The Two-Faced Cytokine IL-6 in Host Defense and Diseases

1
Department of Respiratory Medicine and Clinical Immunology, Graduate School of Medicine, Osaka University, Osaka 565-0871, Japan
2
Department of Immunopathology, World Premier International Immunology Frontier Research Center, Osaka University, Osaka 565-0871, Japan
3
Laboratory of Immune Regulation, World Premier International Immunology Frontier Research Center, Osaka University, Osaka 565-0871, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(11), 3528; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113528
Submission received: 13 October 2018
/
Revised: 30 October 2018
/
Accepted: 6 November 2018
/
Published: 9 November 2018
(This article belongs to the Special Issue The Interleukins in Health and Disease)
Abstract
:Interleukein-6 (IL-6), is produced locally from infectious or injured lesions and is delivered to the whole body via the blood stream, promptly activating the host defense system to perform diverse functions. However, excessive or sustained production of IL-6 is involved in various diseases. In diseases, the IL-6 inhibitory strategy begins with the development of the anti-IL-6 receptor antibody, tocilizumab (TCZ). This antibody has shown remarkable effects on Castleman disease, rheumatoid arthritis and juvenile idiopathic arthritis. In 2017, TCZ was proven to work effectively against giant cell arteritis, Takayasu arteritis and cytokine releasing syndrome, initiating a new era for the treatment of these diseases. In this study, the defensive functions of IL-6 and various pathological conditions are compared. Further, the diseases of which TCZ has been approved for treatment are summarized, the updated results of increasing off-label use of TCZ for various diseases are reviewed and the conditions for which IL-6 inhibition might have a beneficial role are discussed. Given the involvement of IL-6 in many pathologies, the diseases that can be improved by IL-6 inhibition will expand. However, the important role of IL-6 in host defense should always be kept in mind in clinical practice.
Keywords:
cytokine; interleukin-6 (IL-6); IL-6 receptor; tocilizumab; inflammation; host defense; pathology
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Interleukein-6 (IL-6) is an essential cytokine that transmits defense signals from a pathogen invasion or tissue damage site to stimulate acute phase reactions, immune responses, hematopoiesis and various internal organs to prepare for host defense [1]. However, excessive and sustained production of IL-6 is associated with various inflammatory diseases [2,3,4,5]. A healthy serum IL-6 level is lower than 4 pg/mL but this level increases to several tens or even hundreds of pg/mL in chronic diseases depending on the disease severity and location. Values of more than 1000 pg/mL can occur during septic shock or a cytokine storm and in severe cases, a level measured in µg/mL can be reached, in which excessive defense signaling can threaten survival [6]. As IL-6 is involved in various diseases, it was proposed that its inhibition could improve the pathology of such conditions. The creation of an IL-6 inhibitory strategy started with the development of the anti-IL-6 receptor antibody, tocilizumab (TCZ). After observation of a drastic clinical effect of TCZ on Castleman disease, which is a typical inflammatory disease caused by IL-6 overproduction, TCZ was shown to have significant effects on rheumatoid arthritis (RA) and juvenile idiopathic arthritis (JIA). The safety profile of IL-6-targeting therapy has been confirmed through its use by a large number of patients with arthritis. Consequently, in 2017, TCZ was approved for treatment of giant cell arteritis, Takayasu arteritis and an artificial T cell therapy-induced cytokine releasing syndrome in leukemia/lymphoma. The successful results achieved with TCZ have changed the therapeutic strategies for these conditions and now, it is expected that IL-6-targeting therapy could be used in many refractory diseases; thus, clinical trials are underway.
2. Production of IL-6
Expression of IL-6 is regulated by various mechanisms that are gene polymorphism, chromatin remodeling, transcriptional and posttranscriptional levels [3]. A G/C polymorphism at −174 in the human IL-6 flanking region affects the transcriptional level and is associated with inflammatory diseases [7,8]. The chromatin structure of the IL-6 promoter is changed into accessible conformation for transcriptional factors during differentiation of immune cells such as monocyte [9]. Functional cis-regulatory elements in the human IL-6 gene include nuclear factor kappa B (NF-κB) binding site (−74/−63 from the transcription start site), nuclear factor IL6 (NF-IL6) (also known as CCAAT/enhancer-binding protein beta or C/EBPβ) (−87/−76 and −159/−145), specificity protein 1 (SP1) (−109/−104 and −123/−119), cyclic AMP response element binding protein (CREB) (−165/−158), interferon regulatory factor 1 (IRF-1) (−267/−254) and activator protein 1 (AP-1) (−284/−277) [3]. Among these factors, NF-κB is a main transcriptional factor commonly activated by Toll-like receptors (TLRs)-mediated signals and pro-inflammatory cytokines including tumor necrosis factor α (TNFα), IL-1β and IL-17 [10,11].
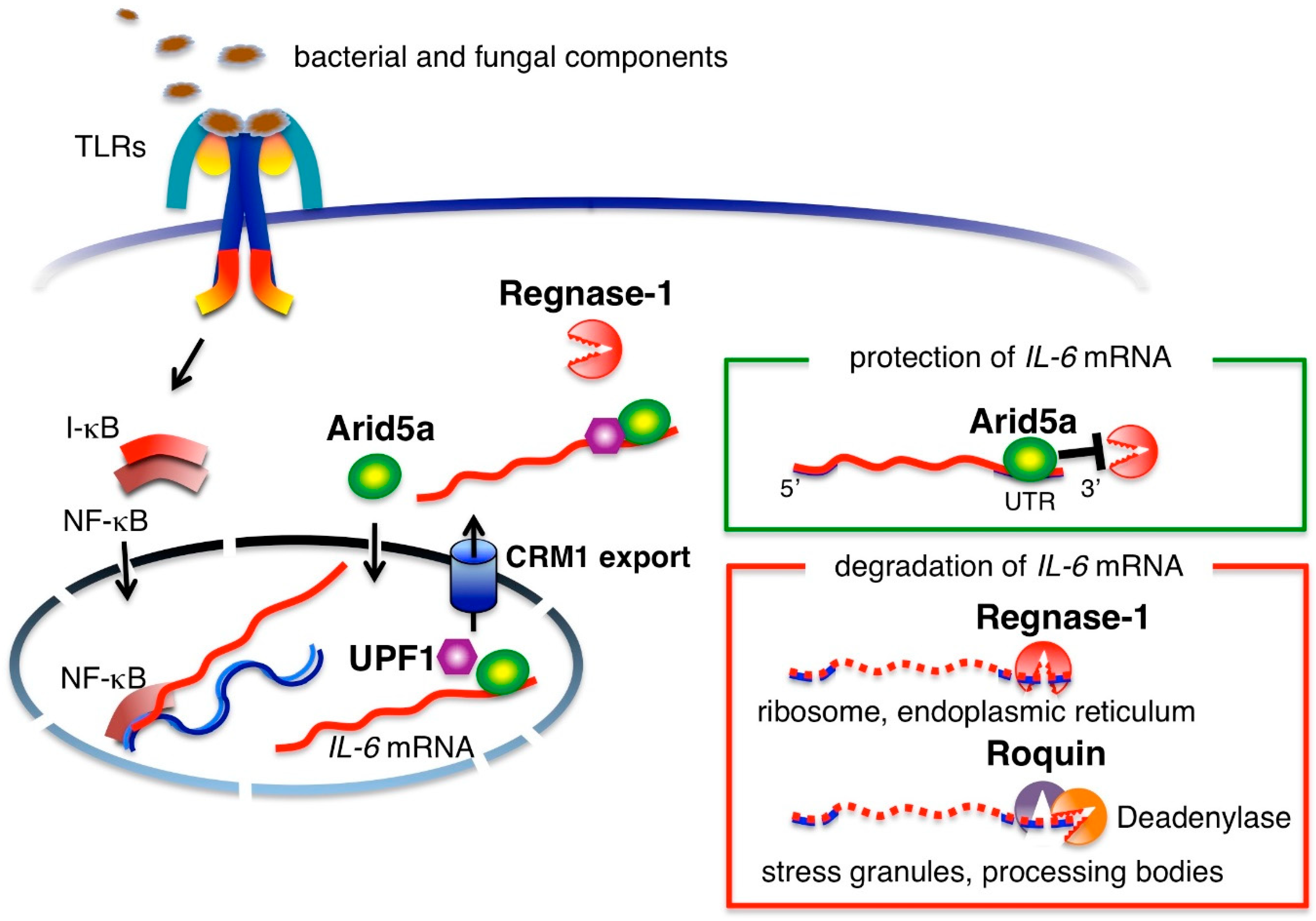
In infected lesions, bacterial and fungal components are recognized by cell surface TLRs and their nucleic acids are recognized by intracellular TLRs [12]. Noninfectious inflammation due to burn, trauma and even sterile surgical operations produces damaged, necrotic cells and a degraded extracellular matrix. The molecules derived from tissue injury are also recognized by TLRs [12,13]. Activation of cell surface and intracellular TLRs in monocytes and macrophages induces mRNA-transcription of IL-6 and other pro-inflammatory cytokines, such as TNFα and IL-1β, via nuclear factor-kappa B (NF-κB) [10]. Signaling from TNFα or IL-1β also induces IL-6 mRNA transcription [14]. After transcription, IL-6 mRNA is regulated at the post-transcriptional level by adenylate-uridylate-rich elements (AREs) located in the 3′UTR region of IL-6 mRNA (Figure 1). A nuclease known as regulatory RNase-1 (Regnase-1) degrades transcriptionally active IL-6 mRNA by binding to IL-6 3′UTR in the cytoplasm, endoplasmic reticulum and ribosomes [15]. Macrophages from Regnase-1 knockout mice produce increased levels of IL-6 and the mice have been shown to spontaneously develop autoimmune diseases [16], indicating that post-transcriptional regulation of IL-6 is critically linked to IL-6 production. Another RNA-binding protein, Roquin, degrades inactive mRNA in stress granules and processing bodies [15]. In contrast, RNA-binding protein, AT-rich interactive domain-containing protein 5a (Arid5a), is expressed in response to the presence of lipopolysaccharides (LPS), IL-1β and IL-6 in macrophages and under Th17 polarizing conditions in T cells. Arid5a is imported into the nucleus via an importin-α/β1 pathway and binds to the 3′UTR of IL-6 mRNA. Then, Arid5a exports IL-6 mRNA to the cytoplasm via the chromosomal region maintenance 1 (CRM1) pathway by binding to up-frameshift protein 1 (UPF1). The IL-6 mRNA bound to Arid5a is protected from degradation by Regnase-1 in the cytoplasm [17,18]. The counteracting roles of Arid5a and Regnase-1 in regard to the stability of IL-6 mRNA indicates that the balance between the two governs the quantity of IL-6 production [19].
3. IL-6 Receptor System and Signal Activation Modes
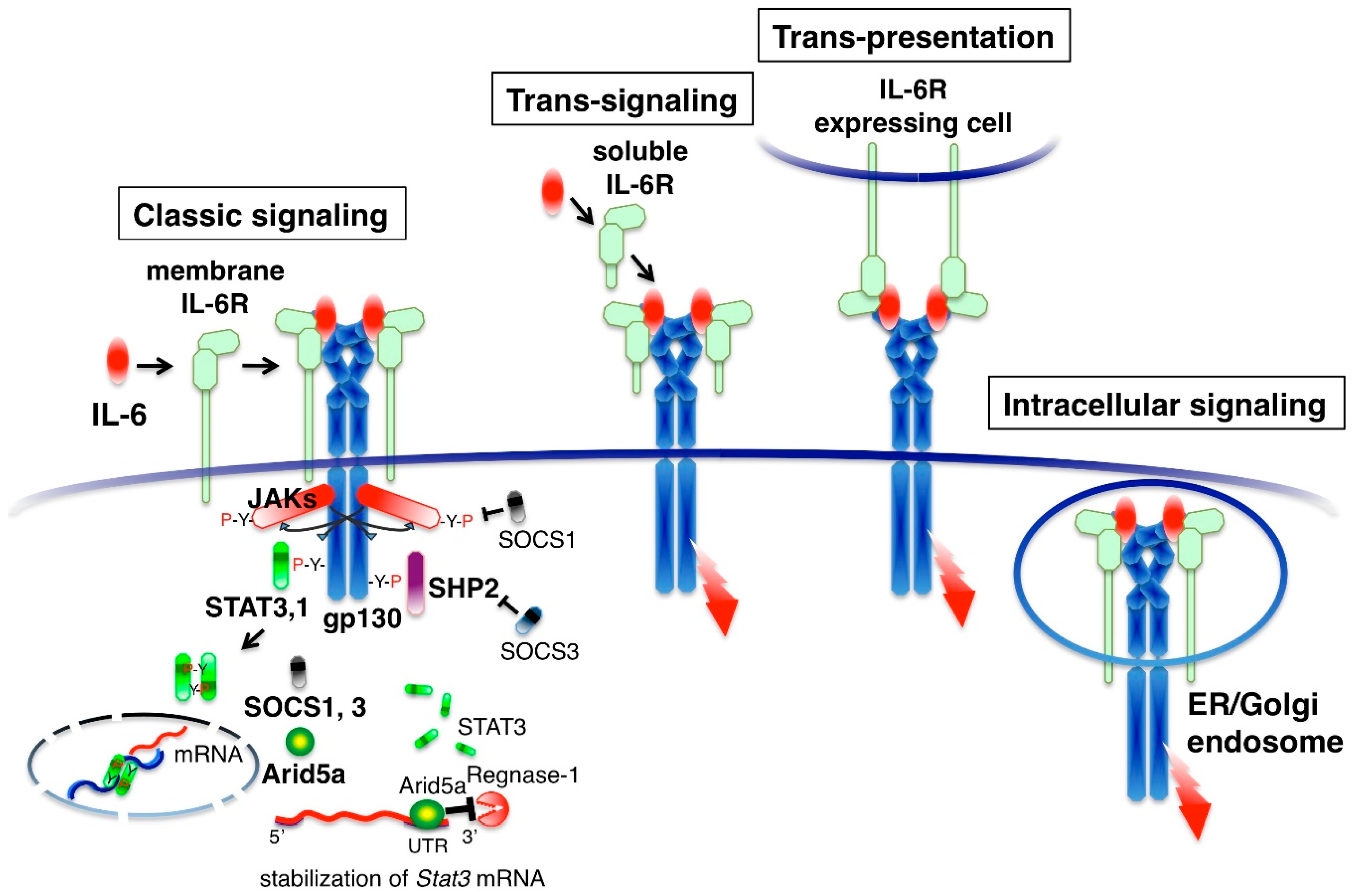
IL-6 binds to its specific membrane receptor (IL-6R, also designated CD126) and the associated IL-6/IL-6R complex, inducing homodimerization of the signaling receptor component gp130 (also designated CD130). This results in the hexamer structure of the IL-6-signaling complex that consists of two molecules each of IL-6, IL-6R and gp130 (Figure 2). IL-6-intracellular signaling is initiated by the activation of cytoplasmic Janus kinases (JAKs: JAK1, JAK2 and TYK2) by homo-dimerization of gp130 and subsequent phosphorylation of tyrosine residues in the cytoplasmic region of gp130 [20,21]. Downstream signaling molecules, the signal transducer and activator of transcription 3 (STAT3), STAT1 and SH2 domain-containing protein-tyrosine phosphatase 2 (SHP2) are recruited to the tyrosine-phosphorylated motifs of gp130 [22]. STAT3 and STAT1 are then phosphorylated by the JAKs and translocated to the nucleus, generating the transcriptional output. SHP-2 activates the Ras-MAP kinase pathway [21]. The intracellular IL-6 signal is regulated by inhibitory or promoting molecules. STAT3 induces negative feedback molecules, which are suppressor of cytokine signaling 1 (SOCS1) and SOCS3 proteins [23,24]. SOCS1 binds to and inhibits JAK [25]. SOCS3 binds to gp130 and inhibits the SHP2 pathway [26]. STAT3 also induces the positive feedback molecule Arid5a, which selectively stabilizes Stat3 mRNA by binding to Stat3 3′UTR and enhancing the STAT3 signal [27]. These negative or positive feedback mechanisms determine the magnitude of the IL-6 response.
Several modes of gp130 activation are known. IL-6 binds with high affinity to cells expressing both membrane IL-6R and gp130, such as hepatocytes, neutrophils, monocytes, activated B cells and CD4 T cells [2]. The activation of gp130 by membrane IL-6R is called classic signaling (Figure 2). Serum contains 60–100 ng/mL of soluble IL-6R, which is derived from the shedding of membrane IL-6R by the metalloproteases ADAM10 and ADAM17 [28]. IL-6 can also bind to the soluble form of IL-6R (sIL-6R) in serum. The IL-6/sIL-6R complex activates gp130-expressing cells, even if cells lack membrane IL-6R, a process known as trans-signaling [29]. Although cell types expressing membrane IL-6R are limited, gp130 is expressed in many cell types and it is thought that some aspects of the pleiotropic effect of IL-6 are derived from trans-signaling. Soluble gp130 (sgp130) is present in human serum and interacts with the IL-6/sIL-6R complex to inhibit trans-signaling [30]. When cells with membrane IL-6R and those with gp130 are in close proximity, IL-6 is presented by membrane IL-6R-expressing cells to the other gp130 expressing cells. This type of signaling is called trans-presentation [31,32]. IL-6 also activates gp130 in the endosomal compartment. Extracellular IL-6 is taken up by IL-6R and moves to the endosomal compartment by endocytosis where it activates gp130. Alternatively, newly synthesized IL-6 can transduce its signals in the endoplasmic reticulum or Golgi body before secretion [33]. When IL-6-producing cells express both IL-6R and gp130, autocrine stimulation may occur in cells as intracellular signaling [34].
The signaling component gp130 is shared with the cytokines of the IL-6 family, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), cardiotrophin 1 (CT-1), IL-11, cardiotrophin-like cytokine factor 1 (CLCF1), IL-27 and IL-35 [35,36]. The concept that signal transducer gp130 is shared with IL-6 family cytokines explains the redundant actions of IL-6 family cytokines; however, the function of IL-6 is not always compensated by the other IL-6 family members. For example, some cytokines induce acute phase proteins in hepatocytes in vitro but acute phase proteins almost normalize when only IL-6 is suppressed in patients with inflammatory diseases. This suggests that the regulation of cytokine expression is different among IL-6 family members.
4. IL-6 in Host Defense and Disease
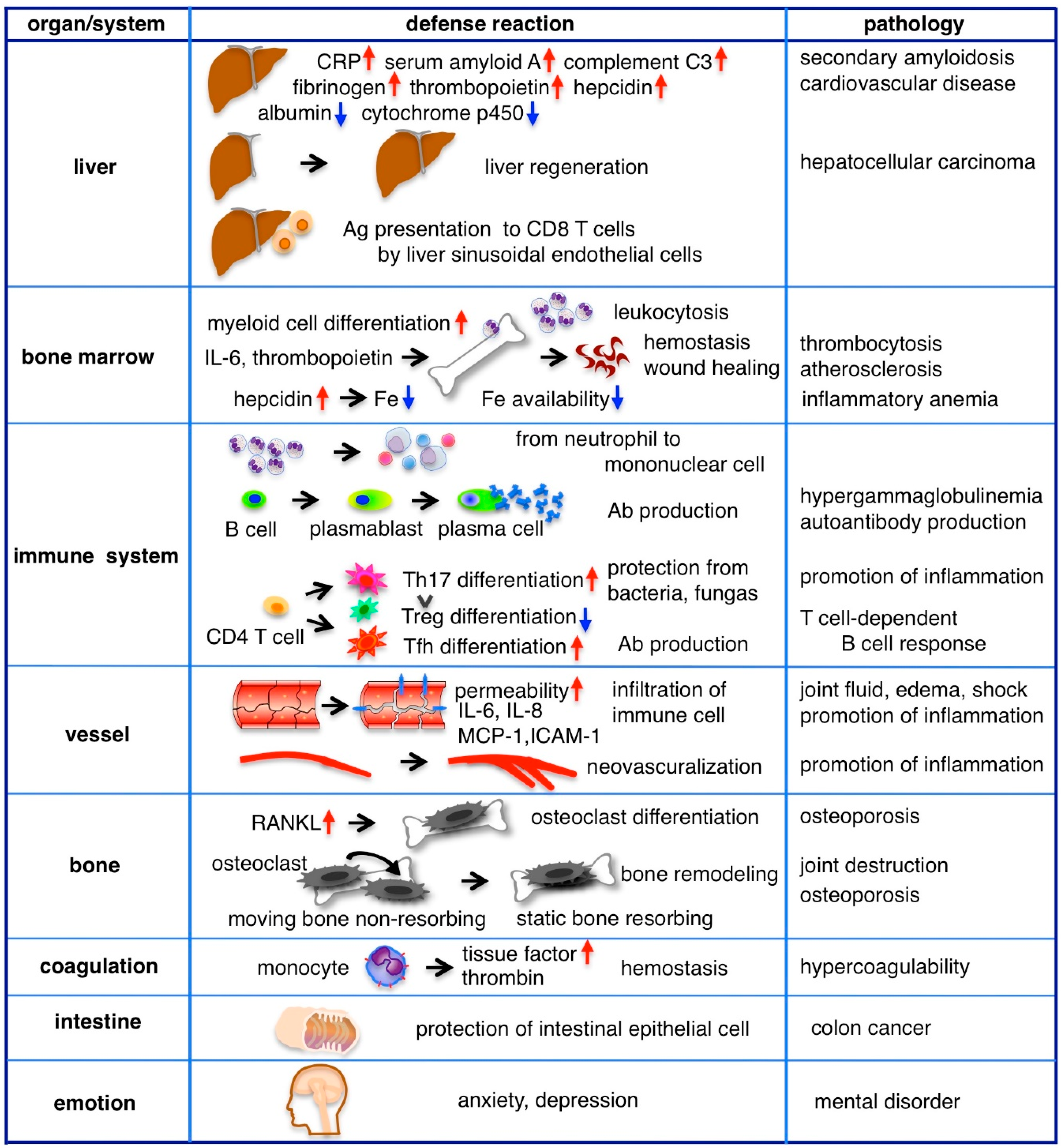
4.1. Acute Phase Protein Production, Regeneration and Immunity in the Liver
The liver has long been considered the main target organ of IL-6 and in the past, IL-6 was alternatively called the hepatocyte stimulatory factor [37]. Since hepatocytes express membrane IL-6R and gp130, classic signaling mainly mediates IL-6 signaling in the liver. The liver quickly responds to IL-6 to help with host defense from the early phase of inflammation, producing various proteins, such as C-reactive protein (CRP), serum amyloid protein A, complement C3, haptoglobin, antitrypsin, fibrinogen and hematopoiesis regulatory proteins including hepcidin and thrombopoietin but reduces the production of albumin and cytochrome p450 (Figure 3). For example, CRP is a pattern recognition molecule that binds to specific shapes on the surfaces of pathogens and damaged cells. Ligand-bound CRP functions as an opsonin and can activate the classical complement pathway [38]. In clinical practice, the levels of acute phase proteins in serum are often measured and correlated with the degree of inflammation. In vitro, hepatocytes respond with the other IL-6 family cytokines to produce acute phase proteins; however, it has been shown that IL-6 deficient mice have a severely impaired acute phase response [39]. In addition, blocking the IL-6 receptor in patients with RA reduces acute phase protein levels [40], indicating that IL-6 is an essential cytokine in the induction of the liver’s acute phase response. The single nucleotide polymorphism rs2228145 leads to Asp358Ala substitution of IL-6R and is known to be associated with a reduction in membrane IL-6R, an increase in sIL-6R and reductions in CRP and fibrinogen, supporting the main role of membrane IL-6R-mediated classic signaling in acute phase protein induction [41]. Persistent high levels of serum amyloid A in chronic inflammation, such as RA or bronchiectasis, leads to secondary amyloidosis, which has various symptoms resulting from the deposition of amyloid A in tissues. Intractable diarrhea and malnutrition are caused by the deposition of amyloid A in the intestine. Urine protein and renal failure are caused by renal amyloidosis. Heart failure and arrhythmia are observed in cardiac amyloidosis.
In addition to the induction of acute phase proteins, IL-6 is also involved in liver regeneration and liver-mediated adaptive immunity [42]. The liver has the capacity to regenerate after injury or even hepatectomy. IL-6-deficient mice have been reported to show impaired liver regeneration that is characterized by liver necrosis and liver failure. However, IL-6 treatment rescues these mice by inducing hepatocyte proliferation and preventing liver damage [43]. IL-6 has anti-apoptotic effects on hepatocytes by inducing an anti-apoptotic member of the Bcl-2 family, Mcl-1, to promote hepatocyte growth [44]. However, despite its many positive effects, IL-6 is also associated with the development of hepatocellular carcinoma (HCC) after liver cirrhosis and elevated serum levels of IL-6 predict HCC progression in patients with chronic hepatitis B infection [45]. Thus, IL-6 is considered a serum marker of HCC and its over-expression has been associated with six-month mortality [46,47].
Antigen-stimulated antigen presenting cells (APCs) encounter naïve T cells in both the lymph nodes and spleen. In the peripheral tissues, the liver also stimulates naïve T cells to clear gut-derived pathogens [48]. Liver sinusoidal endothelial cells (LSECs) present antigens to naive CD8 T cells as APCs and mediate the IL-6 signal to CD8 T cells via trans-presentation using IL-6/IL-6R on the surface of LSECs [31]. Cytotoxic CD8 T cells, received with antigen presentation and IL-6 stimulation, rapidly express effector molecule granzyme B to induce apoptosis in target cells.
4.2. Hematopoiesis
Inflammation often accompanies with leukocytosis, thrombocytosis and anemia. IL-6 has an activity of myeloid blood cell differentiation. This activity was studied as macrophage and granulocyte inducer type 2A [49]. Administration of IL-6 to rabbits causes a biphasic pattern of neutrophilia [50]. At the first peak IL-6 mobilizes polymorphonuclear leukocytes (PMNs) from the marginated pool into the circulating pool at 2–6 h with a decrease of L-selectin expression of PMNs. At the second peak IL-6 releases PMNs from the postmitotic pool in the bone marrow at 12–24 h.
In vitro experiment indicates IL-6 acts a potent growth factor for megakaryocytes and promotes maturation of megakaryocytic colony formation together with IL-3 [51]. However, in inflammatory condition IL-6 indirectly controls platelet production by stimulating the liver to release Thrombopoietin (TPO). TPO is a major regulator of thrombopoiesis and is constitutively produced in hepatocytes [52]. Its concentration in plasma is regulated by binding to TPO receptor c-Mpl on the surfaces of platelets [53]. During inflammation, IL-6 induces TPO production in the liver and elevates its plasma level. IL-6-induced thrombocytosis is abrogated by the neutralization of TPO, suggesting that it is mediated by TPO [54]. Elevated levels of platelets contribute not only to primary hemostasis but also to host defense [55]. Platelets express TLR4 and present bound LPS to neutrophils, stimulating them to produce reactive oxygen species (ROS). Platelets also promote neutrophil extracellular traps (NETs) formation [56]. Activated platelets release many proteins that promote angiogenesis and wound healing, including platelet-derived growth factor-A (PDGF-A), B and C, insulin-like growth factor-1 and vascular endothelial growth factor (VEGF). On the other hand, chronic thrombocytosis and platelet activation are risk factors for atherosclerosis and related diseases, including myocardial infarction and stroke.
IL-6 indirectly controls erythropoiesis by stimulating the liver to release hepcidin [57]. Hepcidin is a master regulator of iron homeostasis, which binds to, internalizes and degrades the iron exporter ferroportin on duodenal enterocytes, macrophages and hepatocytes, resulting in hypoferremia. A reduction in plasma iron is thought to be a defense mechanism against extracellular infections by inhibiting iron availability to invading pathogens [58], whereas sustained elevation of hepcidin by chronic inflammation results in anemia due to the lack of iron availability for erythropoiesis. Anemia, together with high levels of fibrinogen and immunoglobulins, accelerates the erythrocyte sedimentation rate (ESR), which is correlated with the severity of inflammation and is often measured in clinical practice.
4.3. Immune Response
The cells responsible for immune reaction, such as neutrophils, monocytes, B cells and CD4 T cells, express membrane IL-6R and are stimulated by IL-6 classic signaling. Neutrophils accumulate early in the site of inflammation, followed by a more sustained population of mononuclear cells including monocytes, macrophages and lymphocytes. IL-6 orchestrates transition from neutrophils to mononuclear cells by enhancing neutrophil migration toward IL-8, which is produced by endothelial cells at the acute inflammation site [59,60]. Then, IL-8 reduces the adhesion molecule P-selectin glycoprotein ligand 1 (PSGL-1/CD162) on the surface of neutrophils, leading to an increase in their circulation [61]. IL-8 also stimulates neutrophils to release sIL-6R by proteolytical cleavage of IL-6R on the cell surface. Subsequently, IL-6/sIL-6R trans-signaling stimulates endothelial cells to secrete monocyte chemotactic protein-1 (MCP-1) and augment intercellular adhesion molecule-1 (ICAM-1) expression [62,63]. Through these processes, monocytes accumulate at inflammatory sites and firmly adhere to vascular endothelial cells.
IL-6 plays a pivotal role in shaping the acquired immune response by promoting the differentiation of B cells into immunoglobulin producing plasma cells and increasing the gamma globulin level in serum. IL-6 also supports the survival of plasmablasts which are precursors of plasma cells. Accordingly, hypergammaglobulinemia is typically observed in patients with Castleman disease, a condition involving the continuous production of IL-6 from affected lymph nodes. Elevated levels of gamma globulin are normalized by treatment with anti-IL-6R Ab, albeit usually not to levels below the lower limit of the normal range [64]. Further, IL-6 deficient mice have been reported to still produce antigen-specific Abs in vivo, despite the reduction of levels to less than half of those found in wild type mice [65]. Unlike acute phase protein synthesis, in which IL-6 plays an essential role, gamma globulin production does not appear to be completely dependent on IL-6.
In T cells, IL-6 regulates the direction of the specific differentiation of naïve CD4 T cells. IL-6 and transforming growth factor β (TGFβ) are coordinately required for Th17 differentiation. Th17 cells produce IL-17, IL-21 and IL-22 and contribute to protection against bacterial or fungal infection [66]. In these cytokines, IL-17 stimulates fibroblasts, epithelial cells and synoviocytes, leading to the secretion of a range of pro-inflammatory cytokines including IL-6, TNFα, IL-1β and various chemokines that play roles in neutrophil trafficking to the site of inflammation. IL-21 and TGFβ induce Th17 differentiation in a positive feedback loop to amplify Th17 cells. IL-22 induces the production of antimicrobial agents in keratinocytes for immune barrier function [67] and is crucial in host defense against Gram-negative bacterial infections in the lung [68]. IL-6 inhibits TGFβ-induced regulatory T cell (Treg) differentiation that plays a role in immune homeostasis. The resulting up-regulation of the Th17/Treg balance is thought to be involved in the development of various autoimmune disorders and chronic inflammation [69,70].
4.4. Blood Vessels
An increase in vascular permeability is a defense mechanism for immune cells to infiltrate tissues, remove pathogens and repair damaged tissues. This also allows immune-related proteins, such as cytokines, antibodies and complements, to be delivered to inflammatory sites. Vascular endothelial (VE)-cadherin is the main protein that mediates the adhesion of adjacent cells by homophilic binding, whereas its disassembly leads to vascular leakage. Endothelial cells do not express membrane IL-6R but do express gp130; thus, IL-6/sIL-6R trans-signaling stimulates endothelial cells to induce phosphorylation and internalization of VE-cadherin [71]. Inflammatory cytokines, including IL-6, induce the potent vascular permeability factor VEGF from adipose tissue or other cells and phosphorylate and internalize VE-cadherin in endothelial cells [72,73]. Swelling and fluid accumulation in the joints of patients with RA and peripheral pitting edema in RS3PE syndrome are explained by the enhanced vascular permeability caused by IL-6 or VEGF [74]. The large amount of IL-6 from affected lymph nodes induces a type of systemic edema, called anasarca, massive ascites fluid and pleural effusions that are observed in Castleman disease and TAFRO syndrome [75]. In addition, edema is enhanced by IL-6-mediated hypoalbuminemia in the liver. If very strong, acute and systemic inflammation occurs, excessive hyperpermeability leads to a shock vital state, as observed in sepsis or cytokine releasing syndrome. In this case, the level of IL-6 is correlated with prognosis. In the cecal ligation and puncture sepsis model, survival rates were improved by sgp130-Fc, which inhibits IL-6 trans-signaling [76], suggesting the importance of IL-6 trans-signaling inhibition in the systemic inflammatory response.
In order to supply oxygen and nutrients to the inflammatory lesion, new vessel formation from pre-existing vasculature to the inflammatory lesion is necessary [77]. IL-6 induces endothelial cell proliferation, migration and vascular vessel sprouting, with similar efficacy as VEGF [78]. New vessel formation in chronic inflammatory lesions of the RA joints is examined by power Doppler ultrasound assessment in clinical practice.
4.5. Bone Homeostasis
Bone fracture healing is a sequential process involving inflammation, regeneration and remodeling phases. Using the femur osteotomy model in mice, it was shown that the inhibition of IL-6 reduces inflammation, recruitment of immune cells and bone regeneration in the early phase and disturbs bone formation and remodeling in the repair phase, suggesting an essential role of IL-6 in bone healing [79]. On the other hand, chronic inflammation results in bone loss. Inflammatory cytokines, including IL-6, induce receptor activator of nuclear factor κB ligand (RANKL), an essential factor for osteoclastogenesis [80]. IL-6 transgenic mice show osteopenia—a decrease in osteoblasts and an increase in osteoclasts—resulting in severe alterations in cortical and trabecular bone microarchitecture [81]. Mature osteoclasts are classified into two different types based on their motility and function: static bone resorptive osteoclasts and moving non-resorptive osteoclasts. Intravital imaging has shown that anti-IL-6R Ab converts static bone resorbing osteoclasts to moving non-resorbing osteoclasts under inflammatory conditions [82]. The osteoclastogenesis-promoting actions of IL-6 have also been indicated in clinical situations where IL-6 inhibition has increased bone formation markers, decreased bone absorption markers and improved bone balance in patients with RA [40,83].
4.6. Coagulation System
IL-6 induces a membrane protein tissue factor (TF, also known as factor III or CD142) on the surfaces of monocytes [84]. TF promotes activation of the coagulation system by initiating the extrinsic coagulation pathway. It activates factors VIIa and Xa, following activating prothrombin to produce thrombin that cleaves fibrinogen to fibrin to generate fibrin clot formation. In an attempt to administer recombinant IL-6 to humans, IL-6 was shown to stimulate coagulation through an increase in thrombin-antithrombin III complexes and the prothrombin activation fragment F1 + 2 [85]. Conversely, activated coagulation factors induce IL-6. In a trial in healthy humans, the activation of coagulation through the administration of recombinant factor VIIa elicited an increase in plasma IL-6 [86]. The actions of IL-6, such as inflammation induction, fibrinogen concentration elevation, an increase in platelets and coagulation promotion, support the role of IL-6 in the development of cardiovascular disease [87,88].
4.7. Intestinal Tract
In addition to the role of IL-6 in the regeneration of the liver, IL-6 is involved in the protection of intestinal epithelial cells. In vitro, IL-6 induces anti-apoptotic proteins in colon epithelial cells against apoptosis. In a murine Citrobactor rodentium infection model, IL-6 deficiency resulted in marked apoptosis and ulcer formation in the colonic epithelium [89]. IL-6 deficiency has also been shown to induce severe colitis in a dextran sodium sulfate-induced colitis model [90]. These mice models suggest that IL-6 maintains intestinal barrier function and limits further inflammation. On the other hand, the effect of IL-6 on intestinal epithelial cells contributes to the promotion of colon cancer growth, invasion and metastasis [91,92].
4.8. Emotion and Behavior
There is a hypothesis that persistent depressive symptoms are based on host defense [93]. Following infection or injury, individuals react to dangerous situations with emotional change, which evokes behavioral alterations. A microarray analysis on whole blood from patients with major depressive disorder revealed that 165 genes were expressed differentially—the 90 overexpressed genes were associated with innate immunity and the 75 under-expressed genes were associated with the adaptive immune response [94]. A meta-analysis indicated that the inflammatory markers associated with depression are TNFα, IL-1, IL-6 and CRP [95] and there are many reports showing the association between serum IL-6 increase and depression [96]. The role of IL-6 in stress was also studied in a mouse model. The level of IL-6 increased with stress in wild-type mice; however, IL-6-deficient mice exhibited resistance to the development of depression-like behaviors by forced swim or tail suspension [97]. The behavioral effect of IL-6 was also demonstrated in mice subjected to continuously high levels of IL-6 through an IL-6-expressing virus. Levels of locomotor activity did not decline but novel object exploration and spontaneous alternation tests were disturbed in all mice with high levels of IL-6 [98].
5. Therapeutic Targeting of IL-6 in Diseases
5.1. Diseases in Which Tocilizumab (TCZ) Was Approved for Use in Japan, the EU or the US Before 2017
5.1.1. Castleman Disease
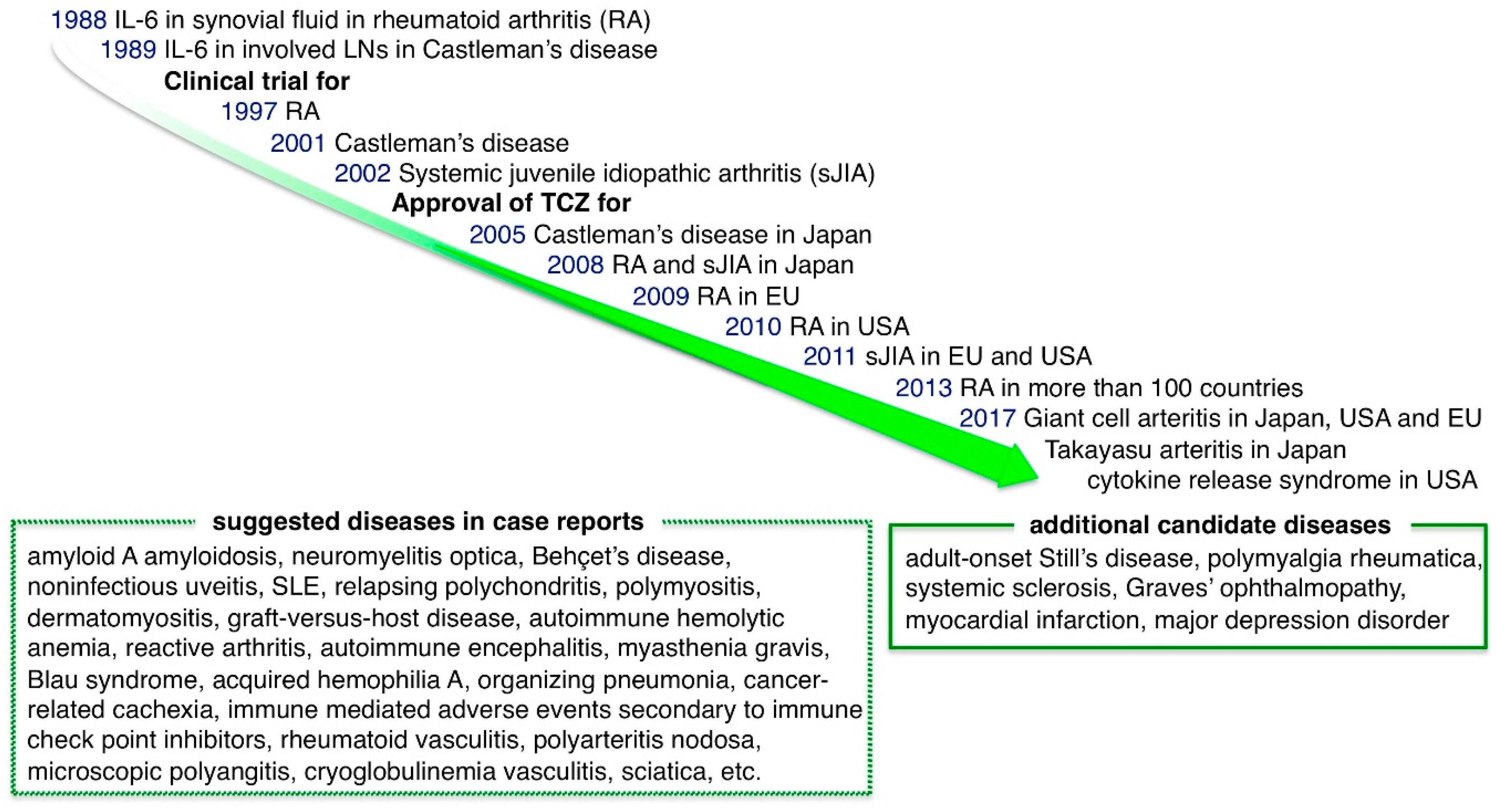
The elucidation of the central role of IL-6 in inflammatory pathology led to the development of a groundbreaking drug that targets IL-6, tocilizumab (TCZ) (Figure 4) [36,99]. TCZ is a humanized anti-IL-6R IgG1 class monoclonal antibody that inhibits IL-6 binding to both membrane IL-6R and sIL-6R, blocking both IL-6 classic and trans-signaling. The first clinical success with TCZ was observed with Castleman disease, which was a good candidate for IL-6 inhibition because continuous production of IL-6 from affected lymph nodes is suggested to be responsible for the systemic inflammation that occurs in this disease [64,100]. A remarkable remission of inflammatory manifestations as well as improvements in anemia and the levels of CRP, fibrinogen and albumin were observed after the TCZ administration, with an accompanying disappearance of fever and fatigue. This provided the initial information on the effect of IL-6 inhibition on inflammatory conditions and TCZ was approved for Castleman disease in 2005 in Japan.
5.1.2. Rheumatoid Arthritis (RA)
RA is a chronic inflammatory disease of the joints and surrounding tissues that is accompanied by joint pain, swelling, destruction and systemic inflammatory complications. Clinical trials of IL-6 inhibition therapy began due to the presence of high levels of IL-6 in synovial fluid in affected joints and the correlation between the serum IL-6 level and disease activity. After the promising results of the first randomized controlled trial of TCZ in RA [101], the efficacy of TCZ in RA was demonstrated by many clinical trials [4,102]. As with the observations in Castleman disease, laboratory parameters associated with RA were markedly improved after TCZ administration [40]. Some crucial studies showed the promising effect of TCZ compared to previous therapies. The AMBITION study, in which TCZ was compared with increasing doses of methotrexate (MTX), showed that TCZ performed better than MTX in terms of the response rate, remission rate and health-related quality of life [103]. In the ADACTA study, TCZ monotherapy improved disease activity scores to a greater extent than anti-TNFα adalimumab (ADA) monotherapy [104]. The U-Act-Early study and post-market surveillance indicated that immediately starting TCZ for patients with less advanced RA produces good results and leads to high rates of drug-free remission, suggesting the importance of stopping disease activity in the early phase [105]. TCZ was approved for RA treatment in 2008, 2009 and 2010 in Japan, the EU and the USA, respectively. The second anti-IL-6R Ab, sarilumab (SAR), is a fully human Ab that showed 10- to 40-fold higher affinity for IL-6R than TCZ. In a phase III study of RA patients, SAR with MTX produced significant improvements in symptoms and radiographic outcomes compared with placebo with MTX groups [106]. In the MONARCH study, SAR monotherapy resulted in greater improvement in disease activity scores than ADA [107]. SAR was approved for RA in 2017 in Japan, the EU and the USA.
5.1.3. Systemic-Onset Juvenile Idiopathic Arthritis (sJIA)
sJIA, a type of chronic childhood arthritis with fever, skin rash, pericarditis, hepatosplenomegaly and growth retardation, was first described by George F. Still and is also known as Still’s disease. It is often treated with glucocorticoids but in the long-term, glucocorticoids cause a variety of serious side effects and growth impairment. IL-6 levels are correlated with the severity of disease manifestation, implicating IL-6 in the pathogenesis of sJIA [108]. A randomized phase III trial in children with refractory sJIA showed drastic improvement in disease activity by TCZ compared to conventional treatment [109]. Furthermore, body growth was restored and bone homeostasis improved in patients who received long-term treatment with TCZ [83,110]. Consequently, TCZ was approved for sJIA treatment in 2008 in Japan and in 2011 in the EU and the USA.
5.2. Diseases in Which TCZ Was Approved for Use in Japan, the EU, or the US in 2017
5.2.1. Giant Cell Arteritis (GCA) and Takayasu Arteritis (TA)
The safety and effectiveness of TCZ for RA encouraged clinical trials of TCZ for various refractory inflammatory diseases. In 2017, the efficacy of TCZ was confirmed for diseases in a field other than inflammatory joint diseases. GCA is the most common large vessel vasculitis and cranial arteritis that affects individuals over the age of 50 and can lead to permanent visual loss. Most patients improve with glucocorticoids; however, some patients show disease flare after the reduction of treatment. A phase III GiACTA study involving 251 patients with GCA compared six months of treatment with prednisone taper plus TCZ (subcutaneous 162 mg) to prednisone taper alone. Fifty-six percent of patients receiving TCZ weekly and 53% receiving it every-other-week achieved sustained remission at 12 months compared to 14% in the six-month prednisone tapering alone group [111]. Treatment with TCZ plus prednisone led to a reduction in the cumulative dose of prednisone required to regulate GCA. In 2017, TCZ became the first GCA therapy to be approved by the USA, Japan and the EU. Another type of large vessel vasculitis is TA, often affecting women under the age of 40, in which arteries and their major branches narrow or cause aneurysms due to chronic inflammation of the vessel walls. Previously, glucocorticoids and immunosuppressive drugs, such as methotrexate or azathioprine, were usually used as treatment. In a phase III TAKT study, 19 patients with relapsed TA randomly received weekly TCZ (subcutaneous 162 mg) or placebo treatment. Although TCZ did not meet the defined primary endpoint for time to relapse TA, the hazard ratio for time to relapse was 0.41 (95.4% CI 0.15–1.10; p = 0.0596), suggesting a favorable effect of TCZ over the placebo [112]. A retrospective study of 46 patients with TA also showed the effectiveness of TCZ through significantly better event-free survival of patients who received TCZ compared with disease-modifying anti-rheumatic drugs (DMARDs) [113]. Japan approved TCZ for the treatment of TA in 2017. However, TA is known to frequently complicate inflammatory bowel disease [114]. Patients with inflammatory bowel disease should receive careful attention when receiving IL-6 inhibition therapy, because IL-6 has a protective effect of the intestinal epithelium.
5.2.2. Cytokine Releasing Syndrome (CRS)
Treatment with TCZ is expected to be effective not only for chronic inflammatory diseases but also for acute inflammatory diseases. T cell-engaged therapy is a promising strategy for leukemia and lymphoma. In chimeric antigen receptor-modified T cell (CAR-T) therapy, for example, CD19-CAR-T is the engineered T cell that expresses an anti-CD19 single-chain variable fragment plus T cell receptor zeta and CD28 signaling domain. The CD19-CAR-T cell attacks B cell leukemia/lymphoma cells with B cell antigen CD19. Another method involves the CD19/CD3-bi-specific antibody (blinatumomab) that links T cells with CD19 cells. These therapies lead to a direct cytotoxic effect of T cells on target tumor cells. However, activated T cells produce massive amounts of IL-6, IL-8, IL-10, MCP-1 and IFNγ and induce an acute systemic inflammatory response, known as a cytokine storm or CRS. Severe cases of CRS are accompanied by life-threatening manifestations such as febrile neutropenia, hypotension, acute vascular leakage syndrome and respiratory distress syndrome. However, a case report showed that IL-6 inhibition with TCZ promptly reversed CRS [115]. A subsequent report showed that 18 of 39 patients with relapsed acute lymphoblastic leukemia who were treated with CAR-T developed grade 3–4 CRS but treatment with TCZ for 13 of them resulted in rapid improvement [116]. The drastic effect of TCZ on CRS opened up the possibility for the use of IL-6 blockade as a new strategy for an emergent fatal condition associated with acute cytokine storm. The USA approved TCZ for the treatment of CRS in 2017.
5.3. Diseases in Which the Efficacy of IL-6 Inhibition Was Demonstrated in Meta-Analysis or Phase II or III Clinical Trial.
5.3.1. Adult-Onset Still’s Disease (AOSD)
The effectiveness of TCZ on some intractable diseases has been supported by a double-blind placebo-controlled randomized study and by numerous cases reporting its favorable effects. AOSD is an inflammatory syndrome involving fever, skin rash, hepatosplenomegaly and arthritis which, due to its similar symptoms, is thought to be the adult form of sJIA. It is characterized by elevated levels of inflammatory cytokines in serum including IL-6, IL-18, IL-1, TNFα and IFNγ. An increasing number of case reports or series has shown the effectiveness of TCZ on AOSD and reduction of glucocorticoid dose [117,118,119]. A meta-analysis of 10 studies with 147 patients showed that TCZ therapy achieved a partial remission rate in 85.38% and complete remission rate in 77.91%, and even in patients with refractory cases, the remission rate was 87.92% [120], suggesting a promising effect of TCZ on AOSD.
5.3.2. Polymyalgia Rheumatica (PMR)
PMR is an inflammatory disease involving stiffness of the shoulders and pelvic girdles that occurs in people older than 50 years and is often associated with GCA. Usually, glucocorticoids are effective for treating PMR but some patients follow a chronic course and have glucocorticoid side effects. In a prospective study of 20 patients with active and recent onset PMR, all patients showed clinical improvement at 12 weeks after three TCZ (8 mg/kg) infusions at 4-week intervals [121]. A phase IIa trial involving newly diagnosed PMR patients also demonstrated the beneficial effects of TCZ on PMR [122]. All nine patients treated with intravenous TCZ (8 mg/kg) with a rapid tapering of glucocorticoids showed relapse-free remission without glucocorticoids after six months. Together, these reports suggest that TCZ is effective and reduces cumulative glucocorticoid doses in patients with PMR. Phase III clinical trial is being conducted (ClinicaTrials.gov NCT03263715).
5.3.3. Systemic Sclerosis (SSc)
SSc is a refractory connective tissue disease characterized by tissue fibrosis that affects the skin, blood vessels, heart, lungs, kidneys and gastrointestinal tract. Currently, no adequate treatment has been developed to stop the progression of SSc; thus, therapeutic development is required [123]. The effects of IL-6 on keratinocyte proliferation promotion [124], collagen generation in dermal fibroblasts and differentiation from dermal fibroblasts into myofibroblasts may account for sclerotic changes in the skin of SSc patients [125]. The effect of IL-6 inhibition on sclerotic skin has been confirmed in bleomycin-induced scleroderma model mice [126]. Case reports of improvements in skin lesions and joint range of motion in SSc patients led to the conduction of a phase II study of 87 SSc patients [127,128]. The study showed that skin scores were not significantly improved following weekly subcutaneous treatment with TCZ but the percentage decrease in the predicted forced vital capacity at 48 weeks was significantly reduced compared to the control group [129]. Pulmonary symptoms associated with SSc are often refractory, thus confirmation of the validity of TCZ treatment is required. TCZ was reported to decrease skin scores in patients with short disease duration, elevated CRP and low IL-33 and CCL5, suggesting that certain types of SSc tend to respond to TCZ well [130]. A particular condition may have to be selected to obtain the effect of TCZ on the skin lesions of SSc. The results of a phase III trial (NCT02453256) are awaited.
5.3.4. Graves’ Ophthalmopathy
There have been reports that TCZ may be an effective treatment for ophthalmic diseases. The clinical manifestations of Graves’ ophthalmopathy include upper eyelid retraction, edema and proptosis. Autoimmune inflammation of extraocular muscles and orbital fat is usually associated with hyperthyroidism [131]. Favorable effects of TCZ on Graves’ ophthalmopathy have been shown in some reports [132]. A randomized phase III clinical trial in 32 patients with glucocorticoid resistant Graves’ ophthalmopathy showed improved clinical activity scores and significant decreases in exophthalmos size following treatment with TCZ compared to a placebo [133]. A decrease in thyroid-stimulating immunoglobulin has also been reported in patients with thyroid-associated ophthalmopathy [134].
5.3.5. Myocardial Infarction
IL-6 blockade therapy is also expected to work effectively against noninfectious local inflammation. IL-6 rapidly increases in response to ischemia in acute myocardial infarction [135]. In addition to CRP and amyloid A, IL-6 is associated with myocardial injury and mortality in acute coronary syndrome, which includes unstable angina and myocardial infarction [136]. This suggests that IL-6 contributes to the pathology of myocardial infarction. A double-blind randomized placebo-controlled phase II trial with 117 patients with non-ST-elevated myocardial infarction showed that a single 280 mg dose of TCZ significantly decreased the release of troponin T in patients who received TCZ within two days of symptom appearance and who were treated with coronary intervention [137]. The single dose of 280 mg used is much lower than that usually used for RA treatment, so further study is needed to clarify the effect of TCZ on myocardial infarction. A phase II clinical trial is being conducted to assess the effects of TCZ on myocardial infarction (NCT03004703).
5.3.6. Depression
Many reports indicate a relationship between inflammatory cytokines and depression. In particular, TNFα and IL-6 have been associated with antidepressant-resistant depression [138]. Currently, it is thought that the next wave of antidepressants will target the immune system [139]. The first clinical trial of anti-cytokine Ab on depression was performed with TNFα inhibitors. A double-blind, placebo-controlled, randomized trial with anti-TNFα Ab infliximab in 60 patients with major depression showed an association between baseline levels of the inflammatory marker CRP and the response to infliximab [140]. Subsequently, a phase II double-blind placebo-controlled trial on depressive symptoms analyzed the effect of anti-IL-6 Ab sirukumab on RA and siltuximab on Castleman disease. Patients were grouped by the presence or absence of a prevalent depressed mood and anhedonia. An analysis of SF-36 questionnaire items on depressed mood and anhedonia indicated that significantly greater improvements in depressive symptoms were achieved with anti-IL-6 Abs compared with a placebo [141]. Another report, in which conventional synthetic DMARDs (csDMARDs) and biological DMARDs (bDMARDs) were indirectly compared using a network meta-analysis, demonstrated that bDMARDs performed better than csDMARDs for improving mental health. Among bDMARDs, IL-6 inhibition had a 90% probability of being the most effective for improving mental health outcomes [142]. Trials of IL-6 inhibition are being conducted in patients with depressive disorder using anti-IL-6 Ab sirukumab phase II (NCT02473289) and anti-IL-6R Ab TCZ phase II (NCT02660528).
5.4. Diseases in Which a Substantial Case Reports Showing TCZ Efficacy
5.4.1. Amyloid A Amyloidosis
Clinical case reports describing the effectiveness of IL-6 inhibition on various diseases are accumulating. IL-6 is required for the production of the amyloid A protein in the liver; thus, IL-6 inhibition is thought to be effective for amyloid A amyloidosis. A typical case report showed that TCZ administration to patients with intestinal amyloidosis stopped diarrhea early and removed amyloid A deposition after three months of treatment [143]. Significantly superior effects of TCZ compared to those of anti-TNF agents were shown in a Japanese nationwide survey of 199 patients with amyloid A amyloidosis, which included RA (60.3%), uncharacterized inflammatory disorders (11.1%), neoplasms (7.0%) and other rheumatic diseases (6.5%) [144]. Case reports have supported TCZ treatment improved renal function and decreased frequency of fever attacks in patients with familial Mediterranean fever accompanied by amyloid A amyloidosis [145].
5.4.2. Neuromyelitis Optica (NMO)
The efficacy of IL-6 inhibitory therapy has also been reported in intractable neurological diseases. NMO is a chronic demyelinating disease that is characterized by autoantibody against the astrocyte water channel protein aquaporin-4 (AQP-4). Autoantibodies induce the activation of complements, demyelination and necrosis in the optic nerve and spinal cord [146]. In vitro study showed that IL-6 enhanced the survival of plasmablasts and their anti-AQP-4 Ab secretion, whereas TCZ inhibited the survival of plasmablasts and the production of anti-AQP-4 Ab [147]. A pilot study of TCZ in seven refractory patients with NMO showed a marked decrease in the relapse rate and intractable pain [148], leading to a subsequent clinical phase II trials (NCT03350633, NCT03062579) and phase III trials (NCT02073279, NCT02028884).
5.4.3. Behçet’s Disease
Behçet’s disease is characterized by the symptoms of oral aphtha, genital ulcers, skin lesions and uveitis. A glucocorticoid and immunosuppressant sparing effect of TCZ was reported for seven patients with refractory vasculo-Behçet’s disease [149]. In addition, case reports have suggested that TCZ has favorable effects on neuro-Behçet’s disease characterized by elevated levels of IL-6 in cerebrospinal fluid [150,151]. There have been several case reports in which TCZ showed effectiveness on refractory uveitis induced by Behçet’s disease [152], noninfectious cases [153], or JIA [154,155]. A phase II trial in Behçet’s uveitis is currently underway (NCT03554161).
5.4.4. Systemic Lupus Erythematosus (SLE)
SLE is a typical autoimmune disease involving the activation of T and B cells that, despite few previous reports, is expected to be affected by IL-6 inhibition. In an open trial of 16 patients with SLE, TCZ improved the disease activity score in eight of the 15 evaluable patients. Arthritis symptoms improved in all seven patients with the condition. Anti-dsDNA Ab levels decreased by 47% in patients treated with 4 or 8 mg/kg of TCZ, whereas a 7.8% decrease in IgG levels was observed, suggesting the suppressive effect of TCZ on autoantibody-producing cells [156]. An analysis of circulating T and B cell subsets in patients with SLE showed that TCZ decreased the frequency of plasmablasts/plasma cells and memory B cells but increased the concentrations of antigen-inexperienced B cells and naïve T cells, suggesting the restoration of B and T cell homeostasis [157]. A case report also indicated that TCZ was effective for recurrent pleural and pericardial effusion in patients with SLE [158]. Further clinical studies are required to confirm the efficacy of TCZ on SLE.
5.4.5. Relapsing Polychondritis (RP)
RP is an orphan disease involving refractory inflammation in cartilage with no standardized treatment. Responses of RP patients to TCZ treatment have varied; in one case report, a sustained response was shown by two patients [159] and others have indicated its effectiveness [160,161,162,163] but one report showed it to be ineffective [164]. A French multicenter retrospective cohort study with 41 patients with RP who were exposed to 105 biologics reported the efficacy of TCZ (n = 17) as well as TNFα inhibitors (n = 60) but a low number of complete responses and concerns about the risk of infection were reported [165].
5.4.6. Inflammatory Myopathy
Inflammatory myopathies are characterized by autoantibody or immune cell-mediated responses. Only a limited number of cases have reported the effectiveness of TCZ for these conditions, including polymyositis [166], dermatomyositis [167] and anti-synthetase syndrome [168]. Among these diseases, a phase II clinical trial for polymyositis and dermatomyositis is underway (NCT02043548).
5.4.7. Graft-Versus-Host Disease (GVHD)
GVHD after hematopoietic cell transplantation sometimes shows refractory manifestations. A retrospective evaluation involving the administration of TCZ (8 mg/kg) every two weeks in 16 patients with severe steroid-refractory acute GVHD of the lower gastrointestinal tract reported that ten patients achieved complete response after a median time of 11 days (range 2 to 28 days) [169]. A phase II trial in acute GVHD is currently being conducted (NCT01475162). GVHD with acquired hemophilia A is characterized by the presence of autoantibodies against coagulation factor VIII after hematopoietic cell transplantation. A case report showed that TCZ treatment was effective for GVHD with acquired hemophilia A [170].
5.5. Diseases in Which a Few Case Reports Showing TCZ Efficacy
Case reports have shown TCZ to be effective for a variety of other diseases, including autoimmune hemolytic anemia [171,172], reactive arthritis [173], autoimmune encephalitis [174,175], myasthenia gravis [176], Blau syndrome [177], anti-Caspre2 syndrome [178], refractory organizing pneumonia associated with Sjögren’s syndrome [179], cancer-related cachexia [180] and steroid refractory immune-mediated adverse events secondary to immune check point inhibitors [181]. Besides large vessel vasculitis, several case reports have indicated that TCZ has effects on rheumatoid vasculitis [182], polyarteritis nodosa [183,184], microscopic polyangiitis [185,186] and cryoglobulinemia vasculitis [187]. A report showed that epidural administration of TCZ reduced pain due to sciatica with lumbar spinal stenosis [188]. Although these case reports showed favorable results for TCZ, the effects have not yet been confirmed by randomized clinical trials. Further clinical studies are required to fully support the use of IL-6 inhibition in the daily clinical practice. Among patients with low frequency diseases, the number of patients who are refractory to conventional therapy is even smaller, sometimes complicating the obtainment of statistical significance in clinical trials, so organized multi-center trials are needed.
6. Safety of TCZ
Given the various roles of IL-6 in the immune system, concerns had been raised about a possible increase in the rate of serious infection associated with an IL-6 blocking strategy. Risk assessment of IL-6 inhibition has mainly been based on experience of TCZ treatment for RA patients. The safety information of TCZ was first reported from Japan [189]. The meta-analysis with 2188 patient-years (PYs) exposure reported the rate of serious infections was 6.22/100PYs. Abnormalities in the laboratory test such as increase in lipid parameters and liver enzymes but most were mild. The ACT-SURE study reported 5.1 serious infections per 100 PYs following TCZ treatment in 1680 patients with RA, which is similar to the rate previously observed for the anti-TNFα antibody adalimumab [190]. The multicenter retrospective cohort in Japan showed comparative risk of hospitalized infection among biologics infliximab (hazard ratio was 1.54, 95% confidence interval (CI): 0.78–3.04), adalimumab (1.72, 95% CI: 0.88–3.34), abatacept (1.11, 95% CI: 0.55–2.21) and tocilizumab (1.02, 95% CI: 0.55–1.87) compared with etanercept in the study with 1596 RA patients. However, RA functional class 3/4 (1.92, 1.20–3.09), body mass index < 18.5 (2.55, 95% CI: 1.57–4.14), prednisolone more than 7.5 mg/day (3.56, 95% CI: 2.15–5.88), prednisolone from 5 mg to 7.5 mg/day (1.88, 95% CI: 1.10–3.22) and chronic lung disease (1.85, 95% CI: 1.17–2.92) contributed more to the risk of hospitalized infection than biologics [191].
Decrease of neutrophil counts have been reported in trial of TCZ in RA patients, however TCZ-induced neutropenia does not appear to be associated with serious infection [192]. It is thought that the decrease of neutrophil by TCZ is not due to myelotoxic effect but due to the increase of margination of circulating neutrophils into the bone marrow [50]. Considering the protective effect of IL-6 on epithelial cells, concerns have been expressed about complications in the intestinal mucosa due to IL-6 inhibition. In the German Biologics Register, IL-6 inhibition by TCZ led to an increase in the incidence of lower intestinal perforation with a hazard ratio of 4.48 (95% CI: 2.0–10.0) in RA patients [193]. Patients with a past history of diverticulitis need to be careful with IL-6 inhibition therapy.
The largest surveillance study of TCZ was performed for 28 weeks in 7901 patients in Japan and reported a rate of serious adverse events of 9.6%, with the most common event being infection (3.8%) [194]. However, the standardized mortality ratio 1.15 (95% CI: 0.83–1.61) did not exceed the ratio between 1.46 (95% CI: 1.32–1.60) and 1.90 (95% CI: 1.75–2.07) previously reported in a Japanese RA cohort. Further, long-term observations of 4527 patients over three years indicated no increases in the proportions of patients with fatal events, serious infection, or malignancy [195].
7. Diseases Where IL-6 Inhibition Is Predicted to Be Effective
Considering the reported effective cases of TCZ to date, the diseases for which IL-6 inhibition might have beneficial role can be predicted. First, IL-6 inhibition is expected to affect diseases that can be pathologically related to long-term IL-6 action, such as Castleman disease and amyloid A amyloidosis. Secondly, diseases for which the IL-6 level is correlated with disease activity may be ameliorated by IL-6 inhibition, as evidenced by Castleman disease, RA, sJIA and large vessel vasculitis. However, IL-6 is involved in liver regeneration and intestinal epithelium protection, so it is possible that elevated levels of IL-6 protect organs from damage or promote recovery from dysfunction. For example, a pilot study involving TCZ treatment in Crohn’s disease is of interest, as a report stated that TCZ caused different outcomes in systemic inflammation and intestinal lesions. The acute phase inflammatory response of Crohn’s disease was completely suppressed by TCZ but endoscopic and histological examinations revealed no changes [196]. The beneficial role of IL-6 inhibition is not always certain, even if the IL-6 level is correlated with disease activity, so careful selection is necessary to decide which disease should be treated. Third, IL-6 inhibition may be useful for diseases caused by autoantibodies. Some case reports of TCZ in NMO and SLE have shown decreases in autoantibody levels, such as anti-AQP-4 Ab or anti-dsDNA Ab. A decrease in autoantibody levels over a reduction in total IgG levels suggests that IL-6 is deeply involved in autoantibody production. Several case reports have shown that TCZ acts effectively on autoantibody-mediated diseases, such as autoimmune hemolytic anemia, myasthenia gravis, acquired hemophilia A and anti-Caspre2 syndrome. Fourth, IL-6 inhibition may be effective for the prevention of cardiovascular disease, since IL-6 elevates platelet count, fibrinogen concentration and activity of coagulation. Atherothrombosis is initiated and progressed by inflammation [197]. Evidence has also shown that high levels of IL-6 are associated with cardiovascular disease risk [87,88]. A meta-analysis showed that the risk of coronary heart disease is reduced by 3.4% for every copy of IL-6R with Asp358Ala inherited, suggesting a relationship between IL-6R and coronary heart disease [198]. Fifth, IL-6 inhibition therapy may be an effective treatment for certain cancers in which IL-6 promotes proliferation [199]. When the growth of cancer depends on IL-6 autocrine signaling, the IL-6 signal may be mediated by a mode of intracellular signaling and inhibitors of IL-6 signal transduction such as JAK inhibitors are possibly effective [34].
8. Conclusions
IL-6 inhibitory therapy is applied worldwide as treatment for RA and JIA and is now being used for the treatment of large vessel vasculitis. IL-6 inhibitory therapy is expected to be used for other inflammatory diseases given its strong inflammation suppressing effect and safety profile. Various biologics that interact with the extracellular domain of the IL-6 receptor complex components and low molecular weight compounds that interact with intracellular signaling molecules are being developed one after the other [200]. A new type of IL-6 inhibitor, the sgp130-Fc fusion protein, that selectively inhibits trans-signaling has also been developed [76]. Options for inhibiting IL-6 action will further increase in the future. The growing effectiveness of IL-6 inhibition therapy against intractable diseases is a reminder of the history of glucocorticoids. The inflammatory suppressive effect of glucocorticoids was revealed when they were administered to patients with RA. Since then, glucocorticoids have been used for a large number of noninfectious inflammatory diseases; however, long-term use of glucocorticoids is accompanied by various side effects and careful attention should be paid in clinical practice. Given the involvement of IL-6 in many pathologies, the diseases that can be improved and treated by IL-6 inhibition therapy will expand. However, the important role of IL-6 in host defense should always be kept in mind.
Author Contributions
Investigation and Writing, M.N.; Review and Editing, T.K.
Funding
This work was supported by JSPS KAKENHI Grand Number 17K10002 to M.N.
Conflicts of Interest
M.N. has received lecture fee from AbbVie Inc., Eisai Co. Ltd., Pfizer Inc. and Mitsubishi Tanabe Pharma Co. T.K. holds a patent for tocilizumab and has received royalties for Actemra.
References
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Masuda, K.; Kishimoto, T. Regulation of IL-6 in Immunity and Diseases. Adv. Exp. Med. Biol. 2016, 941, 79–88. [Google Scholar] [PubMed]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels and an association with systemic-onset juvenile chronic arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Wang, C.; Sun, F.; Zhao, L.; Dun, A.; Sun, Z. Association of interleukin-6 gene polymorphism with coronary artery disease: An updated systematic review and cumulative meta-analysis. Inflamm. Res. 2015, 64, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Poplutz, M.K.; Wessels, I.; Rink, L.; Uciechowski, P. Regulation of the Interleukin-6 gene expression during monocytic differentiation of HL-60 cells by chromatin remodeling and methylation. Immunobiology 2014, 219, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kim, J.Y.; Kim, K.W.; Park, M.K.; Moon, Y.; Kim, W.U.; Kim, H.Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120–R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Taga, T.; Akira, S. Cytokine signal transduction. Cell 1994, 76, 253–262. [Google Scholar] [CrossRef]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Ripley, B.; Nishimura, R.; Mino, T.; Takeuchi, O.; Shioi, G.; Kiyonari, H.; Kishimoto, T. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 9409–9414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higa, M.; Oka, M.; Fujihara, Y.; Masuda, K.; Yoneda, Y.; Kishimoto, T. Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc. Natl. Acad. Sci. USA 2018, 115, E1214–E1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Kishimoto, T. A potential therapeutic target RNA-binding protein, Arid5a for the treatment of inflammatory disease associated with aberrant cytokine expression. Curr. Pharm. Des. 2018. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Witthuhn, B.A.; Yoshida, K.; Silvennoinen, O.; Yasukawa, K.; Ihle, J.N.; Kishimoto, T.; Taga, T. Activation of JAK2 kinase mediated by the interleukin 6 signal transducer gp130. Proc. Natl. Acad. Sci. USA 1994, 91, 2285–2289. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Akira, S.; Narazaki, M.; Taga, T. Interleukin-6 family of cytokines and gp130. Blood 1995, 86, 1243–1254. [Google Scholar] [PubMed]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.E., Jr.; Yancopoulos, G.D. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, S.; Ikegame, K.; Ueno, K.; Narazaki, M.; Naka, T.; Yamamoto, H.; Matsumoto, T.; Saito, H.; Hosoe, S.; Kishimoto, T. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 1997, 237, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Fujimoto, M.; Matsumoto, T.; Morita, Y.; Saito, H.; Kajita, T.; Yoshizaki, K.; Naka, T.; Kishimoto, T. Three distinct domains of SSI-1/SOCS-1/JAB protein are required for its suppression of interleukin 6 signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 13130–13134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, S.E.; De Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Ripley, B.; Nyati, K.K.; Dubey, P.K.; Zaman, M.M.; Hanieh, H.; Higa, M.; Yamashita, K.; Standley, D.M.; Mashima, T.; et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J. Exp. Med. 2016, 213, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Janner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [PubMed]
- Bottcher, J.P.; Schanz, O.; Garbers, C.; Zaremba, A.; Hegenbarth, S.; Kurts, C.; Beyer, M.; Schultze, J.L.; Kastenmuller, W.; Rose-John, S.; et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 2014, 8, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Moller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Verboogen, D.R.J.; Revelo, N.H.; Ter Beest, M.; van den Bogaart, G. Interleukin-6 secretion is limited by self-signaling in endosomes. J. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Narazaki, M.; Yasukawa, K.; Saito, T.; Miki, D.; Hamaguchi, M.; Davis, S.; Shoyab, M.; Yancopoulos, G.D.; Kishimoto, T. Functional inhibition of hematopoietic and neurotrophic cytokines by blocking the interleukin 6 signal transducer gp130. Proc. Natl. Acad. Sci. USA 1992, 89, 10998–11001. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Ogata, A.; Kishimoto, T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin. Immunol. 2014, 26, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Kohler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Yoshizaki, K.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Hashimoto, J.; Azuma, J.; Kishimoto, T. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: A multicenter, double-blind, placebo-controlled trial. Arthritis. Rheum. 2004, 50, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbers, C.; Monhasery, N.; Aparicio-Siegmund, S.; Lokau, J.; Baran, P.; Nowell, M.A.; Jones, S.A.; Rose-John, S.; Scheller, J. The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta. 2014, 1842, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Naseem, S.; Hussain, T.; Manzoor, S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev. 2018, 39, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Lai, S.L.; Chen, C.N.; Lee, P.H.; Peng, F.C.; Kuo, M.L.; Lai, H.S. IL-6 regulates Mcl-1L expression through the JAK/PI3K/Akt/CREB signaling pathway in hepatocytes: Implication of an anti-apoptotic role during liver regeneration. PLoS ONE 2013, 8, e66268. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Yu, J.; Cheng, A.S.; Wong, G.L.; Chan, H.Y.; Chu, E.S.; Ng, E.K.; Chan, F.K.; Sung, J.J.; Chan, H.L. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int. J. Cancer 2009, 124, 2766–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.T.; Feng, C.L.; Yu, C.J.; Tsai, S.M.; Hsu, P.N.; Chen, Y.L.; Wu, Y.Y. IL-6, through p-STAT3 rather than p-STAT1, activates hepatocarcinogenesis and affects survival of hepatocellular carcinoma patients: A cohort study. BMC Gastroenterol. 2015, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Shabo, Y.; Lotem, J.; Rubinstein, M.; Revel, M.; Clark, S.C.; Wolf, S.F.; Kamen, R.; Sachs, L. The myeloid blood cell differentiation-inducing protein MGI-2A is interleukin-6. Blood 1988, 72, 2070–2073. [Google Scholar] [PubMed]
- Suwa, T.; Hogg, J.C.; English, D.; Van Eeden, S.F. Interleukin-6 induces demargination of intravascular neutrophils and shortens their transit in marrow. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2954–H2960. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Kimura, H.; Uchida, T.; Kariyone, S.; Friese, P.; Burstein, S.A. Human interleukin 6 is a direct promoter of maturation of megakaryocytes in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 5953–5957. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science 2018, 360, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Fielder, P.J.; Gurney, A.L.; Stefanich, E.; Marian, M.; Moore, M.W.; Carver-Moore, K.; de Sauvage, F.J. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood 1996, 87, 2154–2161. [Google Scholar] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105, S13–S33. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Wright, H.L.; Cross, A.L.; Edwards, S.W.; Moots, R.J. Effects of IL-6 and IL-6 blockade on neutrophil function in vitro and in vivo. Rheumatology (Oxford) 2014, 53, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, M.; Higuchi, Y.; Uchiyama, Y.; Mihara, M. IL-6 plays an essential role in neutrophilia under inflammation. Cytokine 2011, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, S.; Saeki, Y.; Mima, T.; Sasai, M.; Nishioka, K.; Nomura, S.; Kopf, M.; Katada, Y.; Tanaka, T.; Suemura, M.; et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8222–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.W.; Chen, M.W.; Hsiao, M.; Wang, S.; Chen, C.A.; Hsiao, S.M.; Chang, J.S.; Lai, T.C.; Rose-John, S.; Kuo, M.L.; et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2011, 71, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [PubMed]
- Arima, K.; Origuchi, T.; Tamai, M.; Iwanaga, N.; Izumi, Y.; Huang, M.; Tanaka, F.; Kamachi, M.; Aratake, K.; Nakamura, H.; et al. RS3PE syndrome presenting as vascular endothelial growth factor associated disorder. Ann. Rheum. Dis. 2005, 64, 1653–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, Y.; Kawabata, H.; Takai, K.; Kojima, M.; Tsukamoto, N.; Ishigaki, Y.; Kurose, N.; Ide, M.; Murakami, J.; Nara, K.; et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int. J. Hematol. 2016, 103, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinathan, G.; Milagre, C.; Pearce, O.M.; Reynolds, L.E.; Hodivala-Dilke, K.; Leinster, D.A.; Zhong, H.; Hollingsworth, R.E.; Thompson, R.; Whiteford, J.R.; et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res. 2015, 75, 3098–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prystaz, K.; Kaiser, K.; Kovtun, A.; Haffner-Luntzer, M.; Fischer, V.; Rapp, A.E.; Liedert, A.; Strauss, G.; Waetzig, G.H.; Rose-John, S.; et al. Distinct Effects of IL-6 Classic and Trans-Signaling in Bone Fracture Healing. Am. J. Pathol. 2018, 188, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford) 2008, 47, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Kikuta, J.; Kishi, Y.; Hasegawa, T.; Okuzaki, D.; Hirano, T.; Minoshima, M.; Kikuchi, K.; Kumanogoh, A.; Ishii, M. In vivo visualisation of different modes of action of biological DMARDs inhibiting osteoclastic bone resorption. Ann. Rheum. Dis. 2018, 77, 1219–1225. [Google Scholar] [PubMed]
- De Benedetti, F.; Brunner, H.; Ruperto, N.; Schneider, R.; Xavier, R.; Allen, R.; Brown, D.E.; Chaitow, J.; Pardeo, M.; Espada, G.; et al. Catch-up growth during tocilizumab therapy for systemic juvenile idiopathic arthritis: Results from a phase III trial. Arthritis Rheumatol. 2015, 67, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.J.; Ott, I.; Marx, N.; Luther, T.; Kenngott, S.; Gawaz, M.; Kotzsch, M.; Schomig, A. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb. Vasc. Biol. 1997, 17, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Stouthard, J.M.; Levi, M.; Hack, C.E.; Veenhof, C.H.; Romijn, H.A.; Sauerwein, H.P.; van der Poll, T. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb. Haemost. 1996, 76, 738–742. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, E.; Friederich, P.W.; Vlasuk, G.P.; Rote, W.E.; Vroom, M.B.; Levi, M.; van der Poll, T. Activation of coagulation by administration of recombinant factor VIIa elicits interleukin 6 (IL-6) and IL-8 release in healthy human subjects. Clin. Diagn. Lab. Immunol. 2003, 10, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.M.; Spehlmann, M.E.; Hammond, D.C.; Iimura, M.; Hase, K.; Choi, L.J.; Hanson, E.; Eckmann, L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008, 180, 6816–6826. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Leday, G.G.R.; Vertes, P.E.; Richardson, S.; Greene, J.R.; Regan, T.; Khan, S.; Henderson, R.; Freeman, T.C.; Pariante, C.M.; Harrison, N.A.; et al. Replicable and Coupled Changes in Innate and Adaptive Immune Gene Expression in Two Case-Control Studies of Blood Microarrays in Major Depressive Disorder. Biol. Psychiatry 2018, 83, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Anderson, G.; Kubera, M.; Berk, M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin. Ther. Targets 2014, 18, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Chourbaji, S.; Urani, A.; Inta, I.; Sanchis-Segura, C.; Brandwein, C.; Zink, M.; Schwaninger, M.; Gass, P. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiol. Dis. 2006, 23, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Sakic, B.; Gauldie, J.; Denburg, J.A.; Szechtman, H. Behavioral effects of infection with IL-6 adenovector. Brain Behav. Immun. 2001, 15, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Sasai, M.; Shima, Y.; Nakagawa, M.; Matsumoto, T.; Shirai, T.; Kishimoto, T.; Yoshizaki, K. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000, 95, 56–61. [Google Scholar] [PubMed]
- Choy, E.H.; Isenberg, D.A.; Garrood, T.; Farrow, S.; Ioannou, Y.; Bird, H.; Cheung, N.; Williams, B.; Hazleman, B.; Price, R.; et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: A randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002, 46, 3143–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Ogata, A.; Narazaki, M. Tocilizumab for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2010, 6, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Sebba, A.; Gu, J.; Lowenstein, M.B.; Calvo, A.; Gomez-Reino, J.J.; Siri, D.A.; Tomsic, M.; Alecock, E.; Woodworth, T.; et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: The AMBITION study. Ann. Rheum. Dis. 2010, 69, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Emery, P.; van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): A randomised, double-blind, controlled phase 4 trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef]
- Bijlsma, J.W.J.; Welsing, P.M.J.; Woodworth, T.G.; Middelink, L.M.; Petho-Schramm, A.; Bernasconi, C.; Borm, M.E.A.; Wortel, C.H.; Ter Borg, E.J.; Jahangier, Z.N.; et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): A multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet 2016, 388, 343–355. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; van Hoogstraten, H.; Garg, A.; Fan, C.; et al. Sarilumab Plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmester, G.R.; Lin, Y.; Patel, R.; van Adelsberg, J.; Mangan, E.K.; Graham, N.M.; van Hoogstraten, H.; Bauer, D.; Ignacio Vargas, J.; Lee, E.B. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): A randomised, double-blind, parallel-group phase III trial. Ann. Rheum. Dis. 2017, 76, 840–847. [Google Scholar] [CrossRef] [PubMed]
- de Benedetti, F.; Massa, M.; Robbioni, P.; Ravelli, A.; Burgio, G.R.; Martini, A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Imagawa, T.; Mori, M.; Miyamae, T.; Aihara, Y.; Takei, S.; Iwata, N.; Umebayashi, H.; Murata, T.; Miyoshi, M.; et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 2008, 371, 998–1006. [Google Scholar] [CrossRef]
- Miyamae, T.; Yokoya, S.; Yamanaka, H.; Yokota, S. Effect of tocilizumab on growth impairment in systemic juvenile idiopathic arthritis with long-term corticosteroid therapy. Mod. Rheumatol. 2014, 24, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, Y.; Isobe, M.; Takei, S.; Tanaka, Y.; Ishii, T.; Yokota, S.; Nomura, A.; Yoshida, S.; Nishimoto, N. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: Results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann. Rheum. Dis. 2018, 77, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Resche-Rigon, M.; Comarmond, C.; Soriano, A.; Constans, J.; Alric, L.; Jego, P.; Busato, F.; Cabon, M.; Dhote, R.; et al. Efficacy of tocilizumab in Takayasu arteritis: Multicenter retrospective study of 46 patients. J. Autoimmun. 2018, 91, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Matsumura, T.; Yoshifuji, H.; Kirino, Y.; Maejima, Y.; Nakaoka, Y.; Takahashi, M.; Amiya, E.; Tamura, N.; Nakajima, T.; et al. Takayasu arteritis and ulcerative colitis: High rate of co-occurrence and genetic overlap. Arthritis Rheumatol. 2015, 67, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, E.; Yamashita, H.; Kaneko, S.; Ueda, Y.; Ozaki, T.; Tsuchiya, H.; Takahashi, Y.; Kaneko, H.; Kano, T.; Mimori, A. Successful tocilizumab therapy in seven patients with refractory adult-onset Still’s disease. Mod. Rheumatol. 2016, 26, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gu, L.; Wang, X.; Guo, L.; Shi, H.; Yang, C.; Chen, S. A Pilot Study on Tocilizumab for Treating Refractory Adult-Onset Still’s Disease. Sci. Rep. 2017, 7, 13477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, H.; Tsuboi, H.; Yagishita, M.; Terasaki, T.; Terasaki, M.; Shimizu, M.; Honda, F.; Ohyama, A.; Takahashi, H.; Miki, H.; et al. Severe Adult-onset Still Disease with Constrictive Pericarditis and Pleuritis That Was Successfully Treated with Tocilizumab in Addition to Corticosteroids and Cyclosporin A. Intern. Med. 2018, 57, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wu, M.; Zhang, X.; Xia, Q.; Yang, J.; Xu, S.; Pan, F. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: A meta-analysis. Mod. Rheumatol. 2018, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Berthelot, J.M.; Cornec, D.; Renaudineau, Y.; Marhadour, T.; Jousse-Joulin, S.; Querellou, S.; Garrigues, F.; De Bandt, M.; Gouillou, M.; et al. Efficacy of first-line tocilizumab therapy in early polymyalgia rheumatica: A prospective longitudinal study. Ann. Rheum. Dis. 2016, 75, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Lally, L.; Forbess, L.; Hatzis, C.; Spiera, R. Brief Report: A Prospective Open-Label Phase IIa Trial of Tocilizumab in the Treatment of Polymyalgia Rheumatica. Arthritis Rheumatol. 2016, 68, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.M.; Krueger, J.; Yourish, D.; Granelli-Piperno, A.; Murphy, D.P.; May, L.T.; Kupper, T.S.; Sehgal, P.B.; Gottlieb, A.B. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 6367–6371. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Berman, B. Stimulation of collagen and glycosaminoglycan production in cultured human adult dermal fibroblasts by recombinant human interleukin 6. J. Investig. Dermatol. 1991, 97, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kitaba, S.; Murota, H.; Terao, M.; Azukizawa, H.; Terabe, F.; Shima, Y.; Fujimoto, M.; Tanaka, T.; Naka, T.; Kishimoto, T.; et al. Blockade of interleukin-6 receptor alleviates disease in mouse model of scleroderma. Am. J. Pathol. 2012, 180, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Kuwahara, Y.; Murota, H.; Kitaba, S.; Kawai, M.; Hirano, T.; Arimitsu, J.; Narazaki, M.; Hagihara, K.; Ogata, A.; et al. The skin of patients with systemic sclerosis softened during the treatment with anti-IL-6 receptor antibody tocilizumab. Rheumatology (Oxford) 2010, 49, 2408–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, Y.; Hosen, N.; Hirano, T.; Arimitsu, J.; Nishida, S.; Hagihara, K.; Narazaki, M.; Ogata, A.; Tanaka, T.; Kishimoto, T.; et al. Expansion of range of joint motion following treatment of systemic sclerosis with tocilizumab. Mod. Rheumatol. 2015, 25, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Shima, Y.; Kawaguchi, Y.; Kuwana, M. Add-on tocilizumab versus conventional treatment for systemic sclerosis and cytokine analysis to identify an endotype to tocilizumab therapy. Mod. Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bahn, R.S. Graves’ ophthalmopathy. N. Engl. J. Med. 2010, 362, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.M. Advances in treatment of active, moderate-to-severe Graves’ ophthalmopathy. Lancet Diabetes Endocrinol. 2017, 5, 134–142. [Google Scholar] [CrossRef]
- Perez-Moreiras, J.V.; Gomez-Reino, J.J.; Maneiro, J.R.; Perez-Pampin, E.; Lopez, A.R.; Rodriguez Alvarez, F.M.; Castillo Laguarta, J.M.; Del Estad Cabello, A.; Sorroche, M.G.; Espana Gregori, E.; et al. Efficacy of tocilizumab in patients with moderate to severe corticosteroid resistant Graves orbitopathy: A randomized clinical trial. Am. J. Ophthalmol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.J.; Wagner, L.H.; Seiff, S.R. Tocilizumab as a steroid sparing agent for the treatment of Graves’ orbitopathy. Am. J. Ophthalmol. Case Rep. 2017, 7, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Interleukin-6 Receptor Mendelian Randomisation Analysis Consortium; Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [PubMed]
- Zamani, P.; Schwartz, G.G.; Olsson, A.G.; Rifai, N.; Bao, W.; Libby, P.; Ganz, P.; Kinlay, S. Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering Study, I. Inflammatory biomarkers, death and recurrent nonfatal coronary events after an acute coronary syndrome in the MIRACL study. J. Am. Heart Assoc. 2013, 2, e003103. [Google Scholar] [CrossRef] [PubMed]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Daguanno, A.W.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.M.; Wommack, E.C.; Felger, J.C.; Miller, A.H. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology 2018, 95, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wetsman, N. Inflammatory illness: Why the next wave of antidepressants may target the immune system. Nat. Med. 2017, 23, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, D.; Salvadore, G.; Hsu, B.; Curran, M.; Casper, C.; Vermeulen, J.; Kent, J.M.; Singh, J.; Drevets, W.C.; et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain Behav. Immun. 2017, 66, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Matcham, F.; Galloway, J.; Hotopf, M.; Roberts, E.; Scott, I.C.; Steer, S.; Norton, S. The Impact of Targeted Rheumatoid Arthritis Pharmacologic Treatment on Mental Health: A Systematic Review and Network Meta-Analysis. Arthritis Rheumatol. 2018, 70, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Hagihara, K.; Shima, Y.; Kawai, M.; Kuwahara, Y.; Arimitsu, J.; Hirano, T.; Narazaki, M.; Ogata, A.; Yoshizaki, K.; et al. Rapid improvement of AA amyloidosis with humanised anti-interleukin 6 receptor antibody treatment. Ann. Rheum. Dis. 2009, 68, 1235–1236. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Yamada, T.; Ueda, M.; Ando, Y. First Nationwide Survey of 199 Patients with Amyloid A Amyloidosis in Japan. Intern. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, S.; Hacioglu, A.; Adibnia, Y.; Hamuryudan, V.; Ozdogan, H. Tocilizumab in the treatment of twelve cases with aa amyloidosis secondary to familial mediterranean fever. Orphanet. J. Rare. Dis. 2017, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Matsuoka, T.; Miyamoto, K.; Kusunoki, S.; Okamoto, T.; Murata, M.; Miyake, S.; Aranami, T.; Yamamura, T. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology 2014, 82, 1302–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Li, C.; Liu, J.; Yu, X.; Wang, Y.; Shi, J.; Li, L.; Zhou, J.; Wang, L.; Chen, H.; et al. Tocilizumab in the treatment of severe and/or refractory vasculo-Behcet’s disease: A single-centre experience in China. Rheumatology (Oxford) 2018. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, P.; Hasler, P.; Kretzschmar, S. Refractory neuro-Behcet treated by tocilizumab: A case report. Clin. Exp. Rheumatol. 2012, 30, S73–S75. [Google Scholar] [PubMed]
- Addimanda, O.; Pipitone, N.; Pazzola, G.; Salvarani, C. Tocilizumab for severe refractory neuro-Behcet: Three cases IL-6 blockade in neuro-Behcet. Semin. Arthritis Rheum. 2015, 44, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ohguro, N.; Hohki, S.; Hagihara, K.; Shima, Y.; Narazaki, M.; Ogata, A.; Yoshizaki, K.; Kumanogoh, A.; Kishimoto, T.; et al. A case of Behcet’s disease treated with a humanized anti-interleukin-6 receptor antibody, tocilizumab. Mod. Rheumatol. 2012, 22, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Sepah, Y.J.; Sadiq, M.A.; Chu, D.S.; Dacey, M.; Gallemore, R.; Dayani, P.; Hanout, M.; Hassan, M.; Afridi, R.; Agarwal, A.; et al. Primary (Month-6) Outcomes of the STOP-Uveitis Study: Evaluating the Safety, Tolerability and Efficacy of Tocilizumab in Patients With Noninfectious Uveitis. Am. J. Ophthalmol. 2017, 183, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Tappeiner, C.; Mesquida, M.; Adan, A.; Anton, J.; Ramanan, A.V.; Carreno, E.; Mackensen, F.; Kotaniemi, K.; de Boer, J.H.; Bou, R.; et al. Evidence for Tocilizumab as a Treatment Option in Refractory Uveitis Associated with Juvenile Idiopathic Arthritis. J. Rheumatol. 2016, 43, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rio, V.; Santos-Gomez, M.; Calvo, I.; Gonzalez-Fernandez, M.I.; Lopez-Montesinos, B.; Mesquida, M.; Adan, A.; Hernandez, M.V.; Maiz, O.; Atanes, A.; et al. Anti-Interleukin-6 Receptor Tocilizumab for Severe Juvenile Idiopathic Arthritis-Associated Uveitis Refractory to Anti-Tumor Necrosis Factor Therapy: A Multicenter Study of Twenty-Five Patients. Arthritis Rheumatol. 2017, 69, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Illei, G.G.; Shirota, Y.; Yarboro, C.H.; Daruwalla, J.; Tackey, E.; Takada, K.; Fleisher, T.; Balow, J.E.; Lipsky, P.E. Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010, 62, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Yarboro, C.; Fischer, R.; Pham, T.H.; Lipsky, P.; Illei, G.G. Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2013, 72, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, V.; Haaland, D.; Legault, K.; Mittoo, S.; Aitken, E. Successful treatment of recurrent pleural and pericardial effusions with tocilizumab in a patient with systemic lupus erythematous. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Hagihara, K.; Hirano, T.; Shima, Y.; Kuwahara, Y.; Arimitsu, J.; Narazaki, M.; Ogata, A.; Kawase, I.; Kishimoto, T.; et al. Sustained response to tocilizumab, anti-interleukin-6 receptor antibody, in two patients with refractory relapsing polychondritis. Rheumatology (Oxford) 2009, 48, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Narshi, C.B.; Allard, S.A. Sustained response to tocilizumab, anti-IL-6 antibody, following anti-TNF-alpha failure in a patient with relapsing polychondritis complicated by aortitis. Rheumatology (Oxford) 2012, 51, 952–953. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Stone, J.H. Refractory relapsing polychondritis treated with serial success with interleukin 6 receptor blockade. J. Rheumatol. 2013, 40, 100–101. [Google Scholar] [CrossRef] [PubMed]
- Stael, R.; Smith, V.; Wittoek, R.; Creytens, D.; Mielants, H. Sustained response to tocilizumab in a patient with relapsing polychondritis with aortic involvement: A case based review. Clin. Rheumatol. 2015, 34, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nishioka, H. Successful treatment with tocilizumab for refractory scleritis associated with relapsing polychondritis. Scand. J. Rheumatol. 2017, 46, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Godfrin-Valnet, M.; Prati, C. Treatment of relapsing polychondritis with tocilizumab. J. Rheumatol. 2013, 40, 1232. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Pugnet, G.; Costedoat-Chalumeau, N.; Mathian, A.; Leroux, G.; Boutemy, J.; Espitia, O.; Bouillet, L.; Berthier, S.; Gaultier, J.B.; et al. Efficacy and safety of biologics in relapsing polychondritis: A French national multicentre study. Ann. Rheum. Dis. 2018, 77, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Hagihara, K.; Shima, Y.; Ogata, A.; Kishimoto, T.; Tanaka, T. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxford) 2011, 50, 1344–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Murakawa, Y.; Matsumura, T.; Matsumoto, O.; Taira, M.; Moriyama, M.; Sumita, Y.; Yamaguchi, S. A case of overlap syndrome successfully treated with tocilizumab: A hopeful treatment strategy for refractory dermatomyositis? Rheumatology (Oxford) 2014, 53, 1907–1908. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.M.; Lilleker, J.B.; Helliwell, P.; Chinoy, H. The successful use of tocilizumab as third-line biologic therapy in a case of refractory anti-synthetase syndrome. Rheumatology (Oxford) 2016, 55, 2277–2278. [Google Scholar] [CrossRef] [PubMed]
- Ganetsky, A.; Frey, N.V.; Hexner, E.O.; Loren, A.W.; Gill, S.I.; Luger, S.M.; Mangan, J.K.; Martin, M.E.; Babushok, D.V.; Drobyski, W.R.; et al. Tocilizumab for the treatment of severe steroid-refractory acute graft-versus-host disease of the lower gastrointestinal tract. Bone Marrow Transplant 2018. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Kawasaki, T.; Kashiwagi, H.; Morishima, A.; Hishitani, Y.; Kawai, M.; Hirano, T.; Ishii, T.; Hagihara, K.; Shima, Y.; et al. Successful treatment of acquired hemophilia A, complicated by chronic GVHD, with tocilizumab. Mod. Rheumatol. 2011, 21, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Kunitomi, A.; Konaka, Y.; Yagita, M.; Nishimoto, N.; Kishimoto, T.; Takatsuki, K. Humanized anti-interleukin 6 receptor antibody induced long-term remission in a patient with life-threatening refractory autoimmune hemolytic anemia. Int. J. Hematol. 2004, 80, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, F.J.; Gonzalez-Leon, R.; Castillo-Palma, M.J.; Ocana-Medina, C.; Sanchez-Roman, J. Tocilizumab for treating refractory haemolytic anaemia in a patient with systemic lupus erythematosus. Rheumatology (Oxford) 2012, 51, 1918–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Kuwahara, Y.; Shima, Y.; Hirano, T.; Kawai, M.; Ogawa, M.; Arimitsu, J.; Hagihara, K.; Narazaki, M.; Ogata, A.; et al. Successful treatment of reactive arthritis with a humanized anti-interleukin-6 receptor antibody, tocilizumab. Arthritis Rheum. 2009, 61, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Lee, S.T.; Moon, J.; Sunwoo, J.S.; Byun, J.I.; Lim, J.A.; Kim, T.J.; Shin, Y.W.; Lee, K.J.; Jun, J.S.; et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics 2016, 13, 824–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randell, R.L.; Adams, A.V.; Van Mater, H. Tocilizumab in Refractory Autoimmune Encephalitis: A Series of Pediatric Cases. Pediatr. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, D.I.; Pirskanen, R.; Piehl, F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscul. Disord. 2017, 27, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Shen, M.; Jiang, D.; Li, Y.; Zheng, X.; Li, Y.; Li, Z.; Zhang, L.; Tang, J.; Guo, Y.; et al. Blau syndrome with good Reponses to Tocilizumab: A case report and focused literature review. Semin. Arthritis. Rheum. 2018, 47, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Krogias, C.; Hoepner, R.; Muller, A.; Schneider-Gold, C.; Schroder, A.; Gold, R. Successful treatment of anti-Caspr2 syndrome by interleukin 6 receptor blockade through tocilizumab. JAMA Neurol. 2013, 70, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Justet, A.; Ottaviani, S.; Dieude, P.; Taille, C. Tocilizumab for refractory organising pneumonia associated with Sjogren’s disease. BMJ Case Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Tetsumoto, S.; Kijima, T.; Kida, H.; Kumagai, T.; Takahashi, R.; Otani, Y.; Inoue, K.; Kuhara, H.; Shimada, K.; et al. Favorable responses to tocilizumab in two patients with cancer-related cachexia. J. Pain Symptom Manag. 2013, 46, e9–e13. [Google Scholar] [CrossRef] [PubMed]
- Stroud, C.R.; Hegde, A.; Cherry, C.; Naqash, A.R.; Sharma, N.; Addepalli, S.; Cherukuri, S.; Parent, T.; Hardin, J.; Walker, P. Tocilizumab for the management of immune mediated adverse events secondary to PD-1 blockade. J. Oncol. Pharm. Pract. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Takeuchi, T.; Sawaki, H.; Imai, T.; Makino, S.; Hanafusa, T. Successful treatment with tocilizumab of pericarditis associated with rheumatoid arthritis. Mod. Rheumatol. 2014, 24, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Rajderkar, D.A.; Modica, R.F. A Case of Polyarteritis Nodosa Associated with Vertebral Artery Vasculitis Treated Successfully with Tocilizumab and Cyclophosphamide. Case. Rep. Pediatr. 2016, 2016, 7987081. [Google Scholar] [CrossRef] [PubMed]
- Saunier, A.; Issa, N.; Vandenhende, M.A.; Morlat, P.; Doutre, M.S.; Bonnet, F. Treatment of polyarteritis nodosa with tocilizumab: A new therapeutic approach? RMD Open 2017, 3, e000446. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Kondo, T.; Kikuchi, J.; Shibata, A.; Chino, K.; Okuyama, A.; Takei, H.; Amano, K. Corticosteroid-free treatment of tocilizumab monotherapy for microscopic polyangiitis: A single-arm, single-center, clinical trial. Mod. Rheumatol. 2016, 26, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Kondo, T.; Kurasawa, T.; Nishi, E.; Okuyama, A.; Chino, K.; Shibata, A.; Okada, Y.; Takei, H.; Nagasawa, H.; et al. Current clinical evidence of tocilizumab for the treatment of ANCA-associated vasculitis: A prospective case series for microscopic polyangiitis in a combination with corticosteroids and literature review. Clin. Rheumatol. 2017, 36, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Mekinian, A.; Saidenberg-Kermanac’h, N.; Stirnemann, J.; Fenaux, P.; Gherardi, R.; Fain, O. Efficacy of tocilizumab in rituximab-refractory cryoglobulinemia vasculitis. Ann. Rheum. Dis. 2012, 71, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Ohtori, S.; Miyagi, M.; Eguchi, Y.; Inoue, G.; Orita, S.; Ochiai, N.; Kishida, S.; Kuniyoshi, K.; Nakamura, J.; Aoki, Y.; et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur. Spine J. 2012, 21, 2079–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, N.; Ito, K.; Takagi, N. Safety and efficacy profiles of tocilizumab monotherapy in Japanese patients with rheumatoid arthritis: Meta-analysis of six initial trials and five long-term extensions. Mod. Rheumatol. 2010, 20, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Bykerk, V.P.; Ostor, A.J.; Alvaro-Gracia, J.; Pavelka, K.; Ivorra, J.A.; Graninger, W.; Bensen, W.; Nurmohamed, M.T.; Krause, A.; Bernasconi, C.; et al. Tocilizumab in patients with active rheumatoid arthritis and inadequate responses to DMARDs and/or TNF inhibitors: A large, open-label study close to clinical practice. Ann. Rheum. Dis. 2012, 71, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Yoshitama, T.; Hidaka, T.; Sakai, F.; Hasegawa, M.; Hashiba, Y.; Suematsu, E.; Tatsukawa, H.; Mizokami, A.; Yoshizawa, S.; et al. Comparative risk of hospitalized infection between biological agents in rheumatoid arthritis patients: A multicenter retrospective cohort study in Japan. PLoS ONE 2017, 12, e0179179. [Google Scholar] [CrossRef] [PubMed]
- Moots, R.J.; Sebba, A.; Rigby, W.; Ostor, A.; Porter-Brown, B.; Donaldson, F.; Dimonaco, S.; Rubbert-Roth, A.; van Vollenhoven, R.; Genovese, M.C. Effect of tocilizumab on neutrophils in adult patients with rheumatoid arthritis: Pooled analysis of data from phase 3 and 4 clinical trials. Rheumatology (Oxford) 2017, 56, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Strangfeld, A.; Richter, A.; Siegmund, B.; Herzer, P.; Rockwitz, K.; Demary, W.; Aringer, M.; Meissner, Y.; Zink, A.; Listing, J. Risk for lower intestinal perforations in patients with rheumatoid arthritis treated with tocilizumab in comparison to treatment with other biologic or conventional synthetic DMARDs. Ann. Rheum. Dis. 2017, 76, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Sano, Y.; Yaguramaki, H.; et al. Effectiveness and safety of tocilizumab: Postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J. Rheumatol. 2014, 41, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Goto, H.; Hirao, K.; Nakajima, A.; Origasa, H.; Tanaka, K.; Tomobe, M.; Totsuka, K. Longterm Safety of Tocilizumab: Results from 3 Years of Followup Postmarketing Surveillance of 5573 Patients with Rheumatoid Arthritis in Japan. J. Rheumatol. 2015, 42, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Heo, T.H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Transcriptional and post-transcriptional regulation of interleukein-6 (IL-6) mRNA. Bacterial and fungal components and the molecules derived from tissue injury activate cell surface or intracellular Toll-like receptors (TLRs). Nuclear factor-kappa B (NF-κB) is a main transcriptional factor to induce transcription of IL-6 mRNA. AT-rich interactive domain-containing protein 5a (Arid5a) translocates into the nucleus via an importin-α/β1 pathway and binds to the 3′UTR of IL-6 mRNA. Then, Arid5a exports IL-6 mRNA to the cytoplasm via the chromosomal region maintenance 1 (CRM1) pathway by binding to up-frameshift protein 1 (UPF1) and protects IL-6 mRNA from degradation by Regulatory RNase-1 (Regnase-1). Regnase-1 degrades transcriptionally active IL-6 mRNA by binding to the IL-6 3′UTR in the cytoplasm, endoplasmic reticulum and ribosomes. Another RNA-binding protein Roquin recognizes mRNAs overlapping with Regnase-1. However, Roquin degrades inactive mRNA by recruiting deadenylase in stress granules and processing bodies. The balance between the degradation of IL-6 mRNA by Regnase-1 and the protection of IL-6 mRNA by Arid5a controls the quantity of IL-6 production.
Figure 1.
Transcriptional and post-transcriptional regulation of interleukein-6 (IL-6) mRNA. Bacterial and fungal components and the molecules derived from tissue injury activate cell surface or intracellular Toll-like receptors (TLRs). Nuclear factor-kappa B (NF-κB) is a main transcriptional factor to induce transcription of IL-6 mRNA. AT-rich interactive domain-containing protein 5a (Arid5a) translocates into the nucleus via an importin-α/β1 pathway and binds to the 3′UTR of IL-6 mRNA. Then, Arid5a exports IL-6 mRNA to the cytoplasm via the chromosomal region maintenance 1 (CRM1) pathway by binding to up-frameshift protein 1 (UPF1) and protects IL-6 mRNA from degradation by Regulatory RNase-1 (Regnase-1). Regnase-1 degrades transcriptionally active IL-6 mRNA by binding to the IL-6 3′UTR in the cytoplasm, endoplasmic reticulum and ribosomes. Another RNA-binding protein Roquin recognizes mRNAs overlapping with Regnase-1. However, Roquin degrades inactive mRNA by recruiting deadenylase in stress granules and processing bodies. The balance between the degradation of IL-6 mRNA by Regnase-1 and the protection of IL-6 mRNA by Arid5a controls the quantity of IL-6 production.

Figure 2.
IL-6 signal transduction and modes of the IL-6 receptor system. IL-6 binds to the IL-6 receptor (IL-6R). The IL-6/IL-6R complex is associated with gp130, resulting in a hexamer structure. Homodimerization of gp130 activates Janus kinase (JAK) and subsequently activates Signal transducer and activator of transcription 3 (STAT3), STAT1 and SH2 domain-containing protein-tyrosine phosphatase 2 (SHP2). STAT3 and STAT1 move into the nucleus and SHP-2 activates the Ras-MAP kinase pathway. STAT3 induces the negative feedback molecules suppressor of cytokine signaling 1 (SOCS1) and SOCS3. SOCS1 inhibits JAKs. SOCS3 inhibits the SHP2 pathway. STAT3 also induces Arid5a, which stabilizes Stat3 mRNA by binding to the Stat3 3′UTR. IL-6 signaling via membrane IL-6R or soluble IL-6R is called classic signaling or trans-signaling, respectively. When an IL-6R-expressing cell and a gp130-expressing cell are in close proximity, IL-6 is presented by between cells, a process called trans-presentation. IL-6 also activates gp130 in the endosomal compartment, endoplasmic reticulum (ER), or Golgi body through intracellular signaling. The red arrow indicates signal transduction.
Figure 2.
IL-6 signal transduction and modes of the IL-6 receptor system. IL-6 binds to the IL-6 receptor (IL-6R). The IL-6/IL-6R complex is associated with gp130, resulting in a hexamer structure. Homodimerization of gp130 activates Janus kinase (JAK) and subsequently activates Signal transducer and activator of transcription 3 (STAT3), STAT1 and SH2 domain-containing protein-tyrosine phosphatase 2 (SHP2). STAT3 and STAT1 move into the nucleus and SHP-2 activates the Ras-MAP kinase pathway. STAT3 induces the negative feedback molecules suppressor of cytokine signaling 1 (SOCS1) and SOCS3. SOCS1 inhibits JAKs. SOCS3 inhibits the SHP2 pathway. STAT3 also induces Arid5a, which stabilizes Stat3 mRNA by binding to the Stat3 3′UTR. IL-6 signaling via membrane IL-6R or soluble IL-6R is called classic signaling or trans-signaling, respectively. When an IL-6R-expressing cell and a gp130-expressing cell are in close proximity, IL-6 is presented by between cells, a process called trans-presentation. IL-6 also activates gp130 in the endosomal compartment, endoplasmic reticulum (ER), or Golgi body through intracellular signaling. The red arrow indicates signal transduction.

Figure 3.
Defense reaction and pathological role of IL-6 in each organ. The defensive role of IL-6 in each organ is closely related to the pathological condition present. In the liver, IL-6 induces acute phase proteins, liver regeneration and CD8 T cell activation. Continuous elevation of IL-6 results in secondary amyloidosis, cardiovascular disease and hepatocellular carcinoma. In hematopoiesis, IL-6 induces leukocytosis by mobilization of neutrophils from the marginal pool into the circulation pool. IL-6 or IL-6-induced thrombopoietin induces thrombocytosis. IL-6-induced hepcidin reduces serum iron levels, leading to anemia. IL-6 plays a crucial role in neutrophil to mononuclear cell transition, B and T cell differentiation. High levels of IL-6 bound with sIL-6R induce vascular permeability in vessels and angiogenesis in chronic inflammatory sites. In bone homeostasis, IL-6 induces osteoclastogenesis. IL-6 activates the coagulation system for hemostasis, together with increases in fibrinogen and platelets, leading to an increased risk for the development of cardiovascular diseases. IL-6 also plays a protective role in intestinal epithelial cells. Accumulating evidence shows the association of IL-6 with a depressed mood. MCP-1: monocyte chemotactic protein 1, ICAM-1: intercellular adhesion molecule-1, RANKL: receptor activator of nuclear factor κB ligand.
Figure 3.
Defense reaction and pathological role of IL-6 in each organ. The defensive role of IL-6 in each organ is closely related to the pathological condition present. In the liver, IL-6 induces acute phase proteins, liver regeneration and CD8 T cell activation. Continuous elevation of IL-6 results in secondary amyloidosis, cardiovascular disease and hepatocellular carcinoma. In hematopoiesis, IL-6 induces leukocytosis by mobilization of neutrophils from the marginal pool into the circulation pool. IL-6 or IL-6-induced thrombopoietin induces thrombocytosis. IL-6-induced hepcidin reduces serum iron levels, leading to anemia. IL-6 plays a crucial role in neutrophil to mononuclear cell transition, B and T cell differentiation. High levels of IL-6 bound with sIL-6R induce vascular permeability in vessels and angiogenesis in chronic inflammatory sites. In bone homeostasis, IL-6 induces osteoclastogenesis. IL-6 activates the coagulation system for hemostasis, together with increases in fibrinogen and platelets, leading to an increased risk for the development of cardiovascular diseases. IL-6 also plays a protective role in intestinal epithelial cells. Accumulating evidence shows the association of IL-6 with a depressed mood. MCP-1: monocyte chemotactic protein 1, ICAM-1: intercellular adhesion molecule-1, RANKL: receptor activator of nuclear factor κB ligand.

Figure 4.
IL-6 inhibition therapy for various diseases. Currently, tocilizumab is approved for the treatment of Castleman disease, rheumatoid arthritis, systemic and polyarticular juvenile idiopathic arthritis, giant cell arteritis, Takayasu arteritis and cytokine releasing syndrome. IL-6 inhibition therapy is expected to constitute a novel therapeutic strategy for a wide range of diseases. Many case reports have also indicated that IL-6 inhibition is an effective treatment for various diseases. Clinical trials for some of them are ongoing.
Figure 4.
IL-6 inhibition therapy for various diseases. Currently, tocilizumab is approved for the treatment of Castleman disease, rheumatoid arthritis, systemic and polyarticular juvenile idiopathic arthritis, giant cell arteritis, Takayasu arteritis and cytokine releasing syndrome. IL-6 inhibition therapy is expected to constitute a novel therapeutic strategy for a wide range of diseases. Many case reports have also indicated that IL-6 inhibition is an effective treatment for various diseases. Clinical trials for some of them are ongoing.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Narazaki, M.; Kishimoto, T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int. J. Mol. Sci. 2018, 19, 3528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113528
AMA Style
Narazaki M, Kishimoto T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. International Journal of Molecular Sciences. 2018; 19(11):3528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113528
Chicago/Turabian StyleNarazaki, Masashi, and Tadamitsu Kishimoto. 2018. "The Two-Faced Cytokine IL-6 in Host Defense and Diseases" International Journal of Molecular Sciences 19, no. 11: 3528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113528
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.