Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases

Abstract
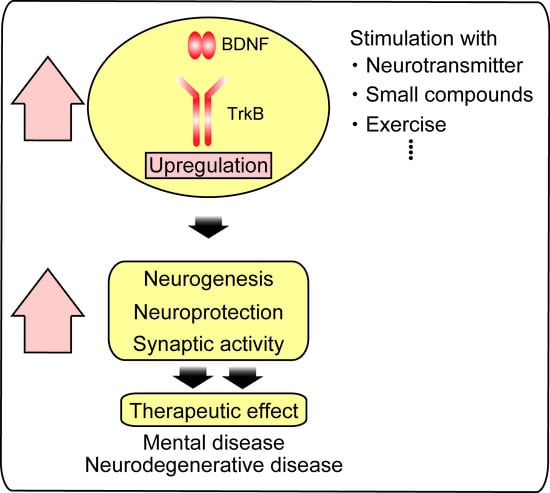
:
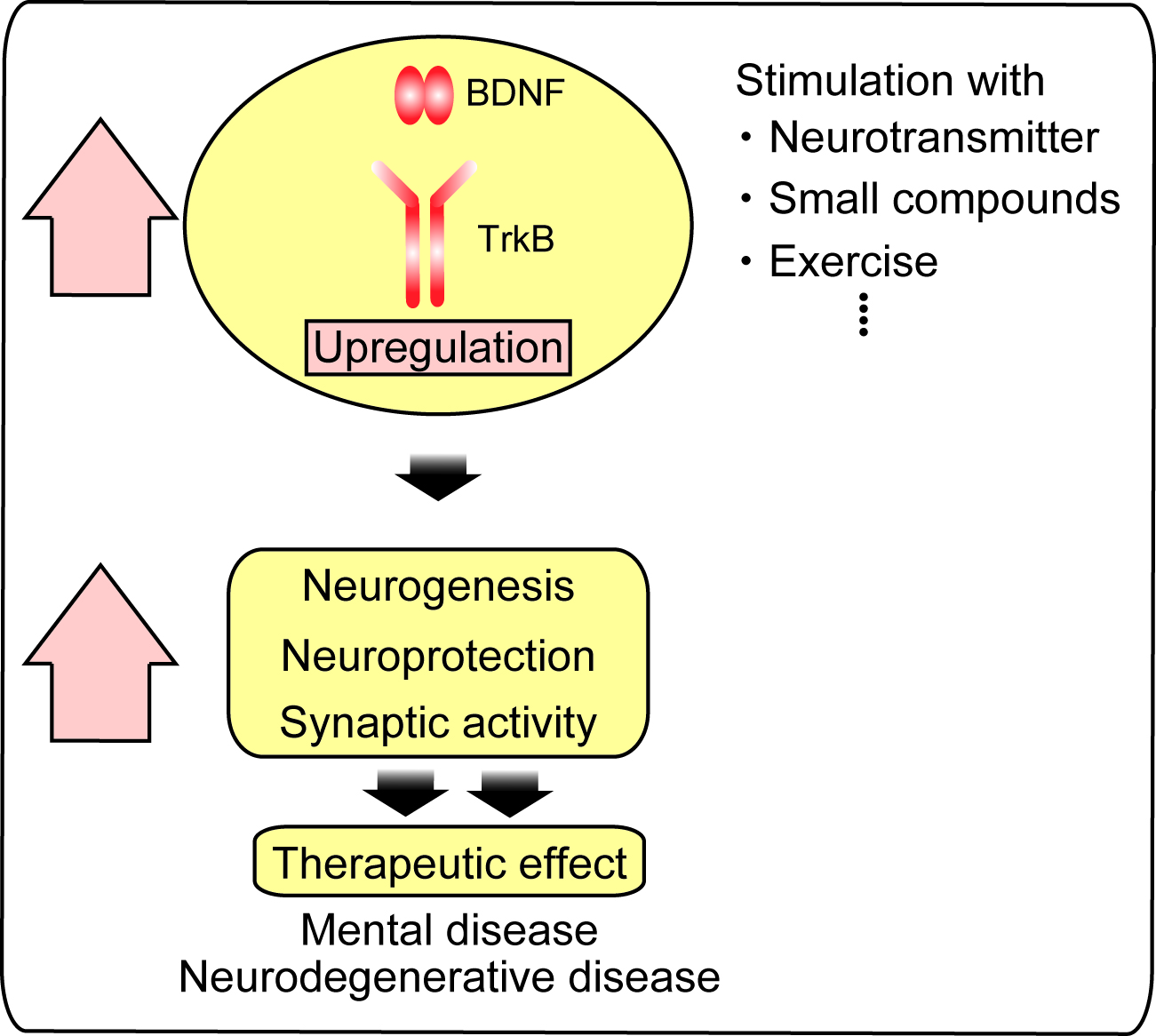{kind=link}
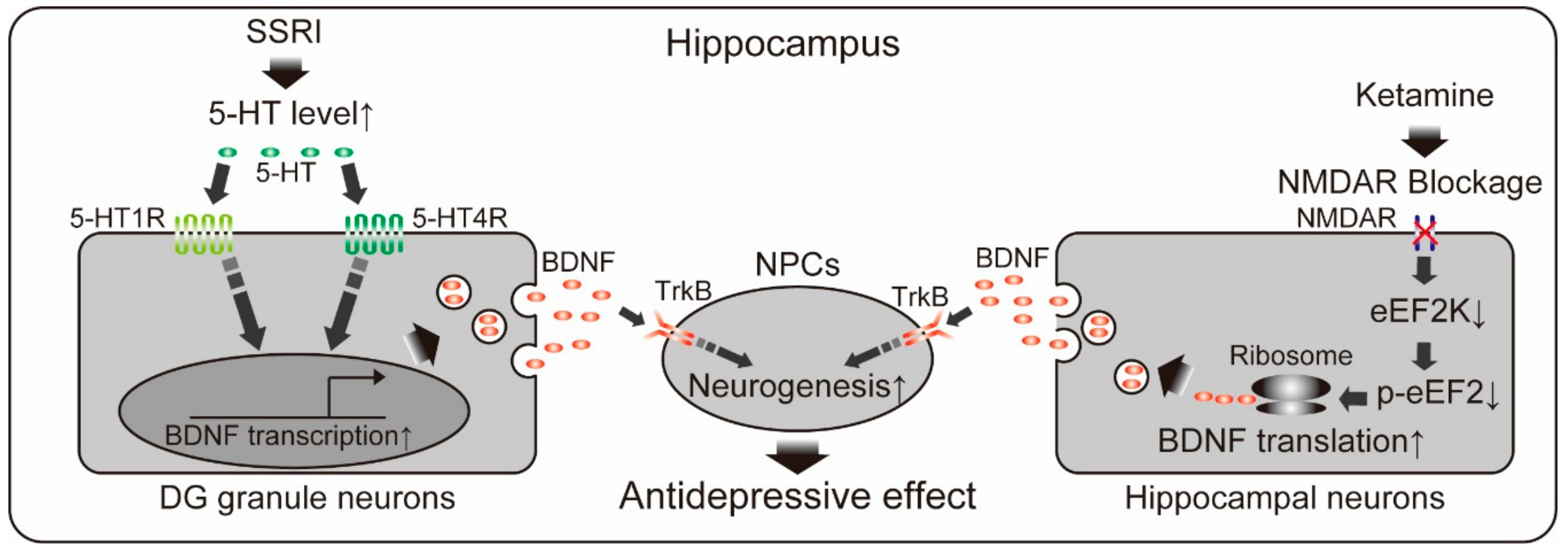{kind=link}
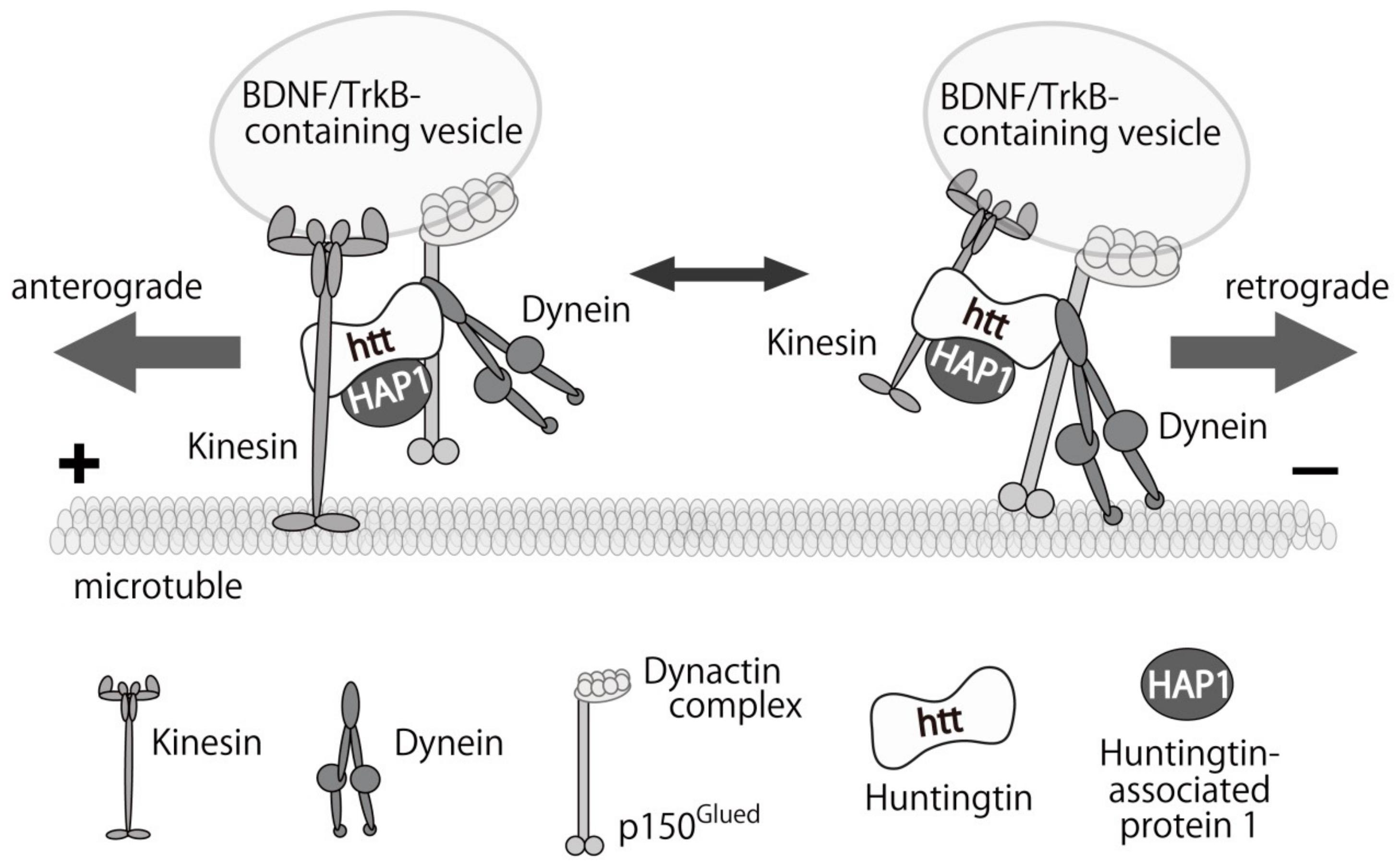{kind=link}
1. Introduction
2. Activation of BDNF/TrkB System by Compounds
3. BDNF and Adult Neurogenesis
4. Monoaminergic System and BDNF-Mediated Neurogenesis
5. Other Antidepressive Therapy and BDNF-Mediated Neurogenesis
6. Influence of Val66Met Polymorphism of BDNF on the Neurogenesis
7. Alzheimer’s Disease and BDNF
8. Parkinson’s Disease and BDNF
9. Huntington’s Disease and BDNF
10. BDNF/TrkB Transport As a Therapeutic Target of Neurodegenerative Diseases
11. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ohira, K.; Hayashi, M. A new aspect of the TrkB signaling pathway in neural plasticity. Curr. Neuropharmacol. 2009, 7, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Zhao, Y.H.; Zeng, M.J.; Fang, F.; Li, M.; Qin, T.T.; Ye, L.Y.; Li, H.W.; Qu, R.; Ma, S.P. Saikosaponin D relieves unpredictable chronic mild stress induced depressive-like behavior in rats: Involvement of HPA axis and hippocampal neurogenesis. Psychopharmacology 2017, 234, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Wang, Y.J.; Song, L.; Wang, J.L.; Huang, C.; Zhang, W.; Jiang, B. Antidepressant-like effects of tetrahydroxystilbene glucoside in mice: Involvement of BDNF signaling cascade in the hippocampus. Neurosci. Ther. 2017, 23, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophic Factor and Glucocorticoid Stress in Neurogenesis. Int. J. Mol. Sci. 2017, 18, e2312. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Mira, H. Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Lu, B. Pro-region of neurotrophins: Role in synaptic modulation. Neuron 2003, 39, 735–738. [Google Scholar] [CrossRef]
- Teng, K.K.; Hempstead, B.L. Neurotrophins and their receptors: Signaling trios in complex biological systems. Cell. Mol. Life Sci. 2004, 61, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef]
- Guo, W.; Nagappan, G.; Lu, B. Differential effects of transient and sustained activation of BDNF-TrkB signaling. Dev. Neurobiol. 2018, 78, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Mitre, M.; Mariga, A.; Chao, M.V. Neurotrophin signalling: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2017, 131, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Afshar, S.; Shahidi, S.; Rohani, A.H.; Komaki, A.; Asl, S.S. The effect of NAD-299 and TCB-2 on learning and memory, hippocampal BDNF levels and amyloid plaques in Streptozotocin-induced memory deficits in male rats. Psychopharmacology 2018, 235, 2809–2822. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, O.; Kloster, E.; Reick, C.; Saft, C.; Gold, R.; Epplen, J.T.; Arning, L.; Ellrichmann, G. Activation of NPY-Y2 receptors ameliorates disease pathology in the R6/2 mouse and PC12 cell models of Huntington's disease. Exp. Neurol. 2018, 302, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Castaldo, S.; Amico, E.; Pepe, G.; Marracino, F.; Capocci, L.; Giovannelli, A.; Madonna, M.; van Bergeijk, J.; Buttari, F.; et al. Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington's disease. Hum. Mol. Genet. 2018, 27, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Anand David, A.V.; Arulmoli, R.; Parasuraman, S. Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacogn. Rev. 2016, 10, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Ademosun, A.O.; Ogunsuyi, O.B. Quercetin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 929, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Rahvar, M.; Owji, A.A.; Mashayekhi, F.J. Effect of quercetin on the brain-derived neurotrophic factor gene expression in the rat brain. Bratisl. Lek. Listy. 2018, 119, 28–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen Res. 2017, 12, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Y.; Zhou, H.H.; Li, X.; Liu, Z.Q. Huperzine A Alleviates Oxidative Glutamate Toxicity in Hippocampal HT22 Cells via Activating BDNF/TrkB-Dependent PI3K/Akt/mTOR Signaling Pathway. Cell Mol. Neurobiol. 2016, 36, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ma, Y.; Pan, Y.L.; Zheng, S.Y.; Wang, J.W.; Huang, G.C. Jisuikang, a Chinese herbal formula, increases neurotrophic factor expression and promotes the recovery of neurological function after spinal cord injury. Neural Regen Res. 2017, 12, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.A.; Gawali, N.B.; Shinde, P.; Munshi, R.; Juvekar, A.R. Imperatorin ameliorates lipopolysaccharide induced memory deficit by mitigating proinflammatory cytokines, oxidative stress and modulating brain-derived neurotropic factor. Cytokine 2018, 110, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, B.; Zhang, B.; Li, S.; Huang, N.; Zhan, G.; Jiang, R.; Yang, L.; Wu, Y.; Miao, L.; et al. Sulforaphane alleviates lipopolysaccharide-induced spatial learning and memory dysfunction in mice: The role of BDNF-mTOR signaling pathway. Neuroscience 2018, 388, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Dhuriya, Y.K.; Kumar, V.; Srivastava, A.; Gupta, R.; Shukla, R.K.; Yadav, R.S.; Dwivedi, H.N.; Pant, A.B.; Khanna, V.K. PI3K/Akt/GSK3β induced CREB activation ameliorates arsenic mediated alterations in NMDA receptors and associated signaling in rat hippocampus: Neuroprotective role of curcumin. Neurotoxicology 2018, 67, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Eimerbrink, M.J.; Pendry, R.J.; Hodges, S.L.; Wiles, J.D.; Peterman, J.L.; White, J.D.; Hayes, H.B.; Chumley, M.J.; Boehm, G.W. The α5-GABAAR inverse agonist MRK-016 upregulates hippocampal BDNF expression and prevents cognitive deficits in LPS-treated mice, despite elevations in hippocampal Aβ. Behav. Brain Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Gong, A.J.; Liang, H.; Wang, Q.; Wu, Y.; Ma, A.; Han, L. Improvement of Symptoms in a Rat Model of Depression through combined Zinc and Folic Acid Administration via Up-regulation of the Trk B and NMDA. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.M.; Jessberger, S. Adult neurogenesis: Mechanisms and functional significance. Development 2014, 141, 1983–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Jinnou, H.; Sawamoto, K.; Hitoshi, S. Adult neurogenesis and its role in brain injury and psychiatric diseases. J. Neurochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Winkler, J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2015, 7, a021287. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiang, L.; Zhang, X.; Chen, H. In-vitro effects of brain-derived neurotrophic factor on neural progenitor/stem cells from rat hippocampus. Neuroreport 2009, 20, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liao, J.; Qi, F.; Meng, Z.; Pan, S. Evidence for the contribution of BDNF-TrkB signal strength in neurogenesis: An organotypic study. Neurosci. Lett. 2015, 606, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 2005, 192, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, M.; Lucas, G.; Ernfors, P.; Castrén, M.; Castrén, E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, X.; She, L.; Xu, D.; Huang, W.; Poo, M.M. Autocrine Action of BDNF on Dendrite Development of Adult-Born Hippocampal Neurons. J. Neurosci. 2015, 35, 8384–8393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergami, M.; Rimondini, R.; Santi, S.; Blum, R.; Götz, M.; Canossa, M. Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc. Natl. Acad. Sci. USA 2008, 105, 15570–15575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.M.; Merson, T.D.; Sotthibundhu, A.; Coulson, E.J.; Bartlett, P.F. p75 neurotrophin receptor expression defines a population of BDNF-responsive neurogenic precursor cells. J. Neurosci. 2007, 27, 5146–5155. [Google Scholar] [CrossRef] [PubMed]
- Galvão, R.P.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J. Neurosci. 2008, 28, 13368–13383. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.A.; Hughes, S.M.; Connor, B. AAV-mediated delivery of BDNF augments neurogenesis in the normal and quinolinic acid-lesioned adult rat brain. Eur. J. Neurosci. 2007, 25, 3513–3525. [Google Scholar] [CrossRef] [PubMed]
- Zigova, T.; Pencea, V.; Wiegand, S.J.; Luskin, M.B. Intraventricular administration of BDNF increases the number of newly generated neurons in the adult olfactory bulb. Mol. Cell. Neurosci. 1998, 11, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Bergami, M.; Vignoli, B.; Motori, E.; Pifferi, S.; Zuccaro, E.; Menini, A.; Canossa, M. TrkB signaling directs the incorporation of newly generated periglomerular cells in the adult olfactory bulb. J. Neurosci. 2013, 33, 11464–11478. [Google Scholar] [CrossRef] [PubMed]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Surget, A.; Saxe, M.; Leman, S.; Ibarguen-Vargas, Y.; Chalon, S.; Griebel, G.; Hen, R.; Belzung, C. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol. Psychiatry 2008, 64, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Hen, R.; Underwood, M.D.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol. Psychiatry 2012, 72, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrini, M.; Santiago, A.N.; Hen, R.; Dwork, A.J.; Rosoklija, G.B.; Tamir, H.; Arango, V.; Mann, J. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 2013, 38, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef] [PubMed]
- Russo-Neustadt, A.A.; Beard, R.C.; Huang, Y.M.; Cotman, C.W. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience 2000, 101, 305–312. [Google Scholar] [CrossRef]
- Chen, B.; Dowlatshahi, D.; MacQueen, G.M.; Wang, J.F.; Young, L.T. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 2001, 50, 260–265. [Google Scholar] [CrossRef]
- Shirayama, Y.; Chen, A.C.; Nakagawa, S.; Russell, D.S.; Duman, R.S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002, 22, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.A.; Anacker, C.; Hu, A.; Levinstein, M.R.; Pickenhagen, A.; Tsetsenis, T.; Madroñal, N.; Donaldson, Z.R.; Drew, L.J.; Dranovsky, A.; et al. 5-HT1A receptors on mature dentate gyrus granule cells are critical for the antidepressant response. Nat. Neurosci. 2015, 18, 1606–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-David, I.; David, D.J.; Darcet, F.; Wu, M.V.; Kerdine-Römer, S.; Gardier, A.M.; Hen, R. Rapid anxiolytic effects of a 5-HT4 receptor agonist are mediated by a neurogenesis-independent mechanism. Neuropsychopharmacology 2014, 39, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Imoto, Y.; Kira, T.; Sukeno, M.; Nishitani, N.; Nagayasu, K.; Nakagawa, T.; Kaneko, S.; Kobayashi, K.; Segi-Nishida, E. Role of the 5-HT4 receptor in chronic fluoxetine treatment-induced neurogenic activity and granule cell dematuration in the dentate gyrus. Mol. Brain 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotheneichner, P.; Lange, S.; O’Sullivan, A.; Marschallinger, J.; Zaunmair, P.; Geretsegger, C.; Aigner, L.; Couillard-Despres, S. Hippocampal neurogenesis and antidepressive therapy: Shocking relations. Neural Plast. 2014, 2014, 723915. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.B.; Dondossola, E.R.; Grande, A.J.; Colonetti, T.; Ceretta, L.B.; Passos, I.C.; Quevedo, J.; da Rosa, M.I. Increased BDNF levels after electroconvulsive therapy in patients with major depressive disorder: A meta-analysis study. J. Psychiatr. Res. 2016, 83, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Angelucci, A.; Costantin, L.; Braschi, C.; Mazzantini, M.; Babbini, F.; Fabbri, M.E.; Tessarollo, L.; Maffei, L.; Berardi, N.; et al. Brain-derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur. J. Neurosci. 2006, 24, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Yang, L.D.; Zhang, K.; Zheng, K.Y.; Wei, X.M.; Yang, Q.; Niu, W.M.; Zhao, M.G.; Wu, Y.M. Silibinin exerts antidepressant effects by improving neurogenesis through BDNF/TrkB pathway. Behav. Brain Res. 2018, 348, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Zang, T.; Birnbaum, S.G.; Wang, Z.; Johnson, J.E.; Zhang, C.L.; Parada, L.F. TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat. Commun. 2017, 8, 1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molero, P.; Ramos-Quiroga, J.A.; Martin-Santos, R.; Calvo-Sánchez, E.; Gutiérrez-Rojas, L.; Meana, J.J. Antidepressant Efficacy and Tolerability of Ketamine and Esketamine: A Critical Review. CNS Drugs 2018. [CrossRef] [PubMed]
- Szczęsny, E.; Slusarczyk, J.; Głombik, K.; Budziszewska, B.; Kubera, M.; Lasoń, W.; Basta-Kaim, A. Possible contribution of IGF-1 to depressive disorder. Pharmacol. Rep. 2013, 65, 1622–1631. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Cheng, X.; Guo.Y., *!!! REPLACE !!!*; Sun, X.; Chen, G.; Li, H.; Li, P.; Lu, X.; Tian, M.; et al. IGF-1 promotes Brn-4 expression and neuronal differentiation of neural stem cells via the PI3K/Akt pathway. PLoS ONE 2014, 9, e113801. [Google Scholar] [CrossRef] [PubMed]
- Aberg, M.A.; Aberg, N.D.; Palmer, T.D.; Alborn, A.M.; Carlsson-Skwirut, C.; Bang, P.; Rosengren, L.E.; Olsson, T.; Gage, F.H.; Eriksson, P.S. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol. Cell Neurosci. 2003, 24, 23–40. [Google Scholar] [CrossRef]
- Mir, S.; Cai, W.; Carlson, S.W.; Saatman, K.E.; Andres, D.A. IGF-1 mediated Neurogenesis Involves a Novel RIT1/Akt/Sox2 Cascade. Sci. Rep. 2017, 7, 3283. [Google Scholar] [CrossRef] [PubMed]
- Odaka, H.; Numakawa, T.; Yoshimura, A.; Nakajima, S.; Adachi, N.; Ooshima, Y.; Inoue, T.; Kunugi, H. Chronic glucocorticoid exposure suppressed the differentiation and survival of embryonic neural stem/progenitor cells: Possible involvement of ERK and PI3K/Akt signaling in the neuronal differentiation. Neurosci. Res. 2016, 113, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberg, M.A.; Aberg, N.D.; Hedbäcker, H.; Oscarsson, J.; Eriksson, P.S. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J. Neurosci. 2000, 20, 2896–2903. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.L.; Carro, E.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 2001, 21, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Lichtenwalner, R.J.; Forbes, M.E.; Bennett, S.A.; Lynch, C.D.; Sonntag, W.E.; Riddle, D.R. Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 2001, 107, 603–613. [Google Scholar] [CrossRef]
- Hoshaw, B.A.; Hill, T.I.; Crowley, J.J.; Malberg, J.E.; Khawaja, X.; Rosenzweig-Lipson, S.; Schechter, L.E.; Lucki, I. Antidepressant-like behavioral effects of IGF-I produced by enhanced serotonin transmission. Eur. J. Pharmacol. 2008, 594, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshaw, B.A.; Malberg, J.E.; Lucki, I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005, 1037, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J. Critical Issues in BDNF Val66Met Genetic Studies of Neuropsychiatric Disorders. Front Mol. Neurosci. 2018, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Balkaya, M.; Cho, S. Genetics of stroke recovery: BDNF val66met polymorphism in stroke recovery and its interaction with aging. Neurobiol. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hajek, T.; Kopecek, M.; Höschl, C. Reduced hippocampal volumes in healthy carriers of brain-derived neurotrophic factor Val66Met polymorphism: Meta-analysis. World J. Biol. Psychiatry 2012, 13, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Harrisberger, F.; Spalek, K.; Smieskova, R.; Schmidt, A.; Coynel, D.; Milnik, A.; Fastenrath, M.; Freytag, V.; Gschwind, L.; Walter, A.; et al. The association of the BDNF Val66Met polymorphism and the hippocampal volumes in healthy humans: A joint meta-analysis of published and new data. Neurosci. Biobehav. Rev. 2014, 42, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Hariri, A.R.; Goldberg, T.E.; Mattay, V.S.; Kolachana, B.S.; Callicott, J.H.; Egan, M.F.; Weinberger, D.R. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J. Neurosci. 2003, 23, 6690–6694. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, C.; Vicario, A.; Li, Z.; Baj, G.; Braiuca, P.; Wu, Y.; Lee, F.S.; Gardossi, L.; Baraban, J.M.; Tongiorgi, E. Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc. Natl. Acad. Sci. USA 2009, 106, 16481–16486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath, K.G.; Jing, D.Q.; Dincheva, I.; Neeb, C.C.; Pattwell, S.S.; Chao, M.V.; Lee, F.S.; Ninan, I. BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology 2012, 37, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Bath, K.G.; Mandairon, N.; Jing, D.; Rajagopal, R.; Kapoor, R.; Chen, Z.Y.; Khan, T.; Proenca, C.C.; Kraemer, R.; Cleland, T.A.; et al. Variant brain-derived neurotrophic factor (Val66Met) alters adult olfactory bulb neurogenesis and spontaneous olfactory discrimination. J. Neurosci. 2008, 28, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Jing, D.; Bath, K.G.; Ieraci, A.; Khan, T.; Siao, C.J.; Herrera, D.G.; Toth, M.; Yang, C.; McEwen, B.S.; et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 2006, 314, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Ieraci, A.; Madaio, A.I.; Mallei, A.; Lee, F.S.; Popoli, M. Brain-Derived Neurotrophic Factor Val66Met Human Polymorphism Impairs the Beneficial Exercise-Induced Neurobiological Changes in Mice. Neuropsychopharmacology 2016, 41. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Lee, F.S.; Li, X.Y.; Bambico, F.; Duman, R.S.; Aghajanian, G.K. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 2012, 71, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, M.; Liu, X.; Su, K.S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping, Y.S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the "amyloid cascade hypothesis. FEBS J. 2017, 284, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta. Neuropathol. 2009, 118, 131–150. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, SW.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer's mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Marín, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Martí, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer's disease parietal cortex. J. Neurochem. 2002, 82, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 7, 846–851. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.S.; Yu, L.; Boyle, P.A.; Schneider, J.A.; De, Jager, P.L.; Bennett, D.A. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology 2016, 86, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Spalletta, G.; di Iulio, F.; Ciaramella, A.; Salani, F.; Colantoni, L.; Varsi, A.E.; Gianni, W.; Sancesario, G.; Caltagirone, C.; et al. Alzheimer's disease (AD) and Mild Cognitive Impairment (MCI) patients are characterized by increased BDNF serum levels. Curr. Alzheimer Res. 2010, 7, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.C.; Gonçalves, G.S.; Rocha, N.P.; Moraes, E.N.; Bicalho, M.A.; Gualberto, C.M.T.; Jardim de Paula, J.; de Miranda, L.F.; Clayton de Souza Ferreira, A.; Teixeira, A.L.; et al. Increased plasma levels of BDNF and inflammatory markers in Alzheimer's disease. J. Psychiatr. Res. 2014, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.Y.; Huang, X.; Bi, C.; Mao, L.L.; Peng, L.J.; Qian, H.R. PGC-1α or FNDC5 Is Involved in Modulating the Effects of Aβ1-42 Oligomers on Suppressing the Expression of BDNF, a Beneficial Factor for Inhibiting Neuronal Apoptosis, Aβ Deposition and Cognitive Decline of APP/PS1 Tg Mice. Front Aging Neurosci. 2017, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, F.; Carboni, L.; Carretta, D.; Candeletti, S.; Romualdi, P. Treatment with the neurotoxic Aβ (25-35) peptide modulates the expression of neuroprotective factors Pin1, Sirtuin 1, and brain-derived neurotrophic factor in SH-SY5Y human neuroblastoma cells. Exp. Toxicol. Pathol. 2016, 68, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [PubMed]
- Moghbelinejad, S.; Nassiri-Asl, M.; Farivar, T.N.; Abbasi, E.; Sheikhi, M.; Taghiloo, M.; Farsad, F.; Samimi, A.; Hajiali, F. Rutin activates the MAPK pathway and BDNF gene expression on beta-amyloid induced neurotoxicity in rats. Toxicol. Lett. 2014, 224, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, Y.; Xu, Y.; Lian, Y.; Xie, N.; Wu, T.; Zhang, H.; Sun, L.; Zhang, R.; Wang, Z. Curcumin Improves Amyloid β-Peptide (1–42) Induced Spatial Memory Deficits through BDNF-ERK Signaling Pathway. PLoS ONE 2015, 10, e0131525. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.Y.; Yu, S.S.; Wang, ZJ.; Zhu, Y.Z. SCM-198 Ameliorates Cognitive Deficits, Promotes Neuronal Survival and Enhances CREB/BDNF/TrkB Signaling without Affecting Aβ Burden in AβPP/PS1 Mice. Int. J. Mol. Sci. 2015, 16, 18544–18563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.C.; Lee, J.; John, J.M.; Gonzalez-Lima, F. Neuroprotective effects of near-infrared light in an in vivo model of mitochondrial optic neuropathy. J. Neurosci. 2008, 28, 13511–13521. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; He, Z.; Xing, D. Low-level laser therapy rescues dendrite atrophy via upregulating BDNF expression: Implications for Alzheimer's disease. J. Neurosci. 2013, 33, 13505–13517. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Castello, N.A.; Nguyen, M.H.; Tran, J.D.; Cheng, D.; Green, K.N.; LaFerla, F.M. 7,8-Dihydroxyflavone, a small molecule TrkB agonist, improves spatial memory and increases thin spine density in a mouse model of Alzheimer disease-like neuronal loss. PLoS ONE 2014, 9, e91453. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Oliff, H.S.; Berchtold, N.C.; Isackson, P.; Cotman, C.W. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Res. Mol. Brain Res. 1998, 61, 147–153. [Google Scholar] [CrossRef]
- Koo, J.H.; Kwon, I.S.; Kang, E.B.; Lee, C.K.; Lee, N.H.; Kwon, M.G.; Cho, I.H.; Cho, J.Y. Neuroprotective effects of treadmill exercise on BDNF and PI3-K/Akt signaling pathway in the cortex of transgenic mice model of Alzheimer’s disease. J. Exerc. Nutr. Biochem. 2013, 17, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.Y.; Li, S.C.; Sun, Y.X.; Zhang, X.S.; Dong, Z.Z.; Zhong, P.; Sun, X.R. Long-term treadmill exercise improves spatial memory of male APPswe/PS1dE9 mice by regulation of BDNF expression and microglia activation. Biol. Sport 2015, 32, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Reighard, C.P.; Crowther, D.C. The pro-domains of neurotrophins, including BDNF, are linked to Alzheimer’s disease through a toxic synergy with Aβ. Hum. Mol. Genet. 2015, 24, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-León, Y.; Pascual, A. Brain-derived neurotrophic factor stimulates beta-amyloid gene promoter activity by a Ras-dependent/AP-1-independent mechanism in SH-SY5Y neuroblastoma cells. J. Neurochem. 2001, 79, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Aleixandre, M.; Linares, C.; Masliah, E.; Moessler, H. Apathy and APOE4 are associated with reduced BDNF levels in Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE isoforms differentially regulates cleavage and secretion of BDNF. Mol. Brain. 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Atlas, R.; Lange, A.; Ginzburg, I. Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 Kinase signalling mechanism. Eur. J. Neurosci. 2005, 22, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Nie, S.; Zhu, W.; Liu, F.; Guo, H.; Chu, J.; Cao, X.B.; Jiang, X.; Zhang, Y.; Li, Y. 7,8-Dihydroxyflavone Ameliorates Cognitive Impairment by Inhibiting Expression of Tau Pathology in ApoE-Knockout Mice. Front Aging Neurosci. 2016, 8, 287. [Google Scholar]
- Zhang, J.; Liu, Z.; Pei, Y.; Yang, W.; Xie, C.; Long, S. MicroRNA-322 Cluster Promotes Tau Phosphorylation via Targeting Brain-Derived Neurotrophic Factor. Neurochem. Res. 2018, 43, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, CH.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.P.; Hamdane, M.; Buée, L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Mazzaro, N.; Barini, E.; Spillantini, M.G.; Goedert, M.; Medini, P.; Gasparini, L. Tau-Driven Neuronal and Neurotrophic Dysfunction in a Mouse Model of Early Tauopathy. J. Neurosci. 2016, 6, 2086–2100. [Google Scholar] [CrossRef] [PubMed]
- Atasoy, I.L.; Dursun, E.; Gezen-Ak, D.; Metin-Armağan, D.; Öztürk, M.; Yılmazer, S. Both secreted and the cellular levels of BDNF attenuated due to tau hyperphosphorylation in primary cultures of cortical neurons. J. Chem. Neuroanat. 2017, 80, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson's Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.M.; Schamne, M.G.; Sampaio, T.B.; Pértile, R.A.; Fernandes, P.A.; Markus, R.P.; Prediger, R.D. Melatoninergic System in Parkinson's Disease: From Neuroprotection to the Management of Motor and Nonmotor Symptoms. Oxid. Med. Cell Longev. 2016, 3472032. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kümmer, A.; Bretas, T.L.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson's disease. J. Neurol. 2010, 257, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Alam, A.; San, C.Y.; Eguchi, S.; Chen, Q.; Lian, Q.; Ma, D. Molecular mechanisms of brain-derived neurotrophic factor in neuro-protection: Recent developments. Brain Res. 2017, 1665, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; Zhang, B.S.; Soares, J.C.; Zhang, X.Y. Low BDNF is associated with cognitive impairments in patients with Parkinson's disease. Parkinsonism Relat. Disord. 2016, 29, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Alomari, M.A.; Khabour, O.F.; Al-Hieshan, A.; Bajwa, J.A. Relationship of circulatory BDNF with cognitive deficits in people with Parkinson's disease. J. Neurol. Sci. 2016, 362, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, M.; Khalid, U.; Klein, A.B.; Aznar, S.; Thomsen, G.; Jensen, P.; Knudsen, G.M. Striatal dopamine transporter binding correlates with serum BDNF levels in patients with striatal dopaminergic neurodegeneration. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Ventriglia, M.; Zanardini, R.; Bonomini, C.; Zanetti, O.; Volpe, D.; Pasqualetti, P.; Gennarelli, M.; Bocchio-Chiavetto, L. Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed. Res. Int. 2013, 901082. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, H.; Wang, C.; Ming, F.; Shi, X.; Yang, M. Serum level of brain-derived neurotrophic factor in Parkinson's disease: A meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 88, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Dung, Ling, Z.; Carvey, P.M.; Fletcher-Turner, A.; Yurek, D.M.; Sladek, J.R., Jr.; Kordower, J.H. Striatal trophic factor activity in aging monkeys with unilateral MPTP-induced parkinsonism. Exp. Neurol. 2005, 191 Suppl. 1, S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Berghauzen-Maciejewska, K.; Wardas, J.; Kosmowska, B.; Głowacka, U.; Kuter, K.; Ossowska, K. Alterations of BDNF and trkB mRNA expression in the 6-hydroxydopamine-induced model of preclinical stages of Parkinson's disease: An influence of chronic pramipexole in rats. PLoS ONE 2015, 10, e0117698. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Pinton, S.; da Rocha, J.T.; Gai, B.M.; Nogueira, C.W. Involvement of BDNF/TrkB signaling in the effect of diphenyl diselenide on motor function in a Parkinson's disease rat model. Eur. J. Pharmacol. 2017, 795, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Peppe, A.; Carlesimo, G.A.; Serafini, F.; Zabberoni, S.; Barban, F.; Shofany, J.; Caltagirone, C.; Costa, A. A pilot study on the effect of cognitive training on BDNF serum levels in individuals with Parkinson's disease. Front. Hum. Neurosci. 2015, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Piermaria, J.; Gelfo, F.; Shofany, J.; Tramontano, M.; Fiore, M.; Caltagirone, C.; Peppe, A. The effects of motor rehabilitation training on clinical symptoms and serum BDNF levels in Parkinson's disease subjects. Can. J. Physiol. Pharmacol. 2016, 94, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, C.; Kvint, S.; Frazzitta, G.; Bera, R.; Ferrazzoli, D.; Di Rocco, A.; Rebholz, H.; Friedman, E.; Pezzoli, G.; Quartarone, A.; et al. Intensive Rehabilitation Enhances Lymphocyte BDNF-TrkB Signaling in Patients With Parkinson's Disease. Neurorehabil. Neural Repair 2016, 30, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.A.; van Wegen, E.E.H.; Newman, M.A.; Heyn, P.C. Exercise-induced increase in brain-derived neurotrophic factor in human Parkinson’s disease: A systematic review and meta-analysis. Transl. Neurodegener. 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Tuon, T.; Valvassori, S.S.; Dal Pont, G.C.; Paganini, C.S.; Pozzi, B.G.; Luciano, T.F.; Souza, P.S.; Quevedo, J.; Souza, C.T.; Pinho, R.A. Physical training prevents depressive symptoms and a decrease in brain-derived neurotrophic factor in Parkinson’s disease. Brain Res. Bull. 2014, 108, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Real, C.C.; Ferreira, A.F.; Chaves-Kirsten, G.P.; Torrão, A.S.; Pire, R.S.; Britto, L.R. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience 2013, 237, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Xu, Y.; Chen, G.; Ma, K.; Han, C.; Guo, Z.; Zhang, Z.; Ye, K.; Cao, X. Small molecule TrkB agonist deoxygedunin protects nigrostriatal dopaminergic neurons from 6-OHDA and MPTP induced neurotoxicity in rodents. Neuropharmacology 2015, 99, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Shi, Y.; Wang, J.; Lin, Q.; Sun, Y.; Ye, K.; Yan, Q.; Zhang, H. 7,8-dihydroxyflavone protects 6-OHDA and MPTP induced dopaminergic neurons degeneration through activation of TrkB in rodents. Neurosci. Lett. 2016, 620, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, N.R.S.; Caesar, J.; Park, A.; Sedgh, S.; Finogenov, G.; Masliah, E.; Davis, J.; Blurton-Jones, M. Neural Stem Cells Rescue Cognitive and Motor Dysfunction in a Transgenic Model of Dementia with Lewy Bodies through a BDNF-Dependent Mechanism. Stem Cell Rep. 2015, 5, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sun, J.; Zhao, M.; Hu, J.; Wang, X.; Du, G.; Chen, N.H. Overexpression of alpha-synuclein down-regulates BDNF expression. Cell Mol. Neurobiol. 2010, 30, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson's disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Goutan, E.; Marín, C.; Rey, M.J.; Ribalta, T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000, 866, 257–261. [Google Scholar] [CrossRef]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Vitali, B.; Tarditi, A.; Mariotti, C.; Valenza, M.; Lahiri, N.; Wild, E.J.; Sassone, J.; Ciammola, A.; et al. Brain-derived neurotrophic factor in patients with Huntington's disease. PLoS ONE 2011, 6, e22966. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Yang, J.; Li, T.; Milner, T.A.; Hempstead, B.L. Selective reduction of striatal mature BDNF without induction of proBDNF in the zQ175 mouse model of Huntington's disease. Neurobiol. Dis. 2015, 82, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.Q.; Rymar, V.V.; Sadikot, A.F. Impaired TrkB Signaling Underlies Reduced BDNF-Mediated Trophic Support of Striatal Neurons in the R6/2 Mouse Model of Huntington’s Disease. Front. Cell Neurosci. 2016, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington's disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Connor, B.; Sun, Y.; von Hieber, D.; Tang, S.K.; Jones, K.S.; Maucksch, C. AAV1/2-mediated BDNF gene therapy in a transgenic rat model of Huntington's disease. Gene Ther. 2016, 23, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Remmers, F.; Lutz, B.; Leschik, J. ESC-Derived BDNF-Overexpressing Neural Progenitors Differentially Promote Recovery in Huntington’s Disease Models by Enhanced Striatal Differentiation. Stem Cell Rep. 2016, 7, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Holzbaur, E.L. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, B.; Eckenstaler, R.; Rönicke, R.; Leschik, J.; Lutz, B.; Reymann, K.; Lessmann, V.; Brigadski, T. Amyloid-Beta Induced Changes in Vesicular Transport of BDNF in Hippocampal Neurons. Neural Plast. 2016, 4145708. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.J.; Silverman, M.A. Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in Aβ oligomer-treated hippocampal neurons. Mol. Biol. Cell 2015, 26, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson's disease. Sci. Rep. 2017, 7, 3868. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain 2012, 135 Pt 7, 2058–2073. [Google Scholar] [CrossRef]
- Adachi, N.; Numakawa, T.; Nakajima, S.; Fukuoka, M.; Odaka, H.; Katanuma, Y.; Ooshima, Y.; Hohjoh, H.; Kunugi, H. Glucocorticoid affects dendritic transport of BDNF-containing vesicles. Sci. Rep. 2015, 5, 12684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meco, A.; Joshi, Y.B.; Lauretti, E.; Praticò, D. Maternal dexamethasone exposure ameliorates cognition and tau pathology in the offspring of triple transgenic AD mice. Mol. Psychiatry 2016, 21, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Kurkowska-Jastrzebska, I.; Litwin, T.; Joniec, I.; Ciesielska, A.; Przybyłkowski, A.; Członkowski, A.; Członkowska, A. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int. Immunopharmacol. 2004, 4, 1307–1318. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113650
Numakawa T, Odaka H, Adachi N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. International Journal of Molecular Sciences. 2018; 19(11):3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113650
Chicago/Turabian StyleNumakawa, Tadahiro, Haruki Odaka, and Naoki Adachi. 2018. "Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases" International Journal of Molecular Sciences 19, no. 11: 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113650