart, and beyond. Cardiovasc. Psychiatry Neurol. 2010, 2010, 656481. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Belli, A. S100B in neuropathologic states: The CRP of the brain? J. Neurosci. Res. 2007, 85, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Kastrisianaki, E.; Giambanco, I.; Donato, R. S100B protein stimulates microglia migration via rage-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 2011, 286, 7214–7226. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Dong, X.Q.; Hu, Y.Y.; Yu, W.H.; Zhang, Z.Y. High S100B levels in cerebrospinal fluid and peripheral blood of patients with acute basal ganglial hemorrhage are associated with poor outcome. World J. Emerg. Med. 2010, 1, 22–31. [Google Scholar] [PubMed]
- Takayasu, M.; Shibuya, M.; Kanamori, M.; Suzuki, Y.; Ogura, K.; Kageyama, N.; Umekawa, H.; Hidaka, H. S-100 protein and calmodulin levels in cerebrospinal fluid after subarachnoid hemorrhage. J. Neurosurg. 1985, 63, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Hardemark, H.G.; Almqvist, O.; Johansson, T.; Pahlman, S.; Persson, L. S-100 protein in cerebrospinal fluid after aneurysmal subarachnoid haemorrhage: Relation to functional outcome, late CT and SPECT changes, and signs of higher cortical dysfunction. Acta Neurochir. 1989, 99, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.; Hårdemark, H.G.; Gustafsson, J.; Rundström, G.; Mendel-Hartvig, I.; Esscher, T.; Påhlman, S. S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: Markers of cell damage in human central nervous system. Stroke 1987, 18, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, M.; Missler, U.; Hagenstrom, H.; Gottmann, D. S-100 protein plasma levels after aneurysmal subarachnoid haemorrhage. Acta Neurochir. 1997, 139, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.; Petzold, A.; Kerr, M.; Keir, G.; Thompson, E.; Nicoll, J. Temporal alterations in cerebrospinal fluid amyloid beta-protein and apolipoprotein e after subarachnoid hemorrhage. Stroke 2003, 34, e240–e243. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.; Petzold, A.; Kerr, M.; Keir, G.; Thompson, E.; Nicoll, J. Decreased cerebrospinal fluid apolipoprotein E after subarachnoid hemorrhage: Correlation with injury severity and clinical outcome. Stroke 2003, 34, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Keir, G.; Lim, D.; Smith, M.; Thompson, E.J. Cerebrospinal fluid (CSF) and serum S100B: Release and wash-out pattern. Brain Res. Bull. 2003, 61, 281–285. [Google Scholar] [CrossRef]
- Sen, J.; Belli, A.; Petzold, A.; Russo, S.; Keir, G.; Thompson, E.J.; Smith, M.; Kitchen, N. Extracellular fluid S100B in the injured brain: A future surrogate marker of acute brain injury? Acta Neurochir. 2005, 147, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Golzarian, J.; Chevalier, C.; DeWitte, O.; Pochet, R.; Heizman, C.; Decaestecker, C.; Brotchi, J.; Salmon, I.; Kiss, R. Expression of members of the calcium-binding S-100 protein family in a rat model of cerebral basilar artery vasospasm. J. Neurosurg. 2002, 97, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Decaestecker, C.; Brotchi, J.; Heizmann, C.W.; Dewitte, O.; Kiss, R.; Mijatovic, T. Co-expression/co-location of S100 proteins (S100B, S100A1 and S100A2) and protein kinase C (PKC-beta, -eta and -zeta) in a rat model of cerebral basilar artery vasospasm. Neuropathol. Appl. Neurobiol. 2005, 31, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Sanchez-Pena, P.; Roche, S.; Beaudeux, J.L.; Colonne, C.; Coriat, P.; Puybasset, L. Prognosis value of plasma S100B protein levels after subarachnoid aneurysmal hemorrhage. Anesthesiology 2006, 104, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Stranjalis, G.; Korfias, S.; Psachoulia, C.; Kouyialis, A.; Sakas, D.E.; Mendelow, A.D. The prognostic value of serum S-100B protein in spontaneous subarachnoid haemorrhage. Acta Neurochir. 2007, 149, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Sanchez-Pena, P.; Biondi, A.; Sourour, N.; Boch, A.L.; Colonne, C.; Lejean, L.; Abdennour, L.; Puybasset, L. Predictors of 1-year outcome after coiling for poor-grade subarachnoid aneurysmal hemorrhage. Neurocrit. Care 2007, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pena, P.; Pereira, A.R.; Sourour, N.A.; Biondi, A.; Lejean, L.; Colonne, C.; Boch, A.L.; Al Hawari, M.; Abdennour, L.; Puybasset, L. S100B as an additional prognostic marker in subarachnoid aneurysmal hemorrhage. Crit. Care Med. 2008, 36, 2267–2273. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, K.; Fujita, M.; Yamashita, S.; Kaneko, T.; Kawamura, Y.; Izumi, T.; Tsuruta, R.; Kasaoka, S.; Maekawa, T. Prognostic value of biochemical markers of brain damage and oxidative stress in post-surgical aneurysmal subarachnoid hemorrhage patients. Brain Res. Bull. 2010, 81, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Moritz, S.; Warnat, J.; Bele, S.; Graf, B.M.; Woertgen, C. The prognostic value of NSE and S100B from serum and cerebrospinal fluid in patients with spontaneous subarachnoid hemorrhage. J. Neurosurg. Anesthesiol. 2010, 22, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, A.; Meissner, S.; Eyupoglu, I.Y.; Parsch, H.; Schmidt, C.; Buchfelder, M. Dynamics of S100B release into serum and cerebrospinal fluid following acute brain injury. Acta Neurochir. Suppl. 2010, 106, 247–250. [Google Scholar] [PubMed]
- Piazza, O.; Venditto, A.; Tufano, R. Neurogenic pulmonary edema in subarachnoid hemorrage. Panminerva Med. 2011, 53, 203–210. [Google Scholar] [PubMed]
- Brandner, S.; Xu, Y.; Schmidt, C.; Emtmann, I.; Buchfelder, M.; Kleindienst, A. Shunt-dependent hydrocephalus following subarachnoid hemorrhage correlates with increased S100B levels in cerebrospinal fluid and serum. In Intracranial Pressure and Brain Monitoring XIV; Schuhmann, U.M., Czosnyka, M., Eds.; Springer: Vienna, Austria, 2012; pp. 217–220. [Google Scholar]
- Hassan, T.; Nassar, M.; Elhadi, S.M.; Radi, W.K. Effect of magnesium sulfate therapy on patients with aneurysmal subarachnoid hemorrhage using serum S100B protein as a prognostic marker. Neurosurg. Rev. 2012, 35, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pena, P.; Nouet, A.; Clarencon, F.; Colonne, C.; Jean, B.; Le Jean, L.; Fonfrede, M.; Aout, M.; Vicaut, E.; Puybasset, L. Atorvastatin decreases computed tomography and S100-assessed brain ischemia after subarachnoid aneurysmal hemorrhage: A comparative study. Crit. Care Med. 2012, 40, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.S.; Lange, B.; Zimmermann, M.; Seifert, V. CSF and serum biomarkers focusing on cerebral vasospasm and ischemia after subarachnoid hemorrhage. Stroke Res. Treat. 2013, 2013, 560305. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Astrand, R.; Romner, B. Can S100B predict cerebral vasospasms in patients suffering from subarachnoid hemorrhage? Front. Neurol. 2013, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- De Azua Lopez, Z.R.; Egea-Guerrero, J.J.; Rivera-Rubiales, G.; Rodriguez-Rodriguez, A.; Vilches-Arenas, A.; Murillo-Cabezas, F. Serum brain injury biomarkers as predictors of mortality after severe aneurysmal subarachnoid hemorrhage: Preliminary results. Clin. Chem. Lab. Med. 2015, 53, e179–e181. [Google Scholar] [CrossRef] [PubMed]
- Oertel, M.; Schumacher, U.; McArthur, D.L.; Kastner, S.; Boker, D.K. S-100B and NSE: Markers of initial impact of subarachnoid haemorrhage and their relation to vasospasm and outcome. J. Clin. Neurosci. 2006, 13, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, I.; Kleindienst, A.; Hore, N.; Buchfelder, M.; Brandner, S. Early CSF and serum S100B concentrations for outcome prediction in traumatic brain injury and subarachnoid hemorrhage. Clin. Neurol. Neurosurg. 2016, 145, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Azurmendi, L.; Degos, V.; Tiberti, N.; Kapandji, N.; Sanchez-Pena, P.; Sarrafzadeh, A.; Puybasset, L.; Turck, N.; Sanchez, J.C. Neopterin plasma concentrations in patients with aneurysmal subarachnoid hemorrhage: Correlation with infection and long-term outcome. J. Neurosurg. 2016, 124, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.M.; Du, R. Association between S100B levels and long-term outcome after aneurysmal subarachnoid hemorrhage: Systematic review and pooled analysis. PLoS ONE 2016, 11, e0151853. [Google Scholar] [CrossRef] [PubMed]
- Changyaleket, B.; Xu, H.; Vetri, F.; Valyi-Nagy, T.; Paisansathan, C.; Chong, Z.Z.; Pelligrino, D.A.; Testai, F.D. Intracerebroventricular application of S100B selectively impairs pial arteriolar dilating function in rats. Brain Res. 2016, 1634, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Turan, N.; Chau, M.; Pradilla, G. Inflammation, vasospasm, and brain injury after subarachnoid hemorrhage. BioMed Res. Int. 2014, 2014, 384342. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Damore, G.; Costa, B.; Gioannini, T.L.; Weiss, J.P.; Peri, F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun. 2011, 17, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [PubMed]
- Kurland, D.B.; Gerzanich, V.; Simard, J.M. DAMPs converging on toll-like receptor 4 in hemorrhagic stroke, a mini-review. Curr. Neurobiol. 2015, 6, 4–8. [Google Scholar]
- Gladwin, M.T.; Ofori-Acquah, S.F. Erythroid damps drive inflammation in SCD. Blood 2014, 123, 3689–3690. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, X.D.; Zhuang, Z.; Xue, Y.J.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Peroxisome proliferator-activated receptor gamma agonist rosiglitazone attenuates oxyhemoglobin-induced toll-like receptor 4 expression in vascular smooth muscle cells. Brain Res. 2010, 1322, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Weir, B.K. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke 1991, 22, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Recinos, P.F.; Pradilla, G.; Thai, Q.-A.; Perez, M.; Hdeib, A.M.; Tamargo, R.J. Controlled release of lipopolysaccharide in the subarachnoid space of rabbits induces chronic vasospasm in the absence of blood. Surg. Neurol. 2006, 66, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Cai, M.; Fang, Z.; Wei, H.; Zhu, F.; Li, G.; Dong, H.; Xiong, L. Hemopexin induces neuroprotection in the rat subjected to focal cerebral ischemia. BMC Neurosci. 2013, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, X.; Chen-Roetling, J.; Regan, R.F. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J. Neurosurg. 2011, 114, 1159–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; Brough, D.; Robinson, E.M.; Girard, S.; Rothwell, N.J.; Allan, S.M. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis. Model. Mech. 2012, 5, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, M.R.; Delanghe, J.R. Biological and clinical significance of haptoglobin polymorphism in humans. Clin. Chem. 1996, 42, 1589–1600. [Google Scholar] [PubMed]
- Rosin, D.L.; Okusa, M.D. Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Seeger, F.H.; Blessing, E.; Gu, L.; Bornhold, R.; Denger, S.; Kreuzer, J. Fibrinogen induces chemotactic activity in endothelial cells. Acta Physiol. Scand. 2002, 176, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed]
- Adhami, F.; Liao, G.; Morozov, Y.M.; Schloemer, A.; Schmithorst, V.J.; Lorenz, J.N.; Dunn, R.S.; Vorhees, C.V.; Wills-Karp, M.; Degen, J.L.; et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006, 169, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Davalos, D.; Akassoglou, K. Fibrinogen signal transduction in the nervous system. J. Thromb. Haemost. JTH 2009, 7 (Suppl. 1), 151–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, M.G. Coagulation abnormalities in subarachnoid hemorrhage. Stroke 1970, 1, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Fodstad, H.; Nilsson, I.M. Coagulation and fibrinolysis in blood and cerebrospinal fluid after aneurysmal subarachnoid haemorrhage: Effect of tranexamic acid (AMCA). Acta Neurochir. 1981, 56, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.D.; Yim, M.B.; Son, E.I.; Kim, D.W.; Lee, J.K.; Kim, I.H.; Jeon, D.S. Blood antithrombin III and cerebrospinal fluid fibrin/fibrinogen degradation products in aneurysmal subarachnoid hemorrhage patients. J. Korean Neurosurg. Soc. 1990, 19, 945–954. [Google Scholar]
- Schisano, G.; Franco, A.; Nina, P.; Papa, M.L.; Iannuzzi, M.; De Biase, R.; Caldora, M. Monitoring of fibrin and fibrinogen degradation products (FDP) in the cerebrospinal fluid of patients with subarachnoid haemorrhage due to ruptured aneurysm. Report of 55 cases. J. Neurosurg. Sci. 1994, 38, 77–86. [Google Scholar] [PubMed]
- Van der Werf, A.J. Vascular spasm and cerebral ischemia after meningeal hemorrhage caused by rupture of an aneurysm. Neuro-Chir. 1986, 32, 1–22. [Google Scholar]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The interleukin-1α precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front. Immunol. 2013, 4, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsiger, S.; Simmen, H.-P.; Werner, C.M.L.; Wanner, G.A.; Rittirsch, D. Danger signals activating the immune response after trauma. Mediat. Inflamm. 2012, 2012, 315941. [Google Scholar] [CrossRef] [PubMed]
- Buryskova, M.; Pospisek, M.; Grothey, A.; Simmet, T.; Burysek, L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J. Biol. Chem. 2004, 279, 4017–4026. [Google Scholar] [CrossRef] [PubMed]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Eigenbrod, T.; Park, J.H.; Harder, J.; Iwakura, Y.; Nunez, G. Cutting edge: Critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J. Immunol. 2008, 181, 8194–8198. [Google Scholar] [CrossRef] [PubMed]
- Aihara, Y.; Kasuya, H.; Onda, H.; Hori, T.; Takeda, J. Quantitative analysis of gene expressions related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke 2001, 32, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kasuya, H.; Onda, H.; Sasahara, A.; Goto, S.; Hori, T.; Inoue, I. Role of p38 mitogen-activated protein kinase on cerebral vasospasm after subarachnoid hemorrhage. Stroke 2004, 35, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.; Dixit, S.; Bonneau, R.H.; Chinchilli, V.M.; Cockroft, K.M. Neutralizing antibody against interleukin-6 attenuates posthemorrhagic vasospasm in the rat femoral artery model. Neurosurgery 2004, 54, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-R.; Milovanović, M.; Allan, D.; Niedbala, W.; Besnard, A.-G.; Fukada, S.Y.; Alves-Filho, J.C.; Togbe, D.; Goodyear, C.S.; Linington, C.; et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur. J. Immunol. 2012, 42, 1804–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.A.; Christophi, G.P.; Gruber, R.C.; Wilmore, J.R.; Lawrence, D.A.; Massa, P.T. Induction of IL-33 expression and activity in central nervous system glia. J. Leukoc. Biol. 2008, 84, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.T.; Li, H.; Sun, Q.; Liu, M.; Li, W.D.; Li, S.; Yu, Z.; Wei, W.T.; Hang, C.H. IL-33 expression in the cerebral cortex following experimental subarachnoid hemorrhage in rats. Cell. Mol. Neurobiol. 2015, 35, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.H.; Chang, W.N.; Tsai, N.W.; Chuang, Y.C.; Huang, C.R.; Wang, H.C. The value of serial plasma nuclear and mitochondrial DNA levels in adult community-acquired bacterial meningitis. QJM Mon. J. Assoc. Phys. 2010, 103, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Perez-Santiago, J.; Schrier, R.D.; de Oliveira, M.F.; Gianella, S.; Var, S.R.; Day, T.R.; Ramirez-Gaona, M.; Suben, J.D.; Murrell, B.; Massanella, M.; et al. Cell-free mitochondrial DNA in CSF is associated with early viral rebound, inflammation, and severity of neurocognitive deficits in HIV infection. J. Neurovirol. 2016, 22, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Llorens, F.; Golanska, E.; Sikorska, B.; Liberski, P.; Zerr, I.; Trullas, R. Mitochondrial DNA differentiates alzheimer’s disease from Creutzfeldt-Jakob disease. Alzheimer Dement. 2016, 12, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Vilas, D.; Taylor, P.; Shaw, L.M.; Tolosa, E.; Trullas, R. Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 2016, 94, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Zollo, O.; Van Deerlin, V.; Trojanowski, J.Q. Analysis of cerebrospinal fluid mitochondrial DNA levels in Alzheimer disease. Ann. Neurol. 2014, 75, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Varhaug, K.N.; Vedeler, C.A.; Myhr, K.M.; Aarseth, J.H.; Tzoulis, C.; Bindoff, L.A. Increased levels of cell-free mitochondrial DNA in the cerebrospinal fluid of patients with multiple sclerosis. Mitochondrion 2017, 34, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Yang, T.M.; Lin, W.C.; Lin, Y.J.; Tsai, N.W.; Liou, C.W.; Kwan, A.L.; Lu, C.H. The value of serial plasma and cerebrospinal fluid nuclear and mitochondrial deoxyribonucleic acid levels in aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2013, 118, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heula, A.L.; Sajanti, J.; Majamaa, K. Glycosaminoglycans in subdural fluid and CSF after meningeal injury. Acta Neurochir. 2015, 157, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke 2010, 41, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kanamaru, K.; Shiba, M.; Fujimoto, M.; Kawakita, F.; Imanaka-Yoshida, K.; Yoshida, T.; Taki, W. Tenascin-C is a possible mediator between initial brain injury and vasospasm-related and -unrelated delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Acta Neurochir. Suppl. 2015, 120, 117–121. [Google Scholar] [PubMed]
- Suzuki, H.; Kinoshita, N.; Imanaka-Yoshida, K.; Yoshida, T.; Taki, W. Cerebrospinal fluid tenascin-C increases preceding the development of chronic shunt-dependent hydrocephalus after subarachnoid hemorrhage. Stroke 2008, 39, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Shiba, M.; Kawakita, F.; Liu, L.; Shimojo, N.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J. Neurosurg. 2016, 124, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Kawakita, F.; Fujimoto, M.; Nakano, F.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Role of periostin in early brain injury after subarachnoid hemorrhage in mice. Stroke 2017, 48, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Kelsh, R.; You, R.; Horzempa, C.; Zheng, M.; McKeown-Longo, P.J. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PLoS ONE 2014, 9, e102974. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, S.; Shiroyama, Y.; Iwamoto, T.; Yamashita, T.; Ito, H. Sequential changes in plasma fibronectin in patients with subarachnoid hemorrhage. Neurol. Medico-Chir. 1993, 33, 225–228. [Google Scholar] [CrossRef]
- Kurogi, R.; Kikkawa, Y.; Matsuo, S.; Nakamizo, A.; Mizoguchi, M.; Sasaki, T. Upregulation of tissue inhibitor of metalloproteinase-1 contributes to restoration of the extracellular matrix in the rabbit basilar artery during cerebral vasospasm after subarachnoid hemorrhage. Brain Res. 2015, 1616, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Alvarez, L.; Ortega, E. The many roles of galectin-3, a multifaceted molecule, in innate immune responses against pathogens. Mediat. Inflamm. 2017, 2017, 9247574. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Zhao, J.; Liu, H.; He, S. Prognostic value of plasma galectin-3 levels after aneurysmal subarachnoid hemorrhage. Brain Behav. 2016, 6, e00543. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-W.; Kim, S.-J.; Cho, H.-I.; Lee, S.-M. Damps activating innate immune responses in sepsis. Ageing Res. Rev. 2015, 24, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Tang, J.; Nanda, A.; Zhang, J.H. Heat shock proteins expression in brain stem after subarachnoid hemorrhage in rats. Acta Neurochir. Suppl. 2003, 86, 477–482. [Google Scholar] [PubMed]
- Matz, P.G.; Sundaresan, S.; Sharp, F.R.; Weinstein, P.R. Induction of HSP70 in rat brain following subarachnoid hemorrhage produced by endovascular perforation. J. Neurosurg. 1996, 85, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Matz, P.; Turner, C.; Weinstein, P.R.; Massa, S.M.; Panter, S.S.; Sharp, F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Turner, C.P.; Panter, S.S.; Sharp, F.R. Anti-oxidants prevent focal rat brain injury as assessed by induction of heat shock proteins (HSP70, HO-1/HSP32, HSP47) following subarachnoid injections of lysed blood. Mol. Brain Res. 1999, 65, 87–102. [Google Scholar] [CrossRef]
- Macomson, S.D.; Brophy, C.M.; Miller, W.; Harris, V.A.; Shaver, E.G. Heat shock protein expression in cerebral vessels after subarachnoid hemorrhage. Neurosurgery 2002, 51, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Tsunoda, H.; Nishimura, Y.; Kirino, T.; Tanaka, T. Potential role for heat shock protein 72 in antagonizing cerebral vasospasm after rat subarachnoid hemorrhage. Circulation 2004, 110, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, F.; Marinelli, C.; Longo, D.; Pugnaghi, M.; Nichelli, P.F.; Meletti, S.; Biagini, G. Hypoxia markers are expressed in interneurons exposed to recurrent seizures. Neuromol. Med. 2013, 15, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Vinet, J.; Meletti, S.; Biagini, G. Ischemic–hypoxic mechanisms leading to hippocampal dysfunction as a consequence of status epilepticus. Epilepsy Behav. 2015, 49, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The hmgb1 receptor rage mediates ischemic brain damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, R.K.; Tamborska, A.; Dorward, D.A.; Ho, G.-T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Research 2017, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A.J. Targeting toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.; Roger, T. Targeting toll-like receptors: Promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front. Immunol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Farooq, A.; Malik, A.; Trawick, B.N.; Berberich, D.W.; McClurg, J.P.; Galen, K.P.; Mehal, W. A novel small molecule enantiomeric analogue of traditional (−)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation induced organ damage. J. Immunol. 2013, 190, 4297–4304. [Google Scholar] [CrossRef] [PubMed]
- Holl, E.K.; Shumansky, K.L.; Borst, L.B.; Burnette, A.D.; Sample, C.J.; Ramsburg, E.A.; Sullenger, B.A. Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proc. Natl. Acad. Sci. USA 2016, 113, 9728–9733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holl, E.K.; Shumansky, K.L.; Pitoc, G.; Ramsburg, E.; Sullenger, B.A. Nucleic acid scavenging polymers inhibit extracellular DNA-mediated innate immune activation without inhibiting anti-viral responses. PLoS ONE 2013, 8, e69413. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.E.; Crack, P.J. Neural injury following stroke: Are toll-like receptors the link between the immune system and the CNS? Br. J. Pharmacol. 2010, 160, 1872–1888. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Sr. # | DAMPs | Receptors |
|---|---|---|
| 1. | HMGB1 | TLR-2, TLR-4, TLR-9, RAGE |
| 2. | IL-1α | IL-1R |
| 3. | IL-33 | ST2 (IL-1RL1) |
| 4. | Heme, Hemin, Oxyhemoglobin, methemoglobin | TLR-4 |
| 5. | mtDNA | TLR-9, NLRP3, NLRC4, AIM-2, cGAS-STING |
| 6. | TFAM | RAGE, TLR-9 |
| 7. | N-formyl peptides | FPR1, FPRL1 |
| 8. | S-100 proteins | TLR-4, RAGE |
| 9. | Fibrinogen | TLR-4 |
| 10. | Fibronectin | TLR-2, TLR-4 |
| 11. | Hyaluronan | TLR-2, TLR-4 |
| 12. | Biglycan | TLR-2, TLR-4, P2X4, P2X7, NLRP3 |
| 13. | Versican | TLR-2, TLR-6, CD14 |
| 14. | Heparan sulfate | TLR-4 |
| 15. | Tenascin C | TLR-4 |
| 16. | Galectin-3 | TLR-2, TLR-4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhry, S.R.; Hafez, A.; Rezai Jahromi, B.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072035
Chaudhry SR, Hafez A, Rezai Jahromi B, Kinfe TM, Lamprecht A, Niemelä M, Muhammad S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). International Journal of Molecular Sciences. 2018; 19(7):2035. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072035
Chicago/Turabian StyleChaudhry, Shafqat Rasul, Ahmad Hafez, Behnam Rezai Jahromi, Thomas Mehari Kinfe, Alf Lamprecht, Mika Niemelä, and Sajjad Muhammad. 2018. "Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH)" International Journal of Molecular Sciences 19, no. 7: 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072035