The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development


Abstract
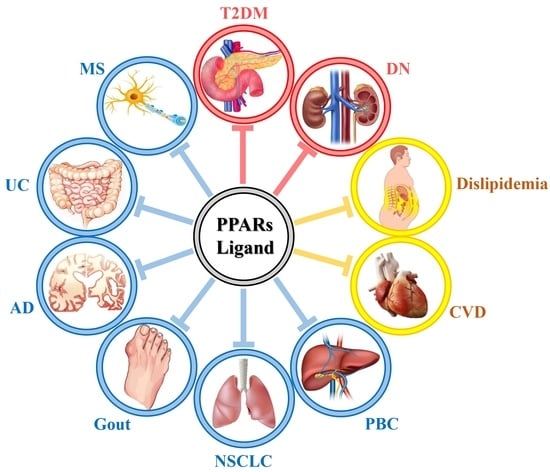
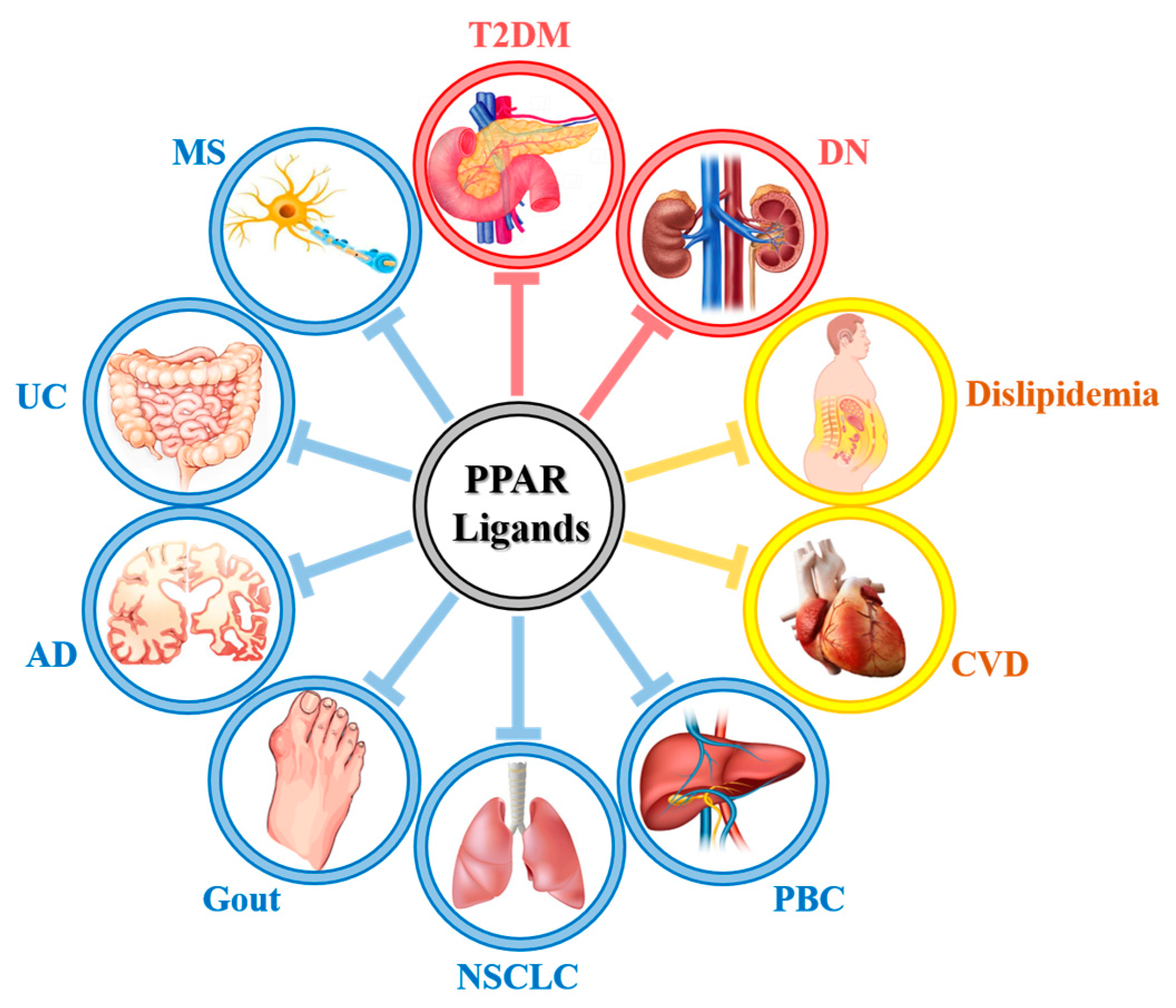
:
1. Introduction
2. PPAR Ligand Therapeutics in Diabetes Mellitus
2.1. Type 2 Diabetes
2.2. Diabetes-Associated Complications
3. PPAR Ligand Therapeutics in Lipid Metabolism Disorder
3.1. Dyslipidemia
3.2. Cardiovascular Diseases (CVDs)
4. PPAR Ligand Therapeutics in Other Diseases
5. Discussion
Acknowledgments
Conflicts of Interest
References
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.; La Vignera, S.; Condorelli, R.A.; Rizzo, G.; et al. Pleiotropic actions of peroxisome proliferator-activated receptors (PPARs) in dysregulated metabolic homeostasis, inflammation and cancer: Current evidence and future perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhai, Y.; Wang, J. The role of PPAR and its cross-talk with car and lxr in obesity and atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef] [PubMed]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The effect of regulating molecules on the structure of the PPAR-RXR complex. Biochim. Biophys. Acta 2016, 1861, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, F.; Ortiz, M.; Valenzuela, R.; Videla, L.A. Long-chain polyunsaturated fatty acids regulation of PPARs, signaling: Relationship to tissue development and aging. Prostaglandins Leukotrienes Essent. Fatty Acids 2016, 114, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. From fat to fat (cd36/sr-b2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Marcus, S.L.; Miyata, K.S.; Zhang, B.; Subramani, S.; Rachubinski, R.A.; Capone, J.P. Diverse peroxisome proliferator-activated receptors bind to the peroxisome proliferator-responsive elements of the rat hydratase/dehydrogenase and fatty acyl-coa oxidase genes but differentially induce expression. Proc. Natl. Acad. Sci. USA 1993, 90, 5723–5727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Marcus, S.L.; Miyata, K.S.; Subramani, S.; Capone, J.P.; Rachubinski, R.A. Characterization of protein-DNA interactions within the peroxisome proliferator-responsive element of the rat hydratase-dehydrogenase gene. J. Biol. Chem. 1993, 268, 12939–12945. [Google Scholar] [PubMed]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. MPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Devine, J.; Beale, E.G.; Spiegelman, B.M. PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 1995, 15, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yang, X.F.; Liu, H.L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-coa synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [PubMed]
- Cronet, P.; Petersen, J.F.; Folmer, R.; Blomberg, N.; Sjoblom, K.; Karlsson, U.; Lindstedt, E.L.; Bamberg, K. Structure of the PPARalpha and -gamma ligand binding domain in complex with az 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Gampe, R.T., Jr.; Montana, V.G.; Lambert, M.H.; Miller, A.B.; Bledsoe, R.K.; Milburn, M.V.; Kliewer, S.A.; Willson, T.M.; Xu, H.E. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell 2000, 5, 545–555. [Google Scholar] [CrossRef]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural peroxisome proliferator-activated receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced t2dm, dyslipidaemia and nafld. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR agonists and metabolic syndrome: An established role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [PubMed]
- DePaoli, A.M.; Higgins, L.S.; Henry, R.R.; Mantzoros, C.; Dunn, F.L.; Group, I.N.T.S. Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 2014, 37, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Koster, I.; Huppertz, E.; Hauner, H.; Schubert, I. Costs of diabetes mellitus (codim) in germany, direct per-capita costs of managing hyperglycaemia and diabetes complications in 2010 compared to 2001. Exp. Clin. Endocrinol. Diabetes 2014, 122, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of cardiovascular diseases: A cohort study in 1.9 million people. Lancet Diabetes Endocrinol. 2015, 3, 105–113. [Google Scholar] [CrossRef]
- Chung, J.W.; Hartzler, M.L.; Smith, A.; Hatton, J.; Kelley, K. Pharmacological agents utilized in patients with type-2 diabetes: Beyond lowering a1c. P & T 2018, 43, 214–227. [Google Scholar]
- Yasmin, S.; Jayaprakash, V. Thiazolidinediones and PPAR orchestra as antidiabetic agents: From past to present. Eur. J. Med. Chem. 2017, 126, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Investigators, D.T.; Gerstein, H.C.; Yusuf, S.; Bosch, J.; Pogue, J.; Sheridan, P.; Dinccag, N.; Hanefeld, M.; Hoogwerf, B.; Laakso, M.; et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: A randomised controlled trial. Lancet 2006, 368, 1096–1105. [Google Scholar]
- Li, Y.; Zhang, Y.; Li, X.; Shi, L.; Tao, W.; Shi, L.; Yang, M.; Wang, X.; Yang, Y.; Yao, Y. Association study of polymorphisms in mirnas with t2dm in chinese population. Int. J. Med. Sci. 2015, 12, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Muneera, M.S.; Thusleem, O.A.; Tahir, M.; Kondaguli, A.V.; Ruckmani, K. Development and validation of a selective online dissolution method for rosiglitazone maleate. J. Chromatogr. Sci. 2007, 45, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; McGraw, T.E. Rosiglitazone, PPARgamma, and type 2 diabetes. N. Engl. J. Med. 2010, 363, 2667–2669. [Google Scholar] [CrossRef] [PubMed]
- Mitka, M. Panel recommends easing restrictions on rosiglitazone despite concerns about cardiovascular safety. JAMA 2013, 310, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Tzanavaras, P.D.; Verdoukas, A.; Themelis, D.G. Development and validation of a flow-injection assay for dissolution studies of the anti-depressant drug venlafaxine. Anal. Sci. 2005, 21, 1515–1518. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, S.; Rosenblatt, S.; Braithwaite, S.; Egan, J.W.; Mathisen, A.L.; Schneider, R.L. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes: A 6-month randomized placebo-controlled dose-response study. The pioglitazone 001 study group. Diabetes Care 2000, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; Bell, S.; Sund, R.; Hartikainen, S.A.; Tuomilehto, J.; Pukkala, E.; Keskimaki, I.; Badrick, E.; Renehan, A.G.; Buchan, I.E.; et al. Pioglitazone and bladder cancer risk: A multipopulation pooled, cumulative exposure analysis. Diabetologia 2015, 58, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Habel, L.A.; Quesenberry, C.P.; Strom, B.L.; Peng, T.; Hedderson, M.M.; Ehrlich, S.F.; Mamtani, R.; Bilker, W.; Vaughn, D.J.; et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA 2015, 314, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. Dipeptidyl peptidase-4 inhibitors: 3 years of experience. Diabetes Technol. Ther. 2012, 14, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Andukuri, R.; Drincic, A.; Rendell, M. Alogliptin: A new addition to the class of dpp-4 inhibitors. Diabetes Metab. Syndr. Obes. 2009, 2, 117–126. [Google Scholar] [PubMed]
- Kaku, K.; Katou, M.; Igeta, M.; Ohira, T.; Sano, H. Efficacy and safety of pioglitazone added to alogliptin in japanese patients with type 2 diabetes mellitus: A multicentre, randomized, double-blind, parallel-group, comparative study. Diabetes Obes. Metab. 2015, 17, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Kim, D.M.; Woo, J.T.; Jang, H.C.; Chung, C.H.; Ko, K.S.; Park, J.H.; Park, Y.S.; Kim, S.J.; Choi, D.S. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: A multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE 2014, 9, e92843. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Bae, H.; Lee, Y.J.; Choi, Y.I.; Kim, H.J.; Park, S.B.; Suh, S.W.; Kim, S.W.; Han, B.W. Structural basis for the enhanced anti-diabetic efficacy of lobeglitazone on PPARgamma. Sci. Rep. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Mittermayer, F.; Caveney, E.; De Oliveira, C.; Gourgiotis, L.; Puri, M.; Tai, L.J.; Turner, J.R. Addressing unmet medical needs in type 2 diabetes: A narrative review of drugs under development. Curr. Diabetes Rev. 2015, 11, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Kim, B.Y.; Ahn, J.B.; Kang, S.K.; Lee, J.H.; Shin, J.S.; Ahn, S.K.; Lee, S.J.; Yoon, S.S. Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione. Eur. J. Med. Chem. 2005, 40, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.S.; Lee, J.E.; Lee, H.S.; Hwang, I.C.; Kim, D.H.; Park, H.K.; Choi, H.J.; Jo, W.; Son, W.C.; Yun, H.I. Ckd-501, a novel selective PPARgamma agonist, shows no carcinogenic potential in icr mice following oral administration for 104 weeks. J. Appl. Toxicol. 2014, 34, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Chang, M.; Lee, J.E.; Kim, W.; Hwang, I.C.; Kim, D.H.; Park, H.K.; Choi, H.J.; Jo, W.; Cha, S.W.; et al. Carcinogenicity study of ckd-501, a novel dual peroxisome proliferator-activated receptors alpha and gamma agonist, following oral administration to sprague dawley rats for 94-101 weeks. Regul. Toxicol. Pharmacol. 2014, 69, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, S.G.; Kim, D.M.; Woo, J.T.; Jang, H.C.; Chung, C.H.; Ko, K.S.; Park, J.H.; Park, Y.S.; Kim, S.J.; et al. Safety and efficacy of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 52 weeks: An open-label extension study. Diabetes Res. Clin. Pract. 2015, 110, e27–e30. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kim, T.E.; Yoon, S.H.; Cho, J.Y.; Shin, S.G.; Jang, I.J.; Yu, K.S. Assessment of the pharmacokinetics of co-administered metformin and lobeglitazone, a thiazolidinedione antihyperglycemic agent, in healthy subjects. Curr. Med. Res. Opin. 2012, 28, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- He, B.K.; Ning, Z.Q.; Li, Z.B.; Shan, S.; Pan, D.S.; Ko, B.C.; Li, P.P.; Shen, Z.F.; Dou, G.F.; Zhang, B.L.; et al. In vitro and in vivo characterizations of chiglitazar, a newly identified PPAR pan-agonist. PPAR Res. 2012, 2012, 546548. [Google Scholar] [CrossRef] [PubMed]
- Konda, V.R.; Desai, A.; Darland, G.; Grayson, N.; Bland, J.S. Kdt501, a derivative from hops, normalizes glucose metabolism and body weight in rodent models of diabetes. PLoS ONE 2014, 9, e87848. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Zhu, B.; Kok, B.P.; Godio, C.; Westgate, P.M.; Grayson, N.; Sims, R.; Bland, J.S.; Saez, E.; Kern, P.A. The influence of a kdt501, a novel isohumulone, on adipocyte function in humans. Front. Endocrinol. 2017, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Finlin, B.S.; Ross, D.; Boyechko, T.; Zhu, B.; Grayson, N.; Sims, R.; Bland, J.S. Effects of kdt501 on metabolic parameters in insulin-resistant prediabetic humans. J. Endocr. Soc. 2017, 1, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Raval, P.; Jain, M.; Goswami, A.; Basu, S.; Gite, A.; Godha, A.; Pingali, H.; Raval, S.; Giri, S.; Suthar, D.; et al. Revisiting glitazars: Thiophene substituted oxazole containing alpha-ethoxy phenylpropanoic acid derivatives as highly potent PPARalpha/gamma dual agonists devoid of adverse effects in rodents. Bioorganic Med. Chem. Lett. 2011, 21, 3103–3109. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.; Mohr, P.; Kuhn, B.; Maerki, H.P.; Hartman, P.; Ruf, A.; Benz, J.; Grether, U.; Wright, M.B. Comparative molecular profiling of the PPARalpha/gamma activator aleglitazar: PPAR selectivity, activity and interaction with cofactors. ChemMedChem 2012, 7, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.R.; Lincoff, A.M.; Mudaliar, S.; Rabbia, M.; Chognot, C.; Herz, M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (synchrony): A phase ii, randomised, dose-ranging study. Lancet 2009, 374, 126–135. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Tardif, J.C.; Schwartz, G.G.; Nicholls, S.J.; Ryden, L.; Neal, B.; Malmberg, K.; Wedel, H.; Buse, J.B.; Henry, R.R.; et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: The alecardio randomized clinical trial. JAMA 2014, 311, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Oleksiewicz, M.B.; Southgate, J.; Iversen, L.; Egerod, F.L. Rat urinary bladder carcinogenesis by dual-acting PPARalpha + gamma agonists. PPAR Res. 2008, 2008, 103167. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, A.; Letowski, J.; Czaika, G.; Ramirez, V.; Nead, M.A.; Jacobs, J.M.; Morais, R. Nucleotide sequence of the hemg gene involved in the protoporphyrinogen oxidase activity of escherichia coli k12. Can. J. Microbiol. 1993, 39, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Stringer, F.; Scott, G.; Valbuena, M.; Kinley, J.; Nishihara, M.; Urquhart, R. The effect of genetic polymorphisms in ugt2b15 on the pharmacokinetic profile of sipoglitazar, a novel anti-diabetic agent. Eur. J. Clin. Pharmacol. 2013, 69, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, L.; Sun, Y.; Muskhelishvili, L.; Blann, E.; Dial, S.; Shi, L.; Schroth, G.; Dragan, Y.P. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR gamma agonists on rat primary hepatocytes and human hepg2 cells. Mol. Divers. 2006, 10, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Byrjalsen, I.; Qvist, P.; Beck-Nielsen, H.; Hansen, G.; Riis, B.J.; Perrild, H.; Svendsen, O.L.; Gram, J.; Karsdal, M.A.; et al. Efficacy and safety of the PPARgamma partial agonist balaglitazone compared with pioglitazone and placebo: A phase iii, randomized, parallel-group study in patients with type 2 diabetes on stable insulin therapy. Diabetes Metab. Res. Rev. 2011, 27, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Minoura, H.; Takeshita, S.; Kimura, C.; Hirosumi, J.; Takakura, S.; Kawamura, I.; Seki, J.; Manda, T.; Mutoh, S. Mechanism by which a novel non-thiazolidinedione peroxisome proliferator-activated receptor gamma agonist, fk614, ameliorates insulin resistance in zucker fatty rats. Diabetes Obes. Metab. 2007, 9, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Minoura, H.; Takeshita, S.; Yamamoto, T.; Mabuchi, M.; Hirosumi, J.; Takakura, S.; Kawamura, I.; Seki, J.; Manda, T.; Ita, M.; et al. Ameliorating effect of fk614, a novel nonthiazolidinedione peroxisome proliferator-activated receptor gamma agonist, on insulin resistance in zucker fatty rat. Eur. J. Pharmacol. 2005, 519, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Minoura, H.; Takeshita, S.; Ita, M.; Hirosumi, J.; Mabuchi, M.; Kawamura, I.; Nakajima, S.; Nakayama, O.; Kayakiri, H.; Oku, T.; et al. Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer, fk614. Eur. J. Pharmacol. 2004, 494, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R. Discontinued drugs in 2005: Endocrine and metabolic. Expert Opin. Investig. Drugs 2007, 16, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R.; Tanis, S.P.; McDonald, W.G.; Kletzien, R.F. Insulin sensitizers in 2013: New insights for the development of novel therapeutic agents to treat metabolic diseases. Expert Opin. Investig. Drugs 2014, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Diani, A.R.; Peterson, T.; Sawada, G.; Jodelis, K.; Wyse, B.M.; Gilchrist, B.J.; Hearron, A.E.; Chang, A.Y. Ciglitazone, a new hypoglycemic agent. 5. Effect on renal lesions in c57bl/ksj-db/db mice. Nephron 1986, 42, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the safety of PPAR agonists—current trends and future prospects. Expert Opin. Drug Saf. 2013, 12, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.S.; de Lusignan, S.; Hinton, W.; Munro, N.; Taylor, S.; McGovern, A. Glycemic control is an important modifiable risk factor for uveitis in patients with diabetes: A retrospective cohort study establishing clinical risk and ophthalmic disease burden. J. Diabetes Its Complicat. 2018, 32, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Prevention Program Research, G. Long-term effects of lifestyle intervention or metformin on diabetes development and microvascular complications over 15-year follow-up: The diabetes prevention program outcomes study. Lancet Diabetes Endocrinol. 2015, 3, 866–875. [Google Scholar]
- Leiter, L.A.; Lundman, P.; da Silva, P.M.; Drexel, H.; Junger, C.; Gitt, A.K.; DYSIS investigators. Persistent lipid abnormalities in statin-treated patients with diabetes mellitus in europe and canada: Results of the dyslipidaemia international study. Diabet. Med. 2011, 28, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Feher, M.; Greener, M.; Munro, N. Persistent hypertriglyceridemia in statin-treated patients with type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2013, 6, 11–15. [Google Scholar] [PubMed]
- Joshi, S.R. Saroglitazar for the treatment of dyslipidemia in diabetic patients. Expert Opin. Pharmacother. 2015, 16, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Bodkin, N.L.; Pill, J.; Meyer, K.; Hansen, B.C. The effects of k-111, a new insulin-sensitizer, on metabolic syndrome in obese prediabetic rhesus monkeys. Horm. Metab. Res. 2003, 35, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Ortmeyer, H.K.; Bodkin, N.L.; Haney, J.; Yoshioka, S.; Horikoshi, H.; Hansen, B.C. A thiazolidinedione improves in vivo insulin action on skeletal muscle glycogen synthase in insulin-resistant monkeys. Int. J. Exp. Diabetes Res. 2000, 1, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Hannah, J.S.; Bodkin, N.L.; Paidi, M.S.; Anh-Le, N.; Howard, B.V.; Hansen, B.C. Effects of acipimox on the metabolism of free fatty acids and very low lipoprotein triglyceride. Acta Diabetol. 1995, 32, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Bodkin, N.L.; Hansen, B.C. Antihypertensive effects of captopril without adverse effects on glucose tolerance in hyperinsulinemic rhesus monkeys. J. Med. Primatol. 1995, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Medicherla, S.; Neogi, P.; Gowri, M.; Cheng, J.; Gross, C.; Sharma, S.D.; Reaven, G.M.; Nag, B. A novel peroxisome proliferator-activated gamma (PPAR gamma) agonist, clx-0921, has potent antihyperglycemic activity with low adipogenic potential. Metabolism 2003, 52, 1012–1018. [Google Scholar] [CrossRef]
- Medicherla, S.; Dey, D.; Neogi, P.; Lakner, F.J.; Nag, B. Clx-0921: A new PPAR-gamma agonist anti-diabetic thiazolidinedione compound. Diabetes 2000, 49, A117. [Google Scholar]
- Soleymanian, T.; Hamid, G.; Arefi, M.; Najafi, I.; Ganji, M.R.; Amini, M.; Hakemi, M.; Tehrani, M.R.; Larijani, B. Non-diabetic renal disease with or without diabetic nephropathy in type 2 diabetes: Clinical predictors and outcome. Ren. Fail. 2015, 37, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Incidence of end-stage renal disease attributed to diabetes among persons with diagnosed diabetes—united states and puerto rico, 1996–2007. MMWR 2010, 59, 1361–1366. [Google Scholar]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henique, C.; Bollee, G.; Lenoir, O.; Dhaun, N.; Camus, M.; Chipont, A.; Flosseau, K.; Mandet, C.; Yamamoto, M.; Karras, A.; et al. Nuclear factor erythroid 2-related factor 2 drives podocyte-specific expression of peroxisome proliferator-activated receptor gamma essential for resistance to crescentic gn. JASN 2016, 27, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Villacorta, L.; Chang, L.; Fan, Z.; Hamblin, M.; Zhu, T.; Chen, C.S.; Cole, M.P.; Schopfer, F.J.; Deng, C.X.; et al. Nitro-oleic acid inhibits angiotensin ii-induced hypertension. Circ. Res. 2010, 107, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.P.; Rudolph, T.K.; Khoo, N.K.; Motanya, U.N.; Golin-Bisello, F.; Wertz, J.W.; Schopfer, F.J.; Rudolph, V.; Woodcock, S.R.; Bolisetty, S.; et al. Nitro-fatty acid inhibition of neointima formation after endoluminal vessel injury. Circ. Res. 2009, 105, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, H.; Jia, Z.; Guan, G.; Yang, T. Effects of endogenous PPAR agonist nitro-oleic acid on metabolic syndrome in obese zucker rats. PPAR Res. 2010, 2010, 601562. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Cole, M.P.; Groeger, A.L.; Chen, C.S.; Khoo, N.K.; Woodcock, S.R.; Golin-Bisello, F.; Motanya, U.N.; Li, Y.; Zhang, J.; et al. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: Selective ligand activity and anti-diabetic signaling actions. J. Biol. Chem. 2010, 285, 12321–12333. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Xue, X.; Li, J.; Liu, X.; Lv, S.; Guan, G.; Liu, H.; Liu, G.; Liu, S.; Chen, Z. Nitro-oleic acid attenuates ogd/r-triggered apoptosis in renal tubular cells via inhibition of bax mitochondrial translocation in a PPAR-gamma-dependent manner. Cell. Physiol. Biochem. 2015, 35, 1201–1218. [Google Scholar] [CrossRef] [PubMed]
- Taygerly, J.P.; McGee, L.R.; Rubenstein, S.M.; Houze, J.B.; Cushing, T.D.; Li, Y.; Motani, A.; Chen, J.L.; Frankmoelle, W.; Ye, G.; et al. Discovery of int131: A selective PPARgamma modulator that enhances insulin sensitivity. Bioorganic Med. Chem. 2013, 21, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Goebel, M. Int-131, a PPARgamma agonist for the treatment of type 2 diabetes. Curr. Opin. Investig. Drugs 2009, 10, 381–387. [Google Scholar] [PubMed]
- Berge, R.K.; Tronstad, K.J.; Berge, K.; Rost, T.H.; Wergedahl, H.; Gudbrandsen, O.A.; Skorve, J. The metabolic syndrome and the hepatic fatty acid drainage hypothesis. Biochimie 2005, 87, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Berge, R.K.; Hvattum, E. Impact of cytochrome p450 system on lipoprotein metabolism. Effect of abnormal fatty acids (3-thia fatty acids). Pharmacol. Ther. 1994, 61, 345–383. [Google Scholar] [CrossRef]
- Vaagenes, H.; Madsen, L.; Asiedu, D.K.; Lillehaug, J.R.; Berge, R.K. Early modulation of genes encoding peroxisomal and mitochondrial beta-oxidation enzymes by 3-thia fatty acids. Biochem. Pharmacol. 1998, 56, 1571–1582. [Google Scholar] [CrossRef]
- Wensaas, A.J.; Rustan, A.C.; Just, M.; Berge, R.K.; Drevon, C.A.; Gaster, M. Fatty acid incubation of myotubes from humans with type 2 diabetes leads to enhanced release of beta-oxidation products because of impaired fatty acid oxidation: Effects of tetradecylthioacetic acid and eicosapentaenoic acid. Diabetes 2009, 58, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafstad, A.D.; Khalid, A.M.; Hagve, M.; Lund, T.; Larsen, T.S.; Severson, D.L.; Clarke, K.; Berge, R.K.; Aasum, E. Cardiac peroxisome proliferator-activated receptor-alpha activation causes increased fatty acid oxidation, reducing efficiency and post-ischaemic functional loss. Cardiovasc. Res. 2009, 83, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Laurent, D.; Gounarides, J.S.; Gao, J.; Boettcher, B.R. Effects of cevoglitazar, a dual PPARalpha/gamma agonist, on ectopic fat deposition in fatty zucker rats. Diabetes Obes. Metabol. 2009, 11, 632–636. [Google Scholar]
- Chen, H.; Dardik, B.; Qiu, L.; Ren, X.; Caplan, S.L.; Burkey, B.; Boettcher, B.R.; Gromada, J. Cevoglitazar, a novel peroxisome proliferator-activated receptor-alpha/gamma dual agonist, potently reduces food intake and body weight in obese mice and cynomolgus monkeys. Endocrinology 2010, 151, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R. Discontinued drugs in 2008: Endocrine and metabolic. Expert Opin. Investig. Drugs 2009, 18, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Tobe, K.; Ide, T.; Mochizuki, T.; Ohashi, M.; Akanuma, Y.; Yazaki, Y.; Kadowaki, T. A novel insulin sensitizer acts as a coligand for peroxisome proliferator-activated receptor-alpha (PPAR-alpha) and PPAR-gamma: Effect of ppar-alpha activation on abnormal lipid metabolism in liver of zucker fatty rats. Diabetes 1998, 47, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Kinoshita, S.; Satoh, H.; Maeda, T.; Murakami, K.; Tsunoda, M.; Miyachi, H.; Awano, K. (3-substituted benzyl)thiazolidine-2,4-diones as structurally new antihyperglycemic agents. Bioorganic Med. Chem. Lett. 1999, 9, 533–538. [Google Scholar] [CrossRef]
- Doebber, T.W.; Kelly, L.J.; Zhou, G.; Meurer, R.; Biswas, C.; Li, Y.; Wu, M.S.; Ippolito, M.C.; Chao, Y.S.; Wang, P.R.; et al. Mk-0767, a novel dual PPARalpha/gamma agonist, displays robust antihyperglycemic and hypolipidemic activities. Biochem. Biophys. Res. Commun. 2004, 318, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Oleksiewicz, M.B.; Thorup, I.; Nielsen, H.S.; Andersen, H.V.; Hegelund, A.C.; Iversen, L.; Guldberg, T.S.; Brinck, P.R.; Sjogren, I.; Thinggaard, U.K.; et al. Generalized cellular hypertrophy is induced by a dual-acting PPAR agonist in rat urinary bladder urothelium in vivo. Toxicol. Pathol. 2005, 33, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Lin, J.J.; Goldfine, I.D. Novel approach to treat insulin resistance, type 2 diabetes, and the metabolic syndrome: Simultaneous activation of PPARalpha, PPARgamma, and PPARdelta. Curr. Diabetes Rev. 2005, 1, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Huang, Y. The peroxisome proliferator-activated receptors in cardiovascular diseases: Experimental benefits and clinical challenges. Br. J. Pharmacol. 2015, 172, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Nakayama, R.; Yamanaka, M.; Takata, M.; Takazawa, T.; Watanabe, K.; Maruta, K.; Nagata, R.; Nagamine, J.; Tsuchida, A.; et al. Effects of dsp-8658, a novel selective peroxisome proliferator-activated receptors a/gamma modulator, on adipogenesis and glucose metabolism in diabetic obese mice. Exp. Clin. Endocrinol. Diabetes 2015, 123, 492–499. [Google Scholar] [PubMed]
- Bray, G.A.; Fruhbeck, G.; Ryan, D.H.; Wilding, J.P. Management of obesity. Lancet 2016, 387, 1947–1956. [Google Scholar] [CrossRef]
- Xu, P.; Dai, S.; Wang, J.; Zhang, J.; Liu, J.; Wang, F.; Zhai, Y. Preventive obesity agent montmorillonite adsorbs dietary lipids and enhances lipid excretion from the digestive tract. Sci. Rep. 2016, 6, 19659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Wang, J.; Hong, F.; Wang, S.; Jin, X.; Xue, T.; Jia, L.; Zhai, Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Carrera, M. Unraveling the effects of PPARbeta/delta on insulin resistance and cardiovascular disease. Trends Endocrinol. Metab. 2016, 27, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Vassilatou, E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J. Gastroenterol. 2014, 20, 8351–8363. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hong, F.; Wang, J.; Cong, Y.; Dai, S.; Wang, S.; Wang, J.; Jin, X.; Wang, F.; Liu, J.; et al. Microbiome remodeling via the montmorillonite adsorption-excretion axis prevents obesity-related metabolic disorders. EBioMedicine 2017, 16, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Teoh, H.; Verma, S. Obesity and atherosclerosis: Mechanistic insights. Can. J. Cardiol. 2015, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Oda, N.; Imamura, S.; Fujita, T.; Uchida, Y.; Inagaki, K.; Kakizawa, H.; Hayakawa, N.; Suzuki, A.; Takeda, J.; Horikawa, Y.; et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 2008, 57, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Lalloyer, F.; Staels, B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Robins, S.J.; Collins, D.; Wittes, J.T.; Papademetriou, V.; Deedwania, P.C.; Schaefer, E.J.; McNamara, J.R.; Kashyap, M.L.; Hershman, J.M.; Wexler, L.F.; et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: Va-hit: A randomized controlled trial. JAMA 2001, 285, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cuenca, S.; Carobbio, S.; Barcelo-Coblijn, G.; Prieur, X.; Relat, J.; Amat, R.; Campbell, M.; Dias, A.R.; Bahri, M.; Gray, S.L.; et al. P465l PPARgamma mutation confers partial resistance to the hypolipidemic action of fibrates. Diabetes Obes. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Suter, T.M.; Hess, O.M. Exercise-induced vasomotion of angiographically normal and stenotic coronary arteries improves after cholesterol-lowering drug therapy with bezafibrate. J. Am. Coll. Cardiol. 1995, 26, 1615–1622. [Google Scholar] [CrossRef]
- Khera, A.V.; Qamar, A.; Reilly, M.P.; Dunbar, R.L.; Rader, D.J. Effects of niacin, statin, and fenofibrate on circulating proprotein convertase subtilisin/kexin type 9 levels in patients with dyslipidemia. Am. J. Cardiol. 2015, 115, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Anderson, R.A.; Graham, J.; Ellis, G.R.; Morris, K.; Davies, S.; Jackson, S.K.; Lewis, M.J.; Frenneaux, M.P.; Rees, A. Ciprofibrate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus. Circulation 2000, 101, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Chu, Z.; Min, L.; Zhen, T.; Li, P.; Han, L.; Bu, S.; Yang, J.; Gonzale, F.J.; Liu, A. Gemfibrozil not fenofibrate decreases systemic glucose level via PPARalpha. Die Pharm. 2016, 71, 205–212. [Google Scholar]
- Parhofer, K.G. The treatment of disorders of lipid metabolism. Deutsch. Arzteblatt Int. 2016, 113, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Committee of Principal Investigators. WHO Cooperative trial on primary prevention of ischaemic heart disease using clofibrate to lower serum cholesterol: Mortality follow-up. Report of the committee of principal investigators. Lancet 1980, 2, 379–385. [Google Scholar]
- Fazio, S.; Linton, M.F. The role of fibrates in managing hyperlipidemia: Mechanisms of action and clinical efficacy. Curr. Atheroscler. Rep. 2004, 6, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Croom, K.F. Fenofibrate: A review of its use in primary dyslipidaemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs 2007, 67, 121–153. [Google Scholar] [CrossRef] [PubMed]
- Moutzouri, E.; Kei, A.; Elisaf, M.S.; Milionis, H.J. Management of dyslipidemias with fibrates, alone and in combination with statins: Role of delayed-release fenofibric acid. Vasc. Health Risk Manag. 2010, 6, 525–539. [Google Scholar] [PubMed]
- Chachad, S.S.; Gole, M.; Malhotra, G.; Naidu, R. Comparison of pharmacokinetics of two fenofibrate tablet formulations in healthy human subjects. Clin. Ther. 2014, 36, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, G.; Zhang, T.; Ma, Z.; Wu, B. Effects of pegylated lipid nanoparticles on the oral absorption of one bcs ii drug: A mechanistic investigation. Int. J. Nanomed. 2014, 9, 5503–5514. [Google Scholar]
- Brown, W.V. Treatment of hypercholesterolaemia with fenofibrate: A review. Curr. Med. Res. Opin. 1989, 11, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Pallottini, V.; Marin, R.; Marino, M. Role of the sex hormone estrogen in the prevention of lipid disorder. Curr. Med. Chem. 2014, 21, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, C.G.; Hamsten, A.; Nilsson, J.; Grip, L.; Svane, B.; de Faire, U. Angiographic assessment of effects of bezafibrate on progression of coronary artery disease in young male postinfarction patients. Lancet 1996, 347, 849–853. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Saku, K.; Gartside, P.S.; Hynd, B.A.; Kashyap, M.L. Mechanism of action of gemfibrozil on lipoprotein metabolism. J. Clin. Investig. 1985, 75, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Mikhailidis, D.P.; Jagroon, I.A. Ciprofibrate versus gemfibrozil in the treatment of mixed hyperlipidemias: An open-label, multicenter study. Metabolism 2001, 50, 1385–1386. [Google Scholar] [CrossRef]
- Rizos, E.; Bairaktari, E.; Ganotakis, E.; Tsimihodimos, V.; Mikhailidis, D.P.; Elisaf, M. Effect of ciprofibrate on lipoproteins, fibrinogen, renal function, and hepatic enzymes. J. Cardiovasc. Pharmacol. Ther. 2002, 7, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C. Selective peroxisome proliferator-activated receptor alpha modulators (sPPARmalpha): The next generation of peroxisome proliferator-activated receptor alpha-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorganic Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARalpha modulator (sPPARmalpha) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Schima, S.M.; Maciejewski, S.R.; Hilleman, D.E.; Williams, M.A.; Mohiuddin, S.M. Fibrate therapy in the management of dyslipidemias, alone and in combination with statins: Role of delayed-release fenofibric acid. Expert Opin. Pharmacother. 2010, 11, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Mikhailidis, D.P.; Papageorgiou, A.A.; Didangelos, T.P.; Peletidou, A.; Kleta, D.; Karagiannis, A.; Kakafika, A.I.; Tziomalos, K.; Elisaf, M. Targeting vascular risk in patients with metabolic syndrome but without diabetes. Metabolism 2005, 54, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, S.M.; Pepine, C.J.; Kelly, M.T.; Buttler, S.M.; Setze, C.M.; Sleep, D.J.; Stolzenbach, J.C. Efficacy and safety of abt-335 (fenofibric acid) in combination with simvastatin in patients with mixed dyslipidemia: A phase 3, randomized, controlled study. Am. Heart J. 2009, 157, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Shek, A.; Ferrill, M.J. Statin-fibrate combination therapy. Ann. Pharmacother. 2001, 35, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Backes, J.M.; Gibson, C.A.; Ruisinger, J.F.; Moriarty, P.M. Fibrates: What have we learned in the past 40 years? Pharmacotherapy 2007, 27, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the american association for the study of liver diseases, american college of gastroenterology, and the american gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on nafld/nash based on the easl 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, gft505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist gft505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist gft505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Hallen, J.; Vige, R.; Fraser, D.; Zhou, R.; Hustvedt, S.O.; Orloff, D.G.; Kastelein, J.J. Icosabutate for the treatment of very high triglycerides: A placebo-controlled, randomized, double-blind, 12-week clinical trial. J. Clin. Lipidol. 2016, 10, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.A.; Skjaeret, T.; Qin, Y.; Larsen, L.N.; Husberg, C.; Hovland, R.; Pieterman, E.J.; van den Hoek, A.M.; Princen, H.M.; Hustvedt, S.O. Icosabutate, a novel structurally enhanced fatty-acid increases hepatic uptake of cholesterol and triglycerides in conjunction with increased hepatic LDL receptor expression. Circulation 2014, 130, A11889. [Google Scholar]
- Billin, A.N. PPAR-beta/delta agonists for type 2 diabetes and dyslipidemia: An adopted orphan still looking for a home. Expert Opin. Investig. Drugs 2008, 17, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Strain, J.D.; Farver, D.K.; Clem, J.R. A review on the rationale and clinical use of concomitant rosuvastatin and fenofibrate/fenofibric acid therapy. Clin. Pharmacol. 2010, 2, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Harchaoui, K.E.; Visser, M.E.; Kastelein, J.J.; Stroes, E.S.; Dallinga-Thie, G.M. Triglycerides and cardiovascular risk. Curr. Cardiol. Rev. 2009, 5, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 acc/aha guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the american college of cardiology/american heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [PubMed]
- Chapman, M.J.; Redfern, J.S.; McGovern, M.E.; Giral, P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol. Ther. 2010, 126, 314–345. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme a (hmg-coa) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Nishimura, H.; Nakagawa, S.; Kojima, J.; Suzuki, H.; Tamaki, T.; Wada, Y.; Yokoo, N.; Sato, F.; Kimata, H.; et al. Pharmacological profile of a novel synthetic inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme a reductase. Arzneimittel-Forschung 1997, 47, 904–909. [Google Scholar] [PubMed]
- Wakida, Y.; Suzuki, S.; Nomura, H.; Isomura, T. Additional treatment with fenofibrate for patients treated with pitavastatin under ordinary medical practice for hypertriglyceridemia in japan (approach-j study). Jpn. Clin. Med. 2011, 2, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Tokuno, A.; Hirano, T.; Hayashi, T.; Mori, Y.; Yamamoto, T.; Nagashima, M.; Shiraishi, Y.; Ito, Y.; Adachi, M. The effects of statin and fibrate on lowering small dense LDL- cholesterol in hyperlipidemic patients with type 2 diabetes. J. Atheroscler. Thromb. 2007, 14, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Bisgaier, C.L.; Oniciu, D.C.; Srivastava, R.A.K. Comparative evaluation of gemcabene and PPAR ligands in transcriptional assays of peroxisome proliferator-activated receptors: Implication for the treatment of hyperlipidemia and cardiovascular disease. J. Cardiovasc. Pharmacol. 2018, 72, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Srivastava, R.A.K.; Cornicelli, J.A.; Markham, B.; Bisgaier, C.L. Gemcabene, a first-in-class lipid-lowering agent in late-stage development, down-regulates acute-phase c-reactive protein via c/ebp-delta-mediated transcriptional mechanism. Mol. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.P.; Zhang, X.S.; Xiang, B.R. Discontinued drugs in 2010: Cardiovascular drugs. Expert Opin. Investig. Drugs 2011, 20, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.L. Lipids, lipoproteins, and peroxisome proliferator activated receptor-delta. Am. J. Cardiol. 2007, 100, S20–S24. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.L.; Massien, C.; Pearce, G.; Billin, A.N.; Perlstein, I.; Willson, T.M.; Hassall, D.G.; Ancellin, N.; Patterson, S.D.; Lobe, D.C.; et al. Triglyceride:High-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Lee, C.H. PPAR delta as a therapeutic target in metabolic disease. FEBS Lett. 2008, 582, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Madrazo, J.A.; Kelly, D.P. The PPAR trio: Regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 2008, 44, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Schmuth, M.; Jiang, Y.J.; Dubrac, S.; Elias, P.M.; Feingold, K.R. Thematic review series: Skin lipids. Peroxisome proliferator-activated receptors and liver x receptors in epidermal biology. J. Lipid Res. 2008, 49, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, A.; Muramatsu, Y.; Yamazaki, T.; Muramoto, M.; Kita, Y.; Yamazaki, S.; Mihara, K.; Moriguchi, A.; Matsuoka, N. Neuroprotective efficacy of the peroxisome proliferator-activated receptor delta-selective agonists in vitro and in vivo. J. Pharmacol. Exp. Ther. 2007, 320, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin. Sci. 2008, 115, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E. The immunobiology and pathophysiology of primary biliary cirrhosis. Ann. Rev. Pathol. 2013, 8, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Abenavoli, L.; Rabahi, N.; Chretien, Y.; Andreani, T.; Johanet, C.; Chazouilleres, O.; Poupon, R. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008, 48, 871–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. Mbx-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (mbx-8025), a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet. Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef]
- Wertheimer, A.; Morlock, R.; Becker, M.A. A revised estimate of the burden of illness of gout. Curr. Ther. Res. Clin. Exp. 2013, 75, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.L.; Sundy, J.S.; Forsythe, A.; Blume, S.; Pan, F.; Becker, M.A. Work productivity loss due to flares in patients with chronic gout refractory to conventional therapy. J. Med. Econ. 2011, 14, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.A.; Forsythe, A.; Smeeding, J.E.; Lawrence Edwards, N. Chronic gout: Epidemiology, disease progression, treatment and disease burden. Curr. Med. Res. Opin. 2010, 26, 2813–2821. [Google Scholar] [CrossRef] [PubMed]
- Aronow, W.S.; Harding, P.R.; Khursheed, M.; Vangrow, J.S.; Papageorge’s, N.P. Effect of halofenate on serum uric acid. Clin. Pharmacol. Ther. 1973, 14, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Aronow, W.S.; Harding, P.R.; Khursheed, M.; Vangrow, J.S.; Papageorge’s, N.P.; Mays, J. Effect of halofenate on serum lipids. Clin. Pharmacol. Ther. 1973, 14, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Poiley, J.; Steinberg, A.S.; Choi, Y.J.; Davis, C.S.; Martin, R.L.; McWherter, C.A.; Boudes, P.F.; Arhalofenate Flare Study, I. A randomized, double-blind, active- and placebo-controlled efficacy and safety study of arhalofenate for reducing flare in patients with gout. Arthritis Rheumatol. 2016, 68, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Deochand, C.; Didsbury, J.; de la Monte, S.M. T3d-959: A multi-faceted disease remedial drug candidate for the treatment of alzheimer’s disease. J. Alzheimer’s Dis. 2016, 51, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Dominguez, C.; Didsbury, J.; de la Monte, S.M. Targeting alzheimer’s disease neuro-metabolic dysfunction with a small molecule nuclear receptor agonist (t3d-959) reverses disease pathologies. J. Alzheimer’s Dis. Parkinsonism 2016, 6, pii:238. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Koyi, H.; Hillerdal, G.; Branden, E. A prospective study of a total material of lung cancer from a county in sweden 1997–1999: Gender, symptoms, type, stage, and smoking habits. Lung Cancer 2002, 36, 9–14. [Google Scholar] [CrossRef]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H.; Eastern Cooperative Oncology, G. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Blanquicett, C.; Roman, J.; Hart, C.M. Thiazolidinediones as anti-cancer agents. Cancer Ther. 2008, 6, 25–34. [Google Scholar] [PubMed]
- Nemenoff, R.A.; Weiser-Evans, M.; Winn, R.A. Activation and molecular targets of peroxisome proliferator-activated receptor-gamma ligands in lung cancer. PPAR Res. 2008, 2008, 156875. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copland, J.A.; Marlow, L.A.; Kurakata, S.; Fujiwara, K.; Wong, A.K.; Kreinest, P.A.; Williams, S.F.; Haugen, B.R.; Klopper, J.P.; Smallridge, R.C. Novel high-affinity PPARgamma agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21waf1/cip1. Oncogene 2006, 25, 2304–2317. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, N.; Togashi, N.; Hanai, M.; Isoyama, T.; Wada, K.; Fujita, T.; Fujiwara, K.; Kurakata, S. Anti-tumour activity of cs-7017, a selective peroxisome proliferator-activated receptor gamma agonist of thiazolidinedione class, in human tumour xenografts and a syngeneic tumour implant model. Eur. J. Cancer 2008, 44, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Budman, D.R.; Calabro, A. Studies of synergistic and antagonistic combinations of conventional cytotoxic agents with the multiple eicosanoid pathway modulator ly 293111. Anti-Cancer Drugs 2004, 15, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Weitzman, A.; O’Reilly, E.; Brail, L.; de Alwis, D.P.; Cleverly, A.; Barile-Thiem, B.; Vinciguerra, V.; Budman, D.R. Phase i and pharmacokinetic study of ly293111, an orally bioavailable ltb4 receptor antagonist, in patients with advanced solid tumors. J. Clin. Oncol. 2005, 23, 5365–5373. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.G.; Ding, X.Z.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. Leukotriene b4 receptor antagonist ly293111 induces s-phase cell cycle arrest and apoptosis in human pancreatic cancer cells. Anti-Cancer Drugs 2007, 18, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar] [CrossRef] [PubMed]
- Gosset, P.; Charbonnier, A.S.; Delerive, P.; Fontaine, J.; Staels, B.; Pestel, J.; Tonnel, A.B.; Trottein, F. Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur. J. Immunol. 2001, 31, 2857–2865. [Google Scholar] [CrossRef]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Bertin, B.; Dubuquoy, L.; Colombel, J.F.; Desreumaux, P. PPAR-gamma in ulcerative colitis: A novel target for intervention. Cur. Drug Targets 2013, 14, 1501–1507. [Google Scholar] [CrossRef]
- Pirat, C.; Farce, A.; Lebegue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.; Speca, S.; Berthelot, P.; Desreumaux, P.; et al. Targeting peroxisome proliferator-activated receptors (PPARs): Development of modulators. J. Med. Chem. 2012, 55, 4027–4061. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Konstantinova, I.; Guignabert, C.; Pezet, S.; Sadoine, J.; Guilbert, T.; Cauvet, A.; Tu, L.; Luccarini, J.M.; Junien, J.L.; et al. Pan-PPAR agonist iva337 is effective in experimental lung fibrosis and pulmonary hypertension. Ann. Rheum. Dis. 2017, 76, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hong, F.; Wang, J.; Wang, J.; Zhao, X.; Wang, S.; Xue, T.; Xu, J.; Zheng, X.; Zhai, Y. DBZ is a putative PPARgamma agonist that prevents high fat diet-induced obesity, insulin resistance and gut dysbiosis. Biochim. Biophys. Acta 2017, 1861, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, P.; Xie, X.; Li, J.; Zhang, J.; Wang, J.; Hong, F.; Li, J.; Zhang, Y.; Song, Y.; et al. DBZ (danshensu bingpian zhi), a novel natural compound derivative, attenuates atherosclerosis in apolipoprotein e-deficient mice. J. Am. Heart Assoc. 2017, 6, pii:e006297. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, A.; Fukushima, M.; Sakai, M.; Tokuyama, K.; Nagata, I.; Fukunaga, A.; Kishimoto, H.; Doi, K.; Yamashita, Y.; Matsuura, T.; et al. Effects of bezafibrate on insulin sensitivity and insulin secretion in non-obese japanese type 2 diabetic patients. Metabolism 2001, 50, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Rose, M.; Ganti, S.S.; Krishan, P.; Singh, M. PPAR dual agonists: Are they opening pandora’s box? Pharmacol. Res. 2007, 56, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.B.; Rodriguez, O.; Martin, P.L.; Albanese, C.; Li, X.; Kopelovich, L.; Glazer, R.I. Induction of metastatic gastric cancer by peroxisome proliferator-activated receptordelta activation. PPAR Res. 2010, 2010, 571783. [Google Scholar] [CrossRef] [PubMed]
- Fievet, C.; Fruchart, J.C.; Staels, B. PPARalpha and PPARgamma dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr. Opin. Pharmacol. 2006, 6, 606–614. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
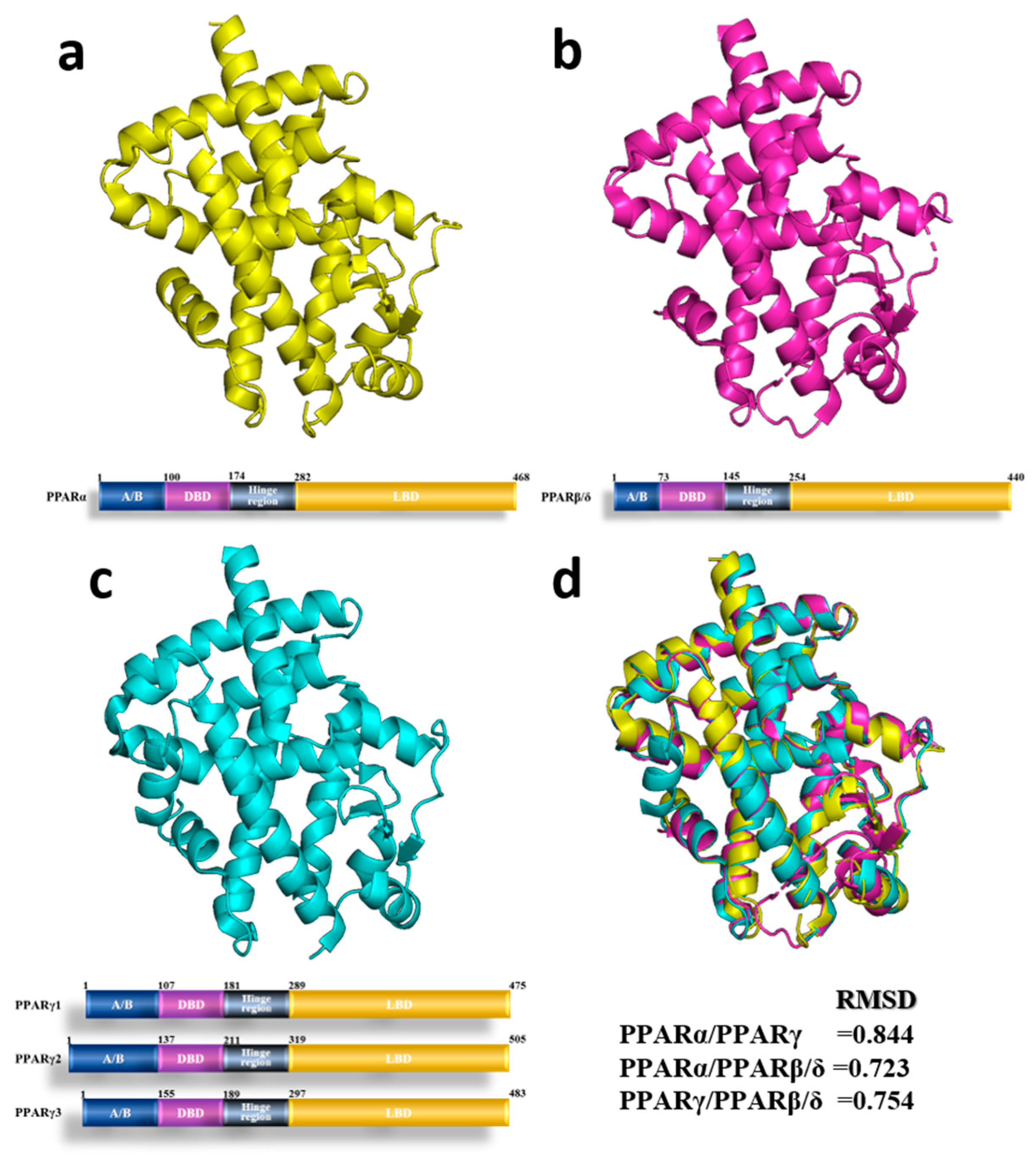{kind=link}
{kind=link}
| Indication | Development Status | Total | |||
|---|---|---|---|---|---|
| In Market | Withdrawn | Clinical Research | Discontinued in Clinical Research | ||
| Type 2 diabetes | 3 | 1 | 5 | 23 | 32 |
| Diabetic diseases | 1 | 0 | 5 | 10 | 16 |
| Dyslipidemia | 7 | 0 | 6 | 8 | 21 |
| CVDs | 0 | 0 | 1 | 1 | 2 |
| Other diseases | 0 | 0 | 12 | 1 | 13 |
| Generic Name | Type of PPAR Agonist | Molecular Weight | Company |
|---|---|---|---|
| Rosiglitazone Maleate | PPARγ agonist | 473.5 | GlaxoSmithKline |
| Pioglitazone Hydrochloride | PPARγ agonist | 392.898 | Takeda(Originator) Lilly |
| Lobeglitazone Sulfate | Dual PPARα/γ agonist | 578.61 | Chong Kun Dang |
| Generic Name | Type of PPAR Agonist | Molecular Weight | Company | Development Status |
|---|---|---|---|---|
| Chiglitazar | PPARs agonist | 594.61 | ChipScreen | Phase Ш active |
| KDT-501 | PPARα agonists | 404.588 | KinDex Pharmaceuticals | Phase II active |
| Naveglitazar | PPAR modulator | 422.477 | Lilly(Originator)Ligand (Originator) | Phase II Pending |
| AVE-0897 | Dual PPARα/γ agonist | 469 | Genfit(Originator)Sanofi | Phase І active |
| ZY-H2 | Dual PPARα/γ agonist | unknown | Zydus cadila | Phase І Pending |
| Generic Name | Type of PPAR Agonist | Molecular Weight | Company | Development Status |
|---|---|---|---|---|
| Aleglitazar | Dual PPARα/γ agonist | 437.51 | Roche | Phase Ш discontinued |
| Ragaglitazar | Dual PPARα/γ agonist | 419.477 | Novo Nordisk Pharmaceutical | Phase Ш discontinued |
| Imiglitazar | Dual PPARα/γ agonist | 470.525 | Takeda | Phase Ш discontinued |
| Tesaglitazar | Dual PPARα/γ agonist | 408.465 | AstraZeneca | Phase Ш discontinued |
| Peliglitazar | Dual PPARα/γ agonist | 530.577 | Bristol-Myers Squibb | Phase II discontinued |
| Farglitazar | Dual PPARα/γ agonist | 546.623 | GlaxoSmithKline | Phase II discontinued |
| Sipoglitazar | Dual PPARα/γ agonist; Insulin sensitizer | 463.552 | Takeda | Phase II discontinued |
| Reglitazar | Dual PPARα/γ agonist | 392.411 | Japan Tbacco(Originator) Pfizer | Phase II discontinued |
| Indeglitazar | Dual PPARα/γ agonist | 389.422 | Pfizer | Phase II discontinued |
| Muraglitazar | Dual PPARα/γ agonist | 516.55 | Bristol-Myers Squibb | NDA Filing US |
| Generic Name | Type of PPAR Agonist | Molecular Weight | Company | Development Status |
|---|---|---|---|---|
| Troglitazone | PPARγ agonists | 441.542 | Daiichi Sankyo (Originator) Pfizer | Withdrawn |
| Rivoglitazone Hydrochloride | PPARγ agonists | 433.907 | Daiichi Sankyo (Originator) Santen | Phase Ш discontinued |
| Balaglitazone | Partial agonist of PPARγ | 395.433 | Dr Reddy’s Laboratories (Originator) Rheoscience | Phase II discontinued |
| FK-614 | PPARγ agonists; Insulin sensitizer | 468.393 | Astellas (Originator) Aestus Therapeutics | Phase II discontinued |
| Ciglitazone | PPAR agonists | 333.446 | Takeda | Phase II discontinued |
| ONO 5129 | Dual PPARα/γ agonist | unknown | Ono | Phase II discontinued |
| EML-4156 | Dual PPARα/γ agonist | 314.381 | Merck Serono | Phase II discontinued |
| Netoglitazone; Isaglitazone | Dual PPARα/γ agonist | 381.421 | Mitsubishi Tanabe Pharma (Originator) Perlegen Sciences | Phase II discontinued |
| PN-2034 | PPARγ agonist | unknown | Wellstat (Originator) Sanofi | Phase II discontinued |
| Edaglitazone | PPARγ agonists | 464.554 | Roche | Phase II discontinued |
| Darglitazone Sodium | Dual PPARα/γ agonist | 442.465 | Pfizer | Phase І discontinued |
| AVE-5376 | Dual PPARα/γ agonist | unknown | Sanofi (Originator) | Phase І discontinued |
| DS-6930 | PPARγ agonists | 136.129 | Daiichi Sankyo | Phase І discontinued |
| E-3030 | Dual PPARα/γ agonist | 481.93 | Eisai | Phase І discontinued |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| Saroglitazar | Dual PPARα/γ agonist | Diabetic dyslipidemia | 439.57 | Zydus cadila | Approved |
| AMG-131 | PPARγ agonist | Type 2 diabetes; Multiple sclerosis (MS) | 672.38 | Amgen (Originator) InteKrin Therapeutics | Phase II active |
| K-111 | PPARα agonists | Type 2 diabetes; Hyperlipidemia | 379.75 | Roche | Phase II Pending |
| CLX-0921 | PPARγ agonist | Type 2 diabetes; Rheumatoid arthritis (RA) | 519.568 | Theracos | Phase II Pending |
| HPP 593 | PPARδ | Diabetes Dyslipidemia | unknown | vTv Therapeutics LLC | Phase II active |
| SAR-351034 | PPAR agonists | Type 2 diabetes; Dyslipidemia | unknown | Sanofi | Phase І active |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| MK-0767 | Dual PPARα/γ agonist | Type 2 diabetes; Dyslipidemia | 422.36 | Kyorin (Originator) Merck Sharp & Dohme | Phase Ш discontinued |
| Cevoglitazar | Dual PPARα/γ agonist | Type 2 diabetes; Lipodystrophy | 558.528 | Novartis | Phase II discontinued |
| Sodelglitazar | Pan–PPAR agonists; Insulin sensitizer | Type 2 diabetes; Hyperlipidemia | 499.539 | GlaxoSmithKline | Phase II discontinued |
| AVE-0847 | Dual PPARα/γ agonist | Type 2 diabetes; Lipodystrophy | unknown | Sanofi | Phase II discontinued |
| KRP-101 | PPARα agonists | Diabetes; Dyslipidemia | 451.49 | Kyorin | Phase II discontinued |
| DSP-8658 | Dual PPARα/γ agonist | Type 2 diabetes; Alzheimer’s disease | unknown | Dainippon Sumitomo | Phase І discontinued |
| ARH-049020 | PPAR agonists | Type 2 diabetes; Insulin resistance | 429.51 | AstraZeneca | Phase I discontinued |
| LY-510929 | Dual PPARα/γ agonist | Type 2 diabetes; Hyperlipidemia | 463.55 | Lilly | Phase I discontinued |
| GSK-376501 | PPARγ agonist | Type 2 diabetes; Hypercholesterolemia | 531.649 | GlaxoSmithKline | Phase I discontinued |
| Tetradecylthioacetic acid | Pan–PPAR agonists; Lipid Peroxidation inhibitors | Type 2 diabetes; Dyslipidemia | 288.49 | Badische Anilin-und-Soda-Fabrik | Phase I discontinued |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company |
|---|---|---|---|---|
| Clofibrate | PPAR agonists | Hyperlipidemia Hypertriglyceridemia Hypercholesterolemia | 242.699 | Pfizer |
| Fenofibrate; Fenomax | PPARα agonists | Hypercholesterolemia Hypertriglyceridemia | 360.834 | Abbvie |
| Choline Fenofibrate | PPARα agonists | Hyperlipidemia | 421.918 | Abbvie |
| Bezafibrate | Pan–PPAR agonists | Hypertriglyceridemia Hypercholesterolemia Mixed hyperlipidemia | 361.822 | Unknown |
| Gemfibrozil | PPAR agonists | Hyperlipidemia; Ischemic heart disorder | 250.338 | Pfizer |
| Ciprofibrate | PPAR agonists | Hyperlipidemia | 289.152 | Unknown |
| Pemafibrate | PPARα agonists | Dyslipidemia | 490.556 | Kowa |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| Elafibranor | Dual PPARα/δ agonist | Non-alcoholic fatty liver disease (NAFLD); Dyslipidemia; Type 2 diabetes | 384.49 | Genfit | Phase III active |
| Icosabutate | PPAR agonists; Cholesterol ester transfer protein inhibitors | Hypertriglyceridemia | 374.565 | BASF | Phase II active |
| ZYH-7 | PPARα agonists | Dyslipidemia | unknown | Zydus cadila | Phase II active |
| CER-002 | PPARδ agonists | Dyslipidemia | unknown | Nippon Chemiphar | Phase І active |
| GSK-625019 | PPAR agonists | Metabolic Syndrome X; Type 2 diabetes | unknown | GlaxoSmithKline | Phase І Pending |
| KD-3010 | PPARα agonists | Obesity; Diabetes; Dyslipidemia | 670.72 | Kalypsys | Phase І Pending |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| GW-501516 | PPARδ agonists | Hyperlipidemia | 453.494 | GlaxoSmithKline | Phase II discontinued |
| GFT 14 | PPARα agonists | Dyslipidemia | unknown | Genfit | Phase II discontinued |
| GW-544 | Dual PPARα/γ agonist | Hyperlipidemia | 510.58 | GlaxoSmithKline (Originator)Ligand | Phase І discontinued |
| DFR-11605 | PPAR agonists | Obesity | unknown | Dr Reddys Laboratories (Originator)Perlecan | Phase І discontinued |
| MP-136 | PPARα agonists | Dyslipidemia | unknown | Mitsubishi Tanabe Pharma | Phase І discontinued |
| DRF-10945 | PPARα agonists | Lipid metabolism disorders | unknown | Dr Reddys Laboratories (Originator)Perlecan | Phase І discontinued |
| NS-220 | PPARα agonists | Lipid metabolism disorders | 373.449 | Nippon Shinyaku Pharma | Phase І discontinued |
| F-16482 | PPAR modulator | Metabolic Syndrome X | unknown | PIERRE FABRE | Phase І discontinued |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| Gemcabene Calcium | PPAR agonists | Hypercholesterolemia | 340.473 | Gemphire Therapeutics | Phase II active |
| KRP-105 | PPARα agonists | Hypercholesterolemia | unknown | Kyorin | Phase І discontinued |
| Generic Name | Type of PPAR Agonist | Indication | Molecular Weight | Company | Development Status |
|---|---|---|---|---|---|
| Seladelpar lysine dihydrate | PPARδ agonists | Primary biliary cirrhosis | 626.685 | Janssen (Originator) CymaBay Therapeutics | Phase Ш active |
| Arhalofenate | Partial PPARγ modulators | Chronic gout | 415.793 | CymaBay Therapeutics | Phase II active |
| T3D-959 | Dual agonist of PPARδ/γ | Alzheimer’s disease | 443.47 | DARA BioSciences | Phase II active |
| Efatutazone hydrochloride | Selectively activates PPARγ | Thyroid cancer; Non-small cell lung cancer; Colorectal cancer | 593.52 | Daiichi Sankyo | Phase II Pending |
| IVA-337 | PPAR agonists | Systemic sclerosis | 434.92 | Abbvie(Originator)Inventiva | Phase II active |
| Fonadelpar | PPAR agonists | Corneal disorders | 504.524 | Senju Pharmaceuticals | Phase II active |
| OMS-403 | PPARγ agonists | Opioid abuse Smoking cessation | unknown | Omeros | Phase II active |
| 10-Nitrooctadec-9-enoic acid | PPARγ ligands; Transcription factor modulators; Inflammation mediator modulators | Acute kidney injury Renal failure | 327.465 | Complexa | Phase І active |
| GED-0507-34 | PPAR modulator | Inflammatory bowel disease | unknown | Giuliani | Phase І active |
| Macuneos | PPARα agonists | Age-related macular degeneration | unknown | Biophytis | Phase І active |
| MA-0211 | PPARδ modulators | Duchenne muscular dystrophy | unknown | Astellas | Phase І active |
| Oxeglitazar | Dual PPARα/γ agonist | Gout | 314.381 | Merck Serono | Phase І Pending |
| Etalocib sodium | PPARγ agonists; 5-Lipoxygenase inhibitor; Leukotriene B4 receptor antagonist | Pancreatic cancer; Non-small cell lung cancer | 566.601 | Lilly(Originator)Vernalis | Phase II discontinued |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082189
Hong F, Xu P, Zhai Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. International Journal of Molecular Sciences. 2018; 19(8):2189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082189
Chicago/Turabian StyleHong, Fan, Pengfei Xu, and Yonggong Zhai. 2018. "The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development" International Journal of Molecular Sciences 19, no. 8: 2189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19082189