WB1, a Regulator of Endosperm Development in Rice, Is Identified by a Modified MutMap Method

 , ,
, ,
Abstract
:
1. Introduction
2. Results
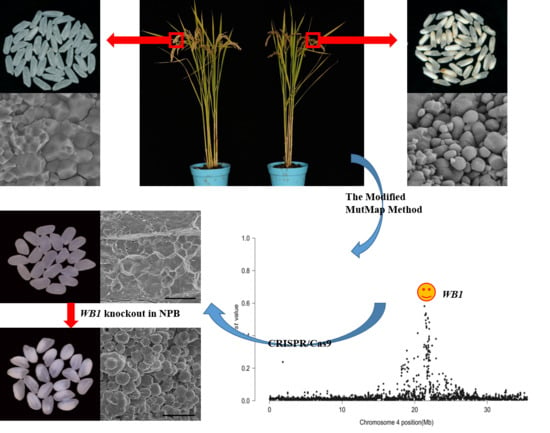
2.1. Phenotypic Characterization and Genetic Analysis of the wb1 Mutant
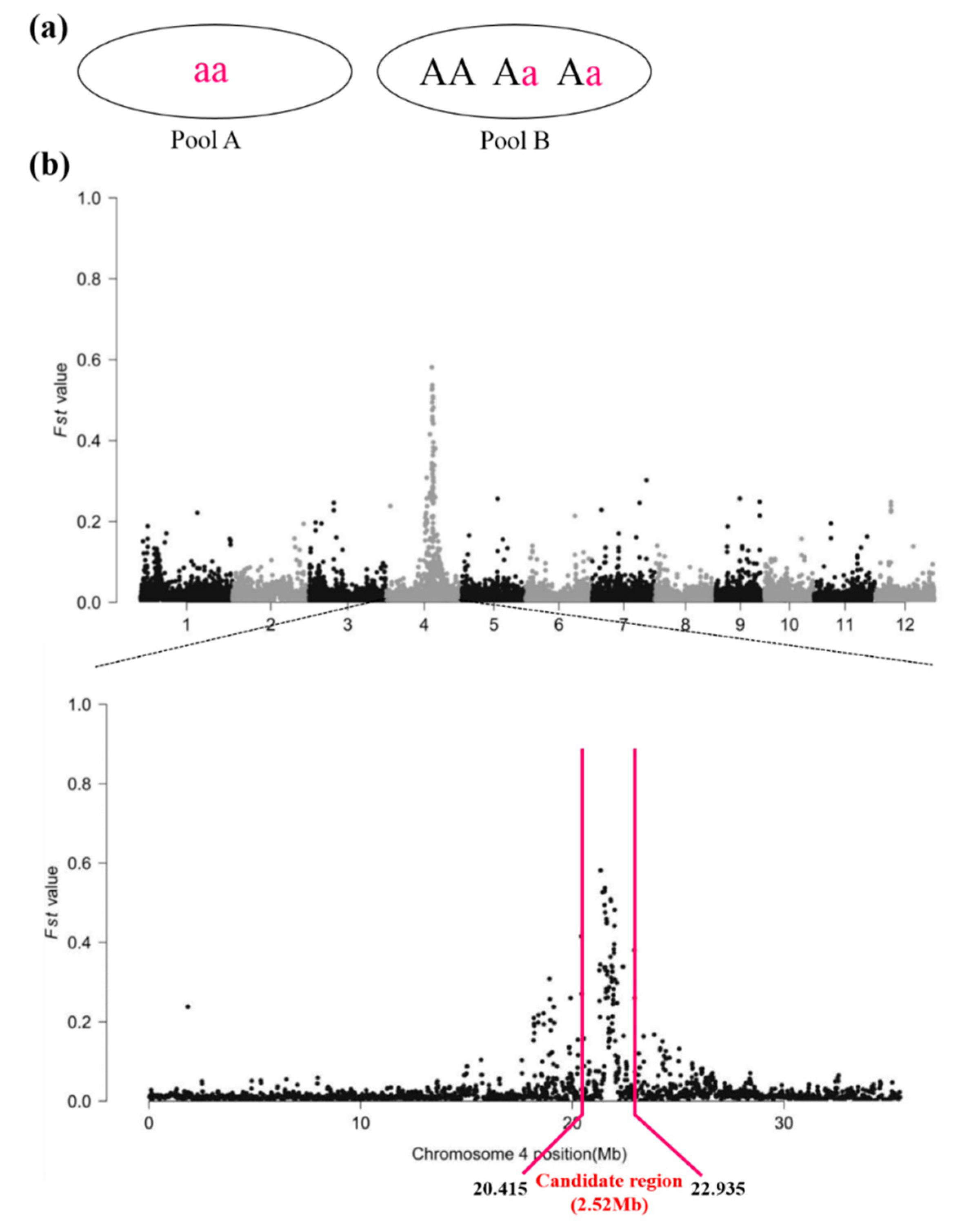
2.2. Candidate Region of the WB1 Gene Obtained through the Modified MutMap Method
2.3. Screening the SNPs Detected in the Candidate Region
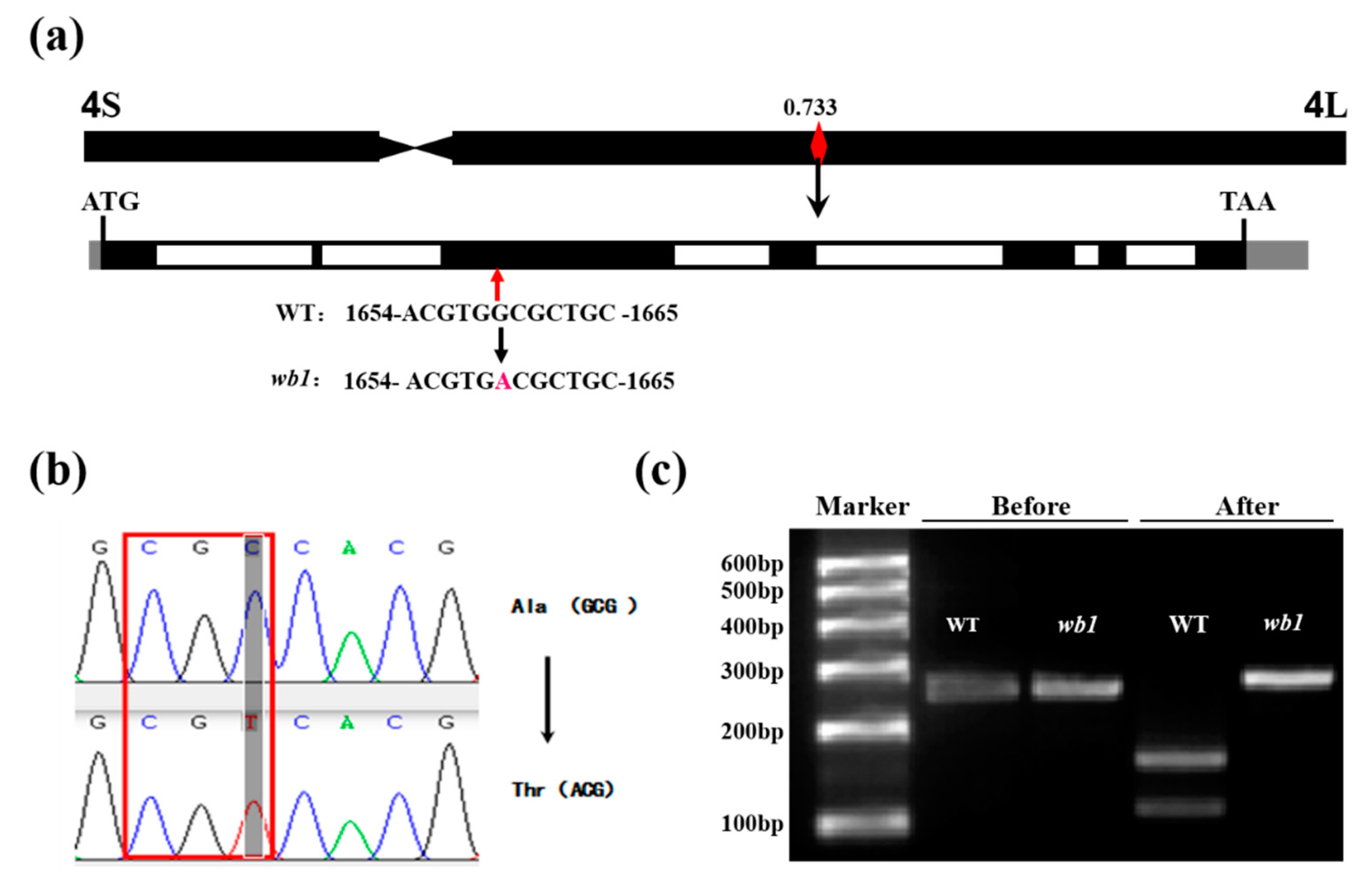
2.4. Identification of the Casual SNP
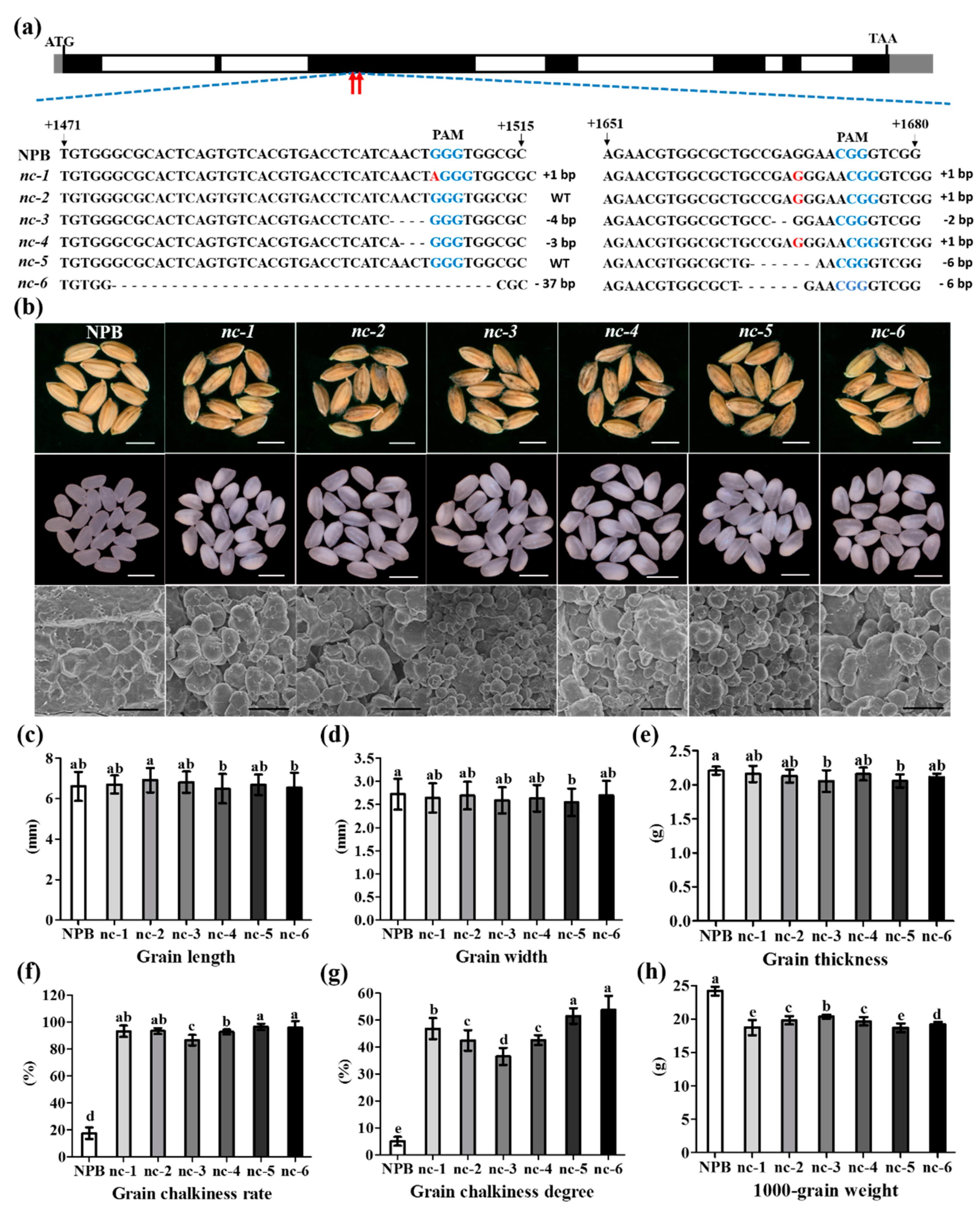
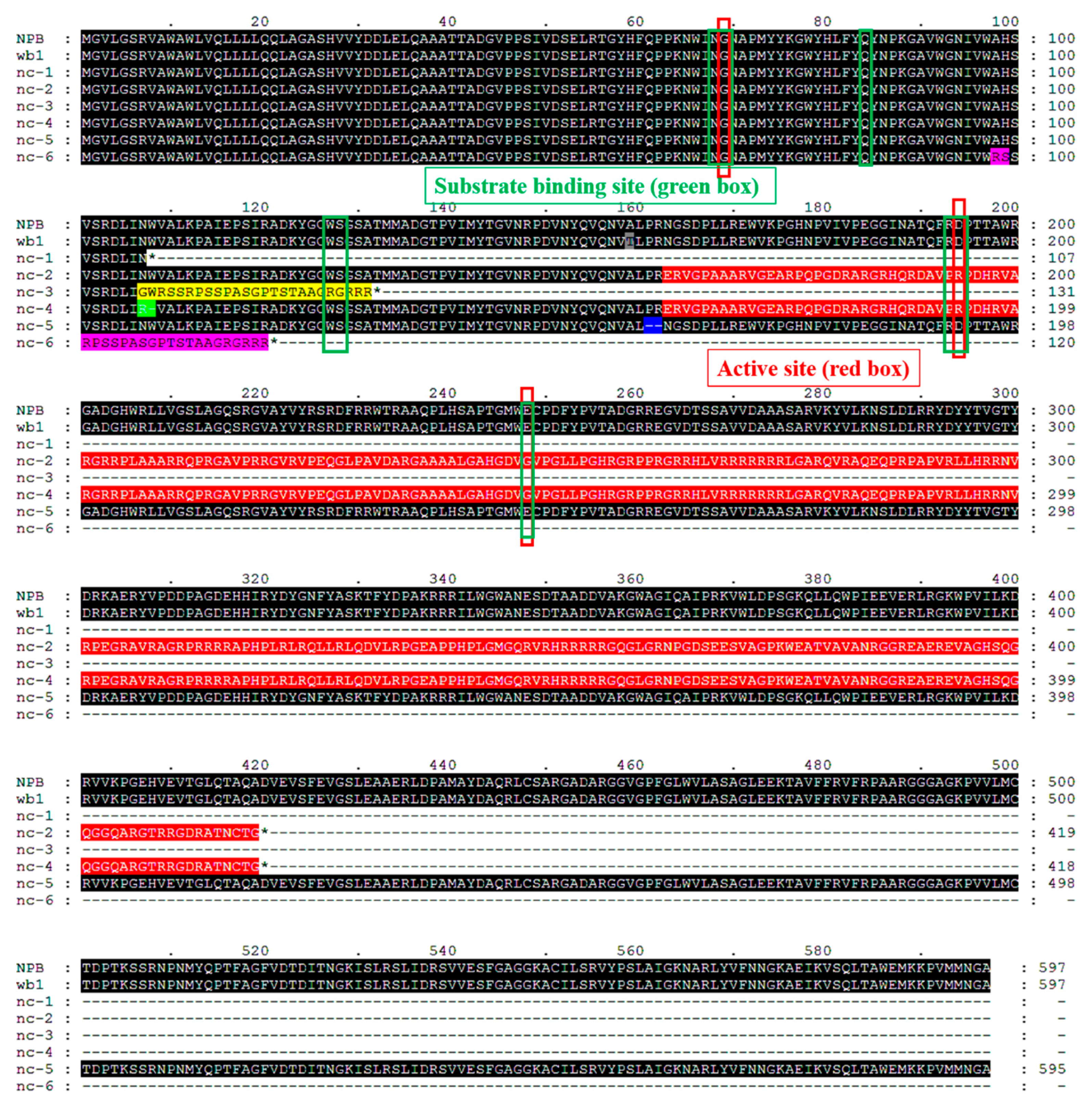
2.5. Function Verification of the WB1 Gene (ORF6) through the CRISPR/Cas9 System in Reverse
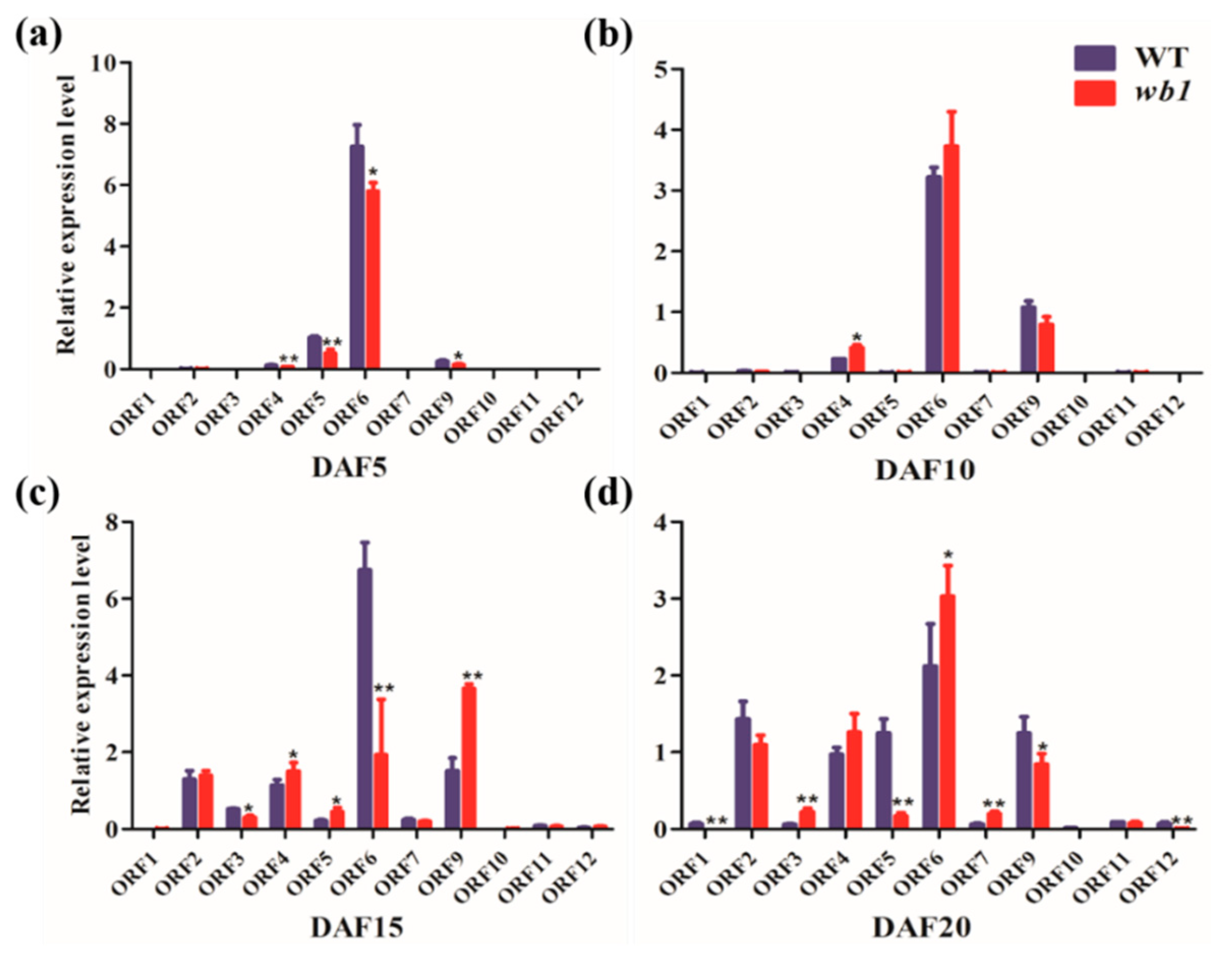
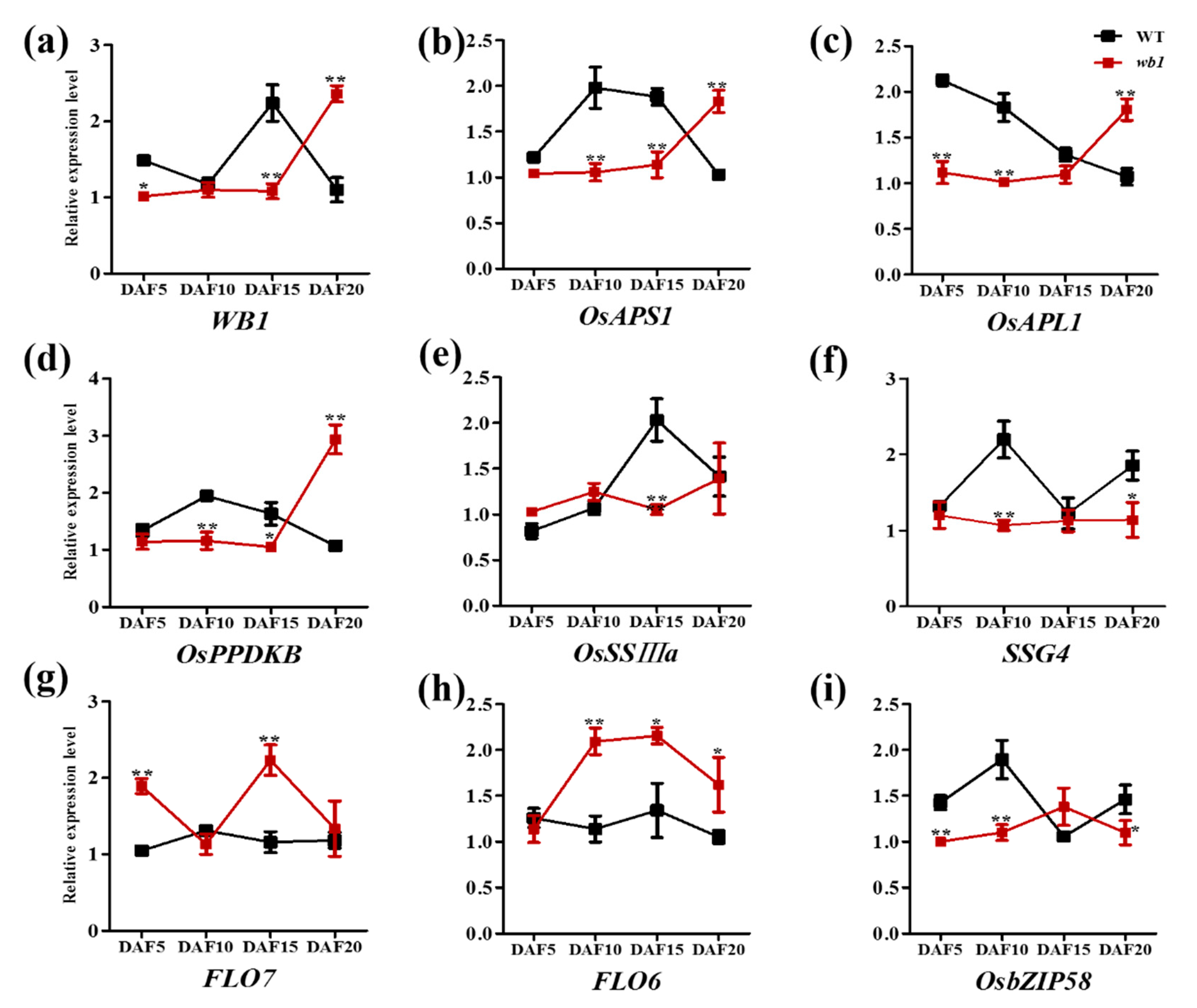
2.6. Expression Analysis of Starch Metabolism-Related Genes in Endosperm
3. Discussion
3.1. WB1 Controls Rice Endosperm Development
3.2. Different Mutations of WB1 Can Disrupt Its Biological Function
3.3. The Modified MutMap Method Applied to Isolate WB1 Is Feasible for Gene Mapping
4. Materials and Methods
4.1. Plant Materials and Growth Condition
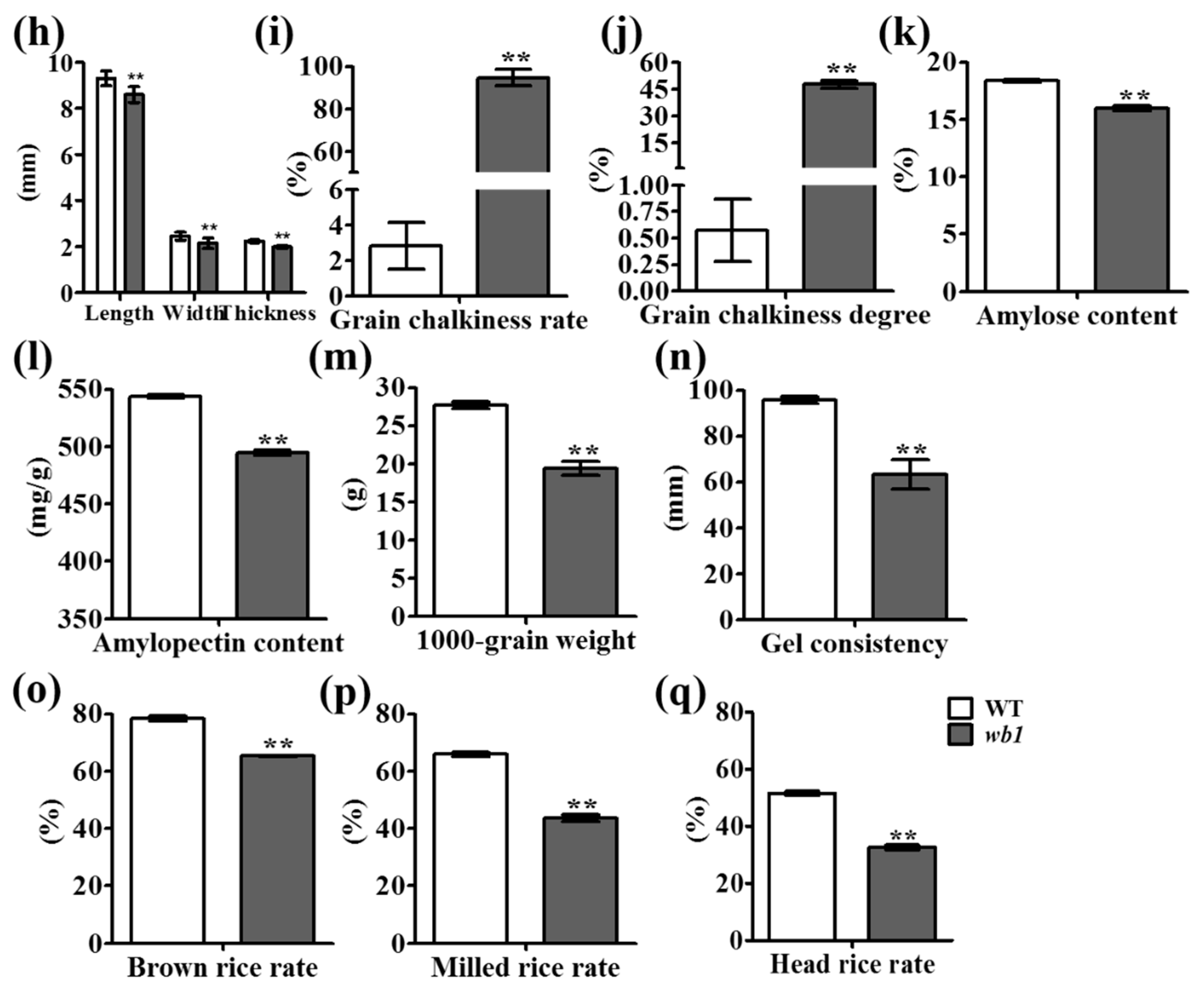
4.2. Grain Quality Analysis
4.3. PCR, RNA Isolation and Real-time Quantitative PCR (qPCR)
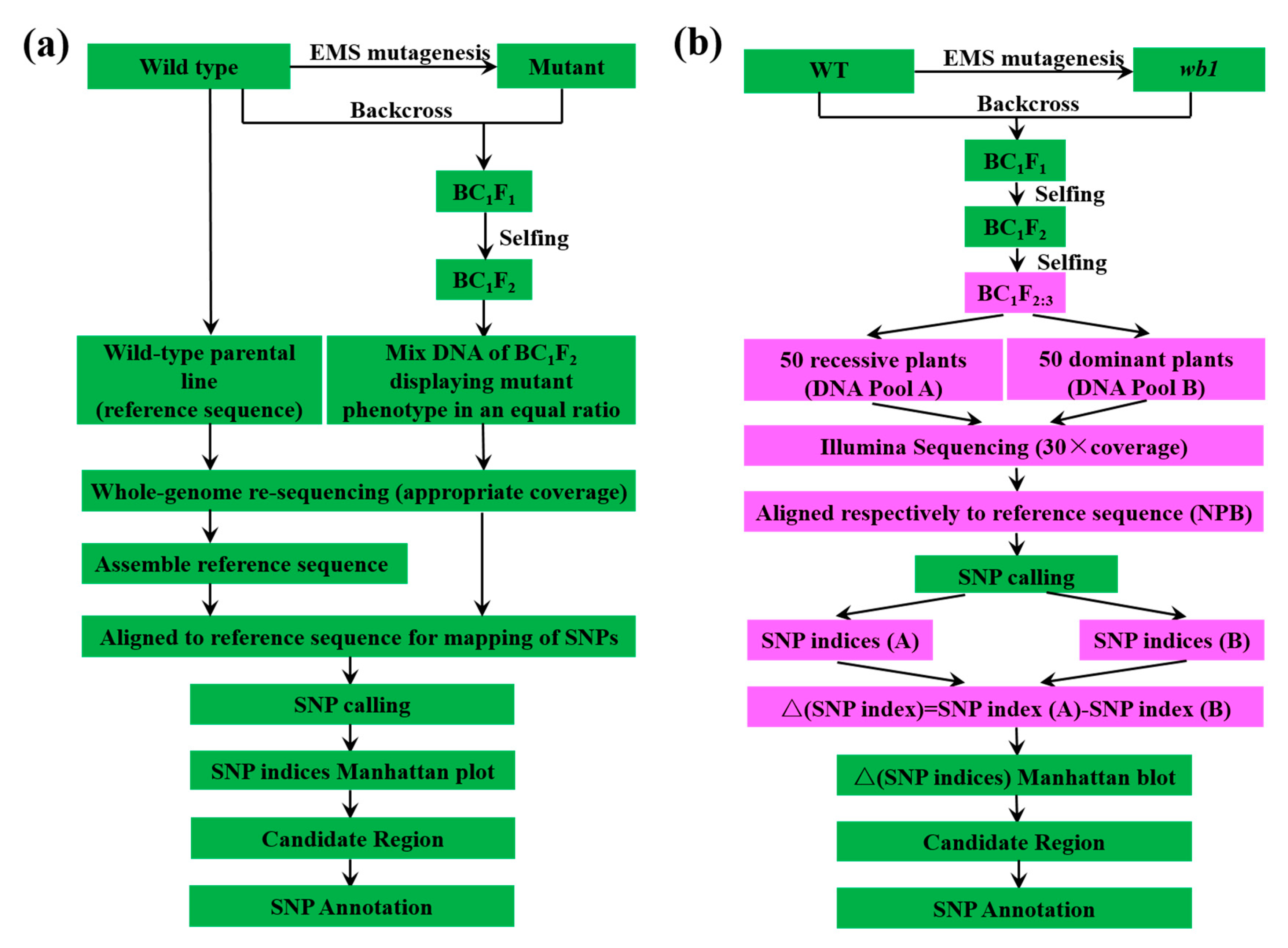
4.4. DNA Template Preparation, DNA Library Construction, and Re-Sequencing
4.5. Re-Sequencing Analysis
4.6. Calculation of ∆ (SNP Indices)
4.7. Restriction Endonuclease Digestion Analysis
4.8. Vector Construction for CRISPR/Cas9-Mediated Mutation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzgerald, M.A.; McCouch, S.R.; Hall, R.D. Not just a grain of rice: The quest for quality. Trends Plant Sci. 2009, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- She, K.C.; Kusano, H.; Koizumi, K.; Yamakawa, H.; Hakata, M.; Imamura, T.; Fukuda, M.; Naito, N.; Tsurumaki, T.; Yaeshima, M.; et al. A novel factor FLOURY ENDOSPERM2 is involved in regulation of rice grain size and starch quality. Plant Cell 2010, 22, 3280–3294. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ren, Y.; Liu, X.; Jiang, L.; Chen, L.; Han, X.; Jin, M.; Liu, S.; Liu, F.; Lv, J.; et al. OsRab5a regulates endomembrane organization and storage protein trafficking in rice endosperm cells. Plant J. 2010, 64, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, Y.H.; Liu, F.; Ren, Y.; Zhou, K.; Lv, J.; Zheng, M.; Zhao, S.; Zhang, L.; Wang, C.; et al. FLOURYE DOSPERM6 encodes a CBM48 domain-containing protein involved in compound granule formation and starch synthesis in rice endosperm. Plant J. 2014, 77, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, Y.; Liu, F.; Zhou, K.; Ding, Y.; Zhou, F.; Wang, Y.; Liu, K.; Gan, L.; Ma, W.; et al. GLUTELIN PRECURSOR ACCUMULATION3 encodes a regulator of post-Golgi vesicular traffic essential for vacuolar protein sorting in rice endosperm. Plant Cell 2014, 26, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, R.; Maekawa, M.; Kusano, M.; Kondo, H.; Fujita, N.; Kawagoe, Y.; Sakamoto, W. Amyloplast-localized SUBSTANDARD STARCH GRAIN4 protein influences the size of starch grains in rice endosperm. Plant Physiol. 2014, 164, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Fukuda, M.; Sunada, M.; Ishino, S.; Ishino, Y.; Okita, T.W.; Ogawa, M.; Ueda, T.; Kumamaru, T. Guanine nucleotide exchange factor 2 for Rab5 proteins coordinated with GLUP6/GEF regulates the intracellular transport of the proglutelin from the Golgi apparatus to the protein storage vacuole in rice endosperm. J. Exp. Bot. 2015, 66, 6137–6147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ren, Y.; Lu, B.; Yang, C.; Feng, Z.; Liu, Z.; Chen, J.; Ma, W.; Wang, Y.; Yu, X.; et al. FLOURY ENDOSPERM 7 encodes a regulator of starch synthesis and amyloplast development essential for peripheral endosperm development in rice. J. Exp. Bot. 2016, 67, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Hwang, S.K.; Han, M.; Eom, J.S.; Kang, H.G.; Han, Y.; Choi, S.B.; Cho, M.H.; Bhoo, S.H.; An, G.; et al. Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.). Plant Mol. Biol. 2007, 65, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Shibahara, K.; Tokunaga, T.; Nishi, A.; Tasaki, M.; Hwang, S.; Okita, T.W.; Kaneko, N.; Fujita, N.; Yoshida, M.; et al. Mutation of the plastidial alpha-glucan phosphorylase gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell 2008, 20, 1833–1849. [Google Scholar] [CrossRef] [PubMed]
- Tuncel, A.; Kawaguchi, J.; Ihara, Y.; Matsusaka, H.; Nishi, A.; Nakamura, T.; Kuhara, S.; Hirakawa, H.; Nakamura, Y.; Cakir, B.; et al. The rice endosperm ADP-glucose pyrophosphorylase large subunit is essential for optimal catalysis and allosteric regulation of the heterotetrameric enzyme. Plant Cell Physiol. 2014, 55, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Shiraishi, S.; Tuncel, A.; Matsusaka, H.; Satoh, R.; Singh, S.; Crofts, N.; Hosaka, Y.; Fujita, N.; Hwang, S.K.; et al. Analysis of the rice ADP-glucose transporter (OsBT1) indicates the presence of regulatory processes in the amyloplast stroma that control ADP-glucose flux into starch. Plant Physiol. 2016, 170, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wei, X.; Ren, Y.; Qiu, J.; Jiao, G.; Guo, X.; Tang, S.; Wan, J.; Hu, P. OsBT1 encodes an ADP-glucose transporter involved in starch synthesis and compound granule formation in rice endosperm. Sci. Rep. 2017, 7, 40124. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, C.; Xing, Y.; Yun, P.; Luo, L.; Yan, B.; Peng, B.; Xie, W.; Wang, G.; Li, X.; et al. Chalk5 encodes a vacuolar H+- translocating pyrophosphatase influencing grain chalkiness in rice. Nat. Genet. 2014, 46, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Hannah, L.C.; James, M. The complexities of starch biosynthesis in cereal endosperms. Curr. Opin. Biotechnol. 2008, 19, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sauer, N. Molecular physiology of higher plant sucrose transporters. FEBS Lett. 2007, 581, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Toyota, K.; Tamura, M.; Ohdan, T.; Nakamura, Y. Expression profiling of starch metabolism-related plastidic translocator genes in rice. Planta 2006, 223, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.S.; Cho, J.I.; Reinders, A.; Lee, S.W.; Yoo, Y.; Tuan, P.Q.; Choi, S.B.; Bang, G.; Park, Y.I.; Cho, M.H.; et al. Impaired function of the tonoplast-localized sucrose transporter in rice, OsSUT2, limits the transport of vacuolar reserve sucrose and affects plant growth. Plant Physiol. 2011, 157, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Sosso, D.; Luo, D.; Li, Q.B.; Sasse, J.; Yang, J.; Gendrot, G.; Suzuki, M.; Koch, K.E.; McCarty, D.R.; Chourey, P.S.; et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 2015, 47, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Tatsuro, H.; Grahamn, S.; Tomio, T. An expression analysis profile for the entire sucrose synthase gene family in rice. Plant Sci. 2008, 174, 534–543. [Google Scholar] [CrossRef]
- Wei, X.J.; Jiao, G.A.; Lin, H.Y.; Sheng, Z.H.; Shao, G.N.; Xie, L.H.; Tang, S.Q.; Xu, Q.; Hu, P.S. GRAIN INCOMPLETE FILLING 2 regulates grain filling and starch synthesis during rice caryopsis development. J. Integr. Plant Biol. 2017, 59, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, S.; Matsuoka, M.; An, G. White-core endosperm floury endosperm-4 in rice is generated by knockout mutations in the C4-type pyruvate orthophosphate dikinase gene (OsPPDKB). Plant J. 2005, 42, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Qian, Q.; Liu, Q.; Yan, M.; Liu, X.; Yan, C.; Liu, G.; Gao, Z.; Tang, S.; Zeng, D.; et al. Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc. Natl. Acad. Sci. USA 2009, 106, 21760–21765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Yoshida, M.; Asakura, N.; Ohdan, T.; Miyao, A.; Hirochika, H.; Nakamura, Y. Function and characterization of starch synthase I using mutants in rice. Plant Physiol. 2006, 140, 1070–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cheng, Z.; Zhang, X.; Gao, X.; Su, N.; Jiang, L.; Mao, L.; Wan, J. Double repression of soluble starch synthase genes SSIIa and SSIIIa in rice (Oryza sativa L.) uncovers interactive effects on the physicochemical properties of starch. Genome 2011, 54, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Yoshida, M.; Kondo, T.; Saito, K.; Utsumi, Y.; Tokunaga, T.; Nishi, A.; Satoh, H.; Park, J.H.; Jane, J.L.; et al. Characterization of SSIIIa-deficient mutants of rice: The function of SSIIIa and pleiotropic effects by SSIIIa deficiency in the rice endosperm. Plant Physiol. 2007, 144, 2009–2023. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, Y.; Kubo, A.; Satoh, H.; Takaiwa, F.; Nakamura, Y. Roles of isoamylase and ADP-glucose pyrophosphorylase in starch granule synthesis in rice endosperm. Plant J. 2005, 42, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, A.; Fujita, N.; Harada, K.; Matsuda, T.; Satoh, H.; Nakamura, Y. The starch-debranching enzymes isoamylase and pullulanase are both involved in amylopectin biosynthesis in rice endosperm. Plant Physiol. 1999, 121, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Fujita, N.; Nish, A.; Satoh, H.; Hosaka, Y.; Ugaki, M.; Kawasaki, S.; Nakamura, Y. The structure of starch can be manipulated by changing the expression levels of starch branching enzyme IIb in rice endosperm. Plant Biotechnol. J. 2004, 2, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.F.; Xue, H.W. Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol. 2010, 154, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Yamamoto, M.P.; Touno, S.M.; Yasuda, H.; Takaiwa, F. Compensation and interaction between RISBZ1 and RPBF during grain filling in rice. Plant J. 2009, 59, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-Generation DNA Sequencing Method. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaeqashi, H.; Obara, T.; Oikawa, K.; Utsushi, H.; Kanzaki, E.; et al. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Uemura, A.; Yaegashi, H.; Tamiru, M.; Abe, A.; Mitsuoka, C.; Utsushi, H.; Natsume, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap-Gap: Whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 2013, 200, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wu, J.; Kanamori, H.; Katayose, Y.; Fujisawa, M.; Namiki, N.; Mizuno, H.; Yamamoto, K.; Antonio, B.A.; Baba, T.; et al. The map-based sequence of the rice genome. Nature 2005, 2436, 793–800. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST). Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Bökel, C. EMS screens: From mutagenesis to screening and mapping. Methods Mol. Biol. 2008, 420, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sodhi, N.S.; Kaur, M.; Saxena, S.K. Physico-chemical, morphological, thermal, cooking and textural properties of chalky and translucent rice kernels. Food Chem. 2003, 82, 433–439. [Google Scholar] [CrossRef]
- Cheng, F.M.; Zhong, L.J.; Wang, F.; Zhang, G.P. Differences in cooking and eating properties between chalky and translucent parts in rice grains. Food Chem. 2005, 90, 39–46. [Google Scholar] [CrossRef]
- Lisle, A.J.; Martin, M.; Fitzgerald, M.A. Chalky and translucent rice grains differ in starch composition and structure and cooking properties. Cereal Chem. 2000, 77, 627–632. [Google Scholar] [CrossRef]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S.; et al. Green revolution: A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional cloning of rice semidwarfing gene, sd-1: Rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 2002, 9, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Spielmeyer, W.; Ellis, M.H.; Chandler, P.M. Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9043–9048. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Chen, L.L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.M.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, Y.; Sun, L.; Sittipun, S.; Yang, Z.; Sun, B.; Xuan, D.; Li, Z.; Yu, P.; Wu, W.; et al. The Rice AAA-ATPase OsFIGNL1 Is Essential for Male Meiosis. Fron. Plant Sci. 2017, 8, 1639. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Kubo, A.; Suh, S.D.; Wong, K.S.; Jane, J.L.; Ozawa, K.; Takaiwa, F.; Inaba, Y.; Nakamura, Y. Antisense inhibition of isoamylase alters the structure of amylopectin and the physicochemical properties of starch in rice endosperm. Plant Cell Physiol. 2003, 44, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hovenkamphermelink, J.H.; Devries, J.N.; Adamse, P.; Jacobsen, E.; Witholt, B.; Feenstra, W.J. Rapid estimation of the amylose/amylopectin ratio in small amounts of tuber and leaf tissue of the potato. Potato Res. 1988, 31, 241–246. [Google Scholar] [CrossRef]
- Zhou, L.; Liang, S.; Ponce, K.; Marundon, S.; Ye, G.; Zhao, X. Factors affecting head rice yield and chalkiness in indica rice. Field Crop. Res. 2015, 172, 1–10. [Google Scholar] [CrossRef]
- Chen, D.H.; Ronald, P.C. A rapid DNA minipreparation method suitable for AFLP and other PCR applications. Plant Mol. Biol. Rep. 1999, 17, 53–57. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1988, 8, 175–185. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The Sequence Alignment/Map (SAM) format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.L.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Δ (SNP Index) | Accession | Location (bp) | Reference Base (WT) | Altered Base in wb1 | Type of Mutation | Gene Annotation |
|---|---|---|---|---|---|---|
| 0.758 | ORF1 | 21550665 | T | G | Missense (T to P) | Helicase conserved C-terminal domain containing protein |
| 0.754 | 21550664 | G | T | Missense (T to K) | ||
| 0.663 | 21550888 | C | T | Intron mutation | ||
| 0.655 | 21550286 | T | A | Missense (T to S) | ||
| 0.649 | 21551279 | G | A | Intron mutation | ||
| 0.754 | ORF2 | 21539737 | A | G | 3′-UTR mutation | Protein of unknown function DUF668 family protein |
| 0.612 | 21539457 | G | T | Splice region mutation | ||
| 0.743 | ORF3 | 21331260 | G | A | Missense (D to N) | 40S ribosomal protein S10 |
| 0.734 | ORF4 | 21514382 | C | T | Intron mutation | Similar to H0315E07.10 protein |
| 0.708 | 21513793 | A | G | Intron mutation | ||
| 0.734 | ORF5 | 21612944 | C | A | Missense (K to N) | CENP-E-like kinetochore protein |
| 0.663 | 21610862 | C | A | Missense (S to I) | ||
| 0.733 | ORF6 | 20423829 | G | A | Missense (A to T) | Glycosyl hydrolases |
| 0.733 | ORF7 | 21795109 | G | A | Missense (L to F) | Expressed protein |
| 0.672 | ORF8 | 21493980 | G | A | Intron mutation | Similar to H0315E07.7 protein |
| 0.639 | ORF9 | 21897538 | C | T | 3′-UTR mutation | Nonsense-mediated decay UPF3 |
| 0.634 | ORF10 | 21970357 | C | T | 5′-UTR mutation | Peptide transporter PTR2 |
| 0.631 | ORF11 | 21710470 | C | G | Intron mutation | Conserved hypothetical protein |
| 0.61 | ORF12 | 21734385 | C | T | Nonsense (R to *) | No apical meristem protein |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhang, Y.; Sun, L.; Xu, P.; Tu, R.; Meng, S.; Wu, W.; Anis, G.B.; Hussain, K.; Riaz, A.; et al. WB1, a Regulator of Endosperm Development in Rice, Is Identified by a Modified MutMap Method. Int. J. Mol. Sci. 2018, 19, 2159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082159
Wang H, Zhang Y, Sun L, Xu P, Tu R, Meng S, Wu W, Anis GB, Hussain K, Riaz A, et al. WB1, a Regulator of Endosperm Development in Rice, Is Identified by a Modified MutMap Method. International Journal of Molecular Sciences. 2018; 19(8):2159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082159
Chicago/Turabian StyleWang, Hong, Yingxin Zhang, Lianping Sun, Peng Xu, Ranran Tu, Shuai Meng, Weixun Wu, Galal Bakr Anis, Kashif Hussain, Aamiar Riaz, and et al. 2018. "WB1, a Regulator of Endosperm Development in Rice, Is Identified by a Modified MutMap Method" International Journal of Molecular Sciences 19, no. 8: 2159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082159