Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets

1
Department of Pharmacology, Faculty of Medicine, University of Valencia, 46010 Valencia, Spain
2
CIBERES, Health Institute Carlos III, 28029 Valencia, Spain
3
Pharmacy Unit, University Clinic Hospital of Valencia, 46010 Valencia, Spain
4
Institute of Health Research-INCLIVA, 46010 Valencia, Spain
5
Research and teaching Unit, University General Hospital Consortium, 46014 Valencia, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2019, 20(3), 593; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030593
Submission received: 20 December 2018
/
Revised: 18 January 2019
/
Accepted: 28 January 2019
/
Published: 30 January 2019
(This article belongs to the Special Issue Links between Fibrogenesis and Cancer: Mechanistic and Therapeutic Challenges)
Abstract
:Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pulmonary disease with a median survival of 2–4 years after diagnosis. A significant number of IPF patients have risk factors, such as a history of smoking or concomitant emphysema, both of which can predispose the patient to lung cancer (LC) (mostly non-small cell lung cancer (NSCLC)). In fact, IPF itself increases the risk of LC development by 7% to 20%. In this regard, there are multiple common genetic, molecular, and cellular processes that connect lung fibrosis with LC, such as myofibroblast/mesenchymal transition, myofibroblast activation and uncontrolled proliferation, endoplasmic reticulum stress, alterations of growth factors expression, oxidative stress, and large genetic and epigenetic variations that can predispose the patient to develop IPF and LC. The current approved IPF therapies, pirfenidone and nintedanib, are also active in LC. In fact, nintedanib is approved as a second line treatment in NSCLC, and pirfenidone has shown anti-neoplastic effects in preclinical studies. In this review, we focus on the current knowledge on the mechanisms implicated in the development of LC in patients with IPF as well as in current IPF and LC-IPF candidate therapies based on novel molecular advances.
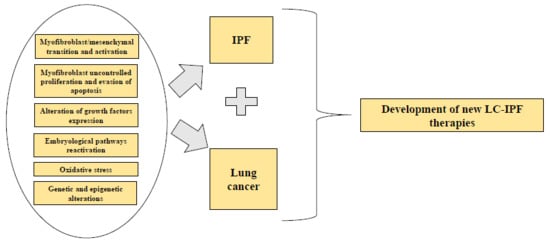
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a form of chronic, progressive fibrosing interstitial pneumonia of unknown cause that occurs primarily in older adults. IPF is associated with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP) that is limited to the lungs [1]. The disease course of IPF is variable and somewhat unpredictable, nevertheless, progression to end-stage respiratory insufficiency and death after the onset of symptoms from diagnosis is 2–4 years [2]. Pulmonary and extrapulmonary comorbid conditions, such as lung cancer (LC), are commonly associated to IPF, altering the disease course and mortality. Links between pulmonary fibrosis and LC have been suggested as early as 1965 [3] and are based on the multiple common genetic, molecular, and cellular processes that connect both diseases and can predispose the patient to develop IPF and LC.
On its own, pulmonary fibrosis is a risk factor for developing lung carcinogenesis [4,5,6,7]. Moreover, elder age, male sex, history of smoking, and coexisting emphysema are also strong risk factors that contribute to developing LC in IPF patients [8,9,10,11,12,13,14,15,16,17,18,19]. The prevalence of LC in IPF patients ranges from 2.7% to 48% [8,9,13,14,15,16,17,18,19,20] (Table 1) and is significantly higher than in the general population [21]. Otherwise, the incidence of LC in IPF patients is reported to be 11.2–36 cases per 1,000 persons per year [8,20,22], which increases with each year following IPF diagnosis [15,18]. Moreover, IPF patients that are diagnosed with LC have a reduced mean survival time (1.6–1.7 years), compared to IPF patients without LC diagnosis [10,17] and Kato et al. reported 53.5%, 78.6%, and 92.9% as 1-, 3-, and 5-year all-cause mortality rates after LC diagnosis in IPF patients [8].
2. Histological Subtypes and Parenchymal Distribution of Lung Cancer in Idiopathic Pulmonary Fibrosis
In the general population, the predominant type of LC is non-small cell lung cancer (NSCLC). Likewise, NSCLC is the predominant type of LC in LC-IPF patients. Furthermore, adenocarcinoma (ADC) is the most common subtype of histological NSCLC in the general population [23]. However, the most frequent histological subtype of LC in IPF has been controversial over the past few years (Table 2). Recently, the majority of studies have shown that squamous cell carcinoma (SQC) is the most frequent type of LC in IPF patients, while ADC is the second most frequent [8,9,10,14,15,17,18,19,24,25,26,27,28,29]. Moreover, some isolated cases of large cell carcinoma and small cell lung cancer (SCLC) have also been reported [8,14,18,27].
Similarly to fibrotic lesions, lung carcinomas are generally more frequently found in the peripheral area of the lungs in IPF patients, i.e., in the lower lobes [3,8,16,25,30,31,32], and are associated with honeycomb lesions, developing from honeycomb areas or in the border between honeycombing and non-fibrotic areas, and epithelial metaplasia [33,34,35]. In general, squamous metaplasia, but not cuboidal cell metaplasia or bronchial cell metaplasia, have been observed more frequently in LC-IPF patients than in IPF patients without lung carcinoma. Then, it is speculated that it might reflect a constitutional susceptibility of IPF patients of developing lung carcinoma [36].
3. Cell Types and Cellular Processes Involved in Lung Cancer Associated with Pulmonary Fibrosis
3.1. Cell Transformations in the Mesenchymal Phenotype
IPF is characterised by an excess of myofibroblasts that are persistently activated in fibrotic lungs [37]. The activated myofibroblasts are stellate- or spindle-shaped, and are characterised by the secretion of extracellular matrix (ECM) components (a characteristic shared with fibroblasts), such as collagen type I, and by the formation of contractile apparatus (a characteristic shared with airway smooth muscle cells), such as α-smooth muscle actin (α-SMA) microfilaments [38]. In IPF lungs, myofibroblasts have heterogeneous phenotypes [39] (Figure 1). The classic concept is that tissue injury induces the activation of resident fibroblasts to proliferate and express constituents of the ECM and α-SMA fibres [40]. One contemporary theory is that tissue injury in the presence of transforming growth factor (TGF-β) induces epithelial to mesenchymal cell transition (EMT) [41,42]. In detail, EMT is part of an unabated form of wound healing in which alveolar type II (ATII) cells [41,42,43] can serve as a source for the increased myofibroblast-like pool that eventually leads to organ destruction if the primary inflammatory insult that triggered the wound healing is not removed or attenuated [44]. Another contemporary theory of myofibroblast activation is that circulating fibrocytes that originate from the bone marrow are mesenchymal progenitor cells that home and extravasate into sites of tissue injury, and differentiate into the myofibroblast-like phenotype [45,46] in response to TGF-β and endothelin 1 [47,48,49]. Pulmonary arterial endothelial cell to mesenchymal transition (EnMT) has been suggested as another source of myofibroblasts that potentially contribute to lung fibrosis and pulmonary hypertension and is often associated with IPF, which portends a poor prognosis [50,51]. Finally, there are two emerging theories that consider pleural mesothelial cells or lung pericytes as significant sources of lung myofibroblasts in IPF [52,53,54,55,56].
Similarly to IPF, cancer-associated fibroblasts (CAFs) are also important players in LC because they exhibit mesenchymal-like features and have heterogeneous phenotypes (Figure 1) [57]. Lung resident fibroblasts surrounding the malignancy are thought to be the first responders to the site of insult that the tumour creates [58]. Resident epithelial cells—generally bronchiolar epithelial cells—can also undergo a partial and possibly reversible EMT during the early steps of carcinogenesis, cancer invasion, and metastasis [44,59]. Likewise, fibrocytes recruited from the peripheral circulation have also been suggested as a potential source of CAFs [58,60,61]. It has also been observed that up to 40% of CAFs arise as a consequence of EndMT in two different murine cancer models, suggesting that EndMT may play a role in tumour angiogenic sprouting into adjacent tissue [62]. In the tumour environment, vascular pericytes have also been associated with tumour vasculature. Pericytes that detach from the tumour microvasculature have been shown to undergo differentiation into stromal fibroblasts via the action of platelet-derived growth factor-BB (PDGF-BB), which significantly contributes to tumour invasion and metastasis [63]. Finally, pleural mesothelial cells are also hypothesised as a source of CAFs, where a mesothelial precursor lineage has been identified as being capable of clonally generating fibroblasts in the lungs, kidney, liver, and gut [64]. Also, it has been reported that an overexpression of mesothelin in lung ADC is induced by tobacco-related carcinogens [57].
3.2. Common Cellular Processes in Lung Cancer Associated with Pulmonary Fibrosis
3.2.1. Apoptosis and Autophagy
Growing evidence indicates a prominent role of enhanced endoplasmic reticulum (ER) stress in IPF, resulting in an unfolded protein response (UPR) [65,66]. This response mechanism activates biochemical pathways to meet the demands of protein folding. However, if that is no longer feasible, a terminal UPR directs alveolar epithelium cells towards apoptosis. By contrast, IPF myofibroblasts and cancer cells escape apoptosis [67,68]. Regarding the role of ER-stress in tumourigenesis, it is controversial [69,70], nevertheless, recent evidence shows that ER stress may attenuate senescence and promote tumorigenesis [71] (Figure 1).
Despite elevated ER stress, there is evidence that autophagy is defective in IPF [72,73,74] (Figure 1), which promotes lung fibroblast differentiation into myofibroblasts via excessive ECM production [72,74,75] and fibroblast resistance to apoptosis [76]. In LC, autophagy functions as a double-edged sword because it suppresses tumorigenesis in a limited number of contexts while facilitating it in most others [77]. In fact, it has been observed that autophagy can promote or inhibit apoptosis under different cellular contexts within the same tumour cell population. Therefore, therapeutic targeting of autophagy in cancer is sometimes viewed as controversial [78].
3.2.2. Cellular Proliferation
Evidence strongly supports the persistent activation of proliferative signalling pathways in IPF (Figure 1). In fact, myofibroblasts sustain their own growth through the autocrine production of TGFβ1 and partly lose their ability to produce anti-fibrotic prostaglandin E2 (PGE2) [79]. Further, they show a lack of response to the inhibitory activity of PGE2 [80], and to other antiproliferative signals [81]. The receptors for platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF) have also recently been implicated in the sustained proliferation signalling of pulmonary fibroblasts [82]. However, this persistent activation is not definitively linked with aberrant fibroblast proliferation in vivo [83,84,85] and the role of excessive fibroblast proliferation as a pathogenic mechanism of IPF is unclear. By contrast, aberrant proliferation of cancer cells and sustained proliferative signalling has been described as “arguably the most fundamental trait of cancer cells” [68].
3.2.3. Altered Cell-Cell Communications
Intercellular channels that are formed by connexins are essential for the synchronisation of cell proliferation and tissue repair [86]. In particular, the expression of connexin 43 (Cx43) is considered crucial in fibroblast-to-fibroblast communication. In IPF fibroblasts, Cx43 expression is strongly down-regulated, leading to a loss of proliferative control [87]. Similarly, cancer cell lines from mouse and human lung carcinoma have low levels or an absence of Cx43 expression [88], which results in reduced cell-to-cell communication (Figure 1); this may explain both the release of cells from contact-inhibition control, and the uncontrolled proliferation that characterises this disease.
3.2.4. Senescence
Analyses of cell types in the lungs of both human IPF and the bleomycin-injured mouse model have demonstrated that fibroblasts and epithelial cells acquire senescent identities [89]. Senescence appears to be a central phenotype that promotes lung fibrosis through increased production of a complex senescence-associated secretory phenotype (SASP) based on growth factors, cytokines, chemokines, and matrix metalloproteinases, as well as acquired apoptosis resistance in IPF fibroblasts [90] (Figure 1). However, therapeutic management of cell senescence is controversial in cancer. On the one hand, cell senescence could limit the replicative capacity of cells and ultimately prevent their proliferation in different stages of malignancy, while providing a protective barrier to neoplastic expansion [91]. On the other hand, it has been proposed that senescent fibroblasts may promote tumour progression, possibly by secreting an SASP based on certain matrix metalloproteases, growth factors, and cytokines [92].
3.2.5. Tissue Invasion
IPF lung fibroblasts are characterised by their ability to invade through the basement membrane and the ECM via the action of metalloproteinases [93,94]. This characteristic is also an important hallmark of cancer (Figure 1). Unlike cancers that can disseminate over long distances because they acquire further invasive mechanisms, fibrotic lung fibroblasts are restricted to local invasion [95]. The capacity of cancer cells to infiltrate the surrounding tissue is strictly related to the expression of laminin, heat shock protein 27, and fascin [96,97,98]. Interestingly, in IPF, it has been shown that bronchiolar basal cells surrounding the fibroblast foci express large amounts of these proteins, which induce cell motility and invasiveness of myofibroblasts [99]. Therefore, targeting these molecules may be a feasible strategy to restrain myofibroblast tissue invasion in LC-IPF patients.
3.2.6. Inflammation
The role of inflammation in IPF is controversial, although evidence shows the existence of a predominant phenotype of fibrosis-associated macrophages (FAMs) that are alternatively activated. These are an M2 phenotype of FAMs [100] that facilitate the enhanced production of FGFs [101], profibrotic cytokines [102,103], and matrix metalloproteinases [104]. Like FAMs, tumour-associated macrophages also display an M2 phenotype and support tumour growth through their ability to promote angiogenesis, activate mesenchymal cells, remodel the matrix, and suppress effector T-cell responses [105,106]. Thus, M2 macrophages could be considered key effectors in the development of LC associated with pulmonary fibrosis.
4. Principal Fibrogenic Molecules and Signal Transduction Pathways Participating in Lung Cancer Associated with Pulmonary Fibrosis
4.1. Growth Factors
TGFβ is a major profibrotic growth factor and is often chronically overexpressed in cancer and fibrosis (Table 3) [107]. TGFβ can be activated by αVβ6 integrin and, in IPF, TGFβ1 mediates fibrogenesis by antiproliferative action and apoptosis in alveolar epithelial cells (AECs) or by stimulation of fibroblast differentiation to myofibroblasts, synthesis of ECM proteins, and inhibition of ECM degradation [108,109]. TGFβ also induces the production of fibrogenic or angiogenic growth factors and is known to strongly elicit EMT and EndMT. In the early stages of cancer pathogenesis, TGFβ acts as a tumour suppressor because it inhibits the growth of many cell types and delays the appearance of primary tumors. However, after the appearance of them, TGFβ promotes tumour progression, because it can induce EMT and EndMT, and suppresses immune surveillance. Therefore, during tumour progression, TGFβ triggers the formation of spontaneous lung metastases. Finally, TGFβ is also central in the development of the tumour stroma because TGFβ1 also activates CAFs [107,110].
Tyrosine kinase receptor ligands, such as PDGF, VEGF, and FGF, are aberrantly expressed in LC and IPF (Table 3) [82]. In IPF, PDGF plays an important role in inducing the secretion of ECM components and growth factors in fibroblasts [111]. It also promotes fibroblast proliferation and recruits fibrocytes to the lung [112,113]. Furthermore, TGFβ1, FGF, and tumour necrosis factor-α exhibit PDGF-dependent profibrotic activity [114,115]. Otherwise, PDGF signalling is also important for tumour growth, angiogenesis, and lymphangiogenesis in cancer [113]. In fact, it has been shown that crenolanib (PDGF receptor inhibitor) is capable of suppressing proliferation and inducing apoptosis in a dose-dependent manner using A549 cells as a NSCLC model system. Moreover, it has been shown that crenolanib-treated A549 cells have reduced migratory activity in response to inducers of chemotaxis, and the antitumor activity of this drug has been confirmed in an NSCLC xenograft tumor model [116]. Finally, it has been observed that PDGF regulates VEGF expression in NSCLC via an autocrine mechanism [117], and is also involved in the recruitment of CAFs to the tumour mass [113]. The contribution of VEGF to IPF is not fully understood because there is still debate on the role of vascular remodelling in IPF [118]. However, in addition to the role of VEGF in tumour angiogenesis, accumulating evidence suggests that it can act directly on cancer cells to regulate growth, migration, and production of several pro-angiogenic factors [119]. FGF is also released by damaged epithelial cells and activated fibroblasts during the remodelling processes [120,121]. It was found that FGF signalling is required for fibroblast expansion within fibrotic areas [122]. FGF can also affect the proliferation, treatment sensitivity, and apoptosis of LC cells [123].
Another important fibrogenic growth factor in LC is connective tissue growth factor (CTGF). In IPF, CTGF induces fibroblast proliferation and ECM deposition [124]. By contrast, it has been observed that CTGF inhibits metastasis and invasion of human lung ADC [125], and its expression is suppressed in many NSCLCs [126] (Table 3).
4.2. Lysophosphatidic Acid (LPA)
LPA, a profibrotic mediator with proinflammatory activity, is released by platelets during epithelial injury [130]. Extracellular production of LPA is catalysed by autotaxin (ATX) and further regulated by phospholipid phosphatases (PLPP). IPF patients have increased LPA levels [131] in their bronchoalveolar lavage fluid (BALF) (Table 3), and recently, it has been shown that LPA signalling mediates both the fibroblast recruitment and vascular leakage induced by lung injury in a bleomycin model of pulmonary fibrosis [131]. Moreover, it has recently been shown that the ATX/PLPP3-LPA/LPA receptor 1 (LPAR1) axis has a procarcinogenic role in lung carcinogenesis [132].
4.3. Cytokines and Chemokines
Epithelial injury causes an imbalance in T-helper type 1 (Th1)/type 2 (Th2) cytokine expression, which results in a stronger Th2 response. In particular, there is evidence that interleukin 13 (IL-13) [133] plays a dominant role in the pathogenesis of fibrosis in the lungs of IPF patients (Table 3) [134]. IL-13 triggers the transformation of fibroblasts to myofibroblasts via the TGFβ-dependent and -independent pathways, while also inducing epithelial apoptosis [135,136,137]. Similarly to IPF, a pattern of Th2 cytokine expression has also been identified in NSCLC [138]. With respect to IL-13, a recent study observed the highest expression level of IL-13 in LC in the SQC subtype, followed by the ADC subtype [139]. Moreover, a clear association between IL-13 receptor subunit alpha-2 overexpression and poor survival in resected NSCLC patients has been shown [140]. There are other profibrotic cytokines besides IL-13 that are also associated with IPF. For example, chemokine ligand 2 (CCL2) has been reported to be present in the BALF of IPF patients at significant concentrations [141] (Table 3). In IPF, CCL2 has been shown to induce the differentiation of developing T-cells into type 2 cells [142], and to stimulate collagen synthesis and TGFβ expression in lung fibroblasts [141]. In LC, the CCL2/CCR2 (chemokine receptor type 2) axis is also important in several aspects of tumorigenesis. One of its most important roles is the generation of new vascular structures that allow tumour growth [140]. However, there is evidence of an association between CCL2 in cancer cells and better survival in NSCLC patients [143].
4.4. Reactive Oxygen Species (ROS)
ROS production by ATII cells results in oxidative stress, which induces apoptosis of epithelial cells, activates intracellular signalling pathways, and upregulates the synthesis of profibrotic cytokines that ultimately leads to tissue injury and fibrosis [144]. In this context, upregulation of NADPH oxidase 4 (NOX4) has been reported in pulmonary fibroblasts and other relevant cells in IPF. In the same way, NOX4 has been reported to be overexpressed in NSCLC (Table 3), contributing to cell proliferation and metastasis [145]. Furthermore, following ROS overproduction, alterations in DNA methylation patterns and specific histone modifications lead to aberrant gene expression, and possibly trigger the multistage process of carcinogenesis [144]. In addition, antioxidant molecules that mitigate oxidative stress, such as Nrf2, have also been reported to be dysregulated in both diseases [146,147] (Table 3). As such, they are proposed to be future targets for anti-IPF/LC treatment.
4.5. Mucins
Significant overexpression of the secreted Mucin 5B (Muc5B) protein has been found in IPF lungs (Table 3) and it is hypothesised that excess Muc5B impairs the mucosal host defence; in turn, this may interfere with alveolar repair and leads to the development of idiopathic interstitial pneumonia [148]. In this context, MUC5B expression has been associated with a high risk of distant metastasis in NSCLC patients and poorer prognosis in ADC patients [149,150].
We have also observed IPF overexpression of the transmembrane mucins, Muc1 [151], Muc4 [152], and Muc16 (unpublished data), which may be involved in the molecular processes that lead to the development of pulmonary fibrosis [151,152,153]. In addition, the extracellular region of Muc1 contains the KL-6 epitope, which is proposed to be a useful biomarker for evaluating disease activity and predicting clinical outcomes in IPF [154]. Similarly, these transmembrane mucins have previously been considered clinically relevant proteins that are aberrantly overexpressed in lung carcinogenesis [155]. In fact, Muc1 is a target in several preclinical and clinical trials for cancer treatment [156,157]. Concurrently, there is evidence that galectin 3 is a promising target for IPF [158] because it has a profibrotic action [159] that is partly mediated by binding to Muc1 [160]. Recently, the potential of galectin-3 as a therapeutic target in cancer has been highlighted since it is capable of modulating anti-tumour immunity [161].
4.6. Embryological Pathways
There is also evidence that some embryological pathways are reactivated or deregulated in fibrotic diseases (Table 3) [162]. For example, the Wnt/β-catenin pathway is overexpressed in the lung tissue of IPF [163] and LC patients [164]. This pathway regulates the expression of molecules involved in tissue invasion, such as matrilysin, laminin, and cyclin-D1, which induces the EMT process. Most importantly, this pathway is involved in biologically relevant cross talk with TGF-β [163].
The Sonic hedgehog (shh) pathway is also aberrantly activated in IPF, mainly in epithelial cells that line honeycomb cysts [165,166]. The overexpression of the shh pathway promotes increased susceptibility to epithelial cell apoptosis and increased resistance to fibroblast apoptosis [167]. This pathway is also reactivated at the early stage of oncogenesis by cancer stem cells and leads to paracrine action on other tumour cells, resulting in tumour growth, tumour spread, and EMT. In LC, reactivation of the shh pathway is involved in the development of resistance to all the main treatments of LC [168].
Finally, the Notch signalling pathway is also reactivated in AECs, induces α-SMA expression in fibroblasts, and mediates EMT in AECs [52]. In the same way, abnormal expression of the members of the Notch signalling pathway is a relatively frequent event in patients with NSCLC [169,170]. It has been demonstrated that members of the Notch signalling pathway may be potential biomarkers for predicting the progression and prognosis of patients with NSCLC. Furthermore, Notch signalling promotes the proliferation of NSCLC cells or inhibits apoptosis of NSCLC cells [171].
4.7. PI3K/AKT/mTOR Pathway
The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR)-dependent pathway is dysregulated in fibroproliferative diseases, like pulmonary fibrosis (Table 3) [172]. In fact, overexpression of class I isoform p110γ in lung homogenates occurs in IPF patients [173], and has been shown to activate the downstream signalling of several key profibrotic growth factors implicated in IPF, including PDGF and TGFβ1 [174,175], as well as abnormal proliferation of epithelial basal cells [173] and TGF-β-induced fibroblast proliferation and differentiation [176]. Moreover, it has been observed that the suppression of phosphatase and tensin homologue mediates matrix-mediated resistance to apoptosis [174]. Phosphatase and tensin homologue are negative regulators of PI3K that in turn activate AKT. De-regulation of the PI3K/AKT/mTOR pathway is also involved in NSCLC and has been associated with high grade tumours and advanced disease. Furthermore, abnormalities in this pathway are more common in SQC than in ADC of the lung [177].
5. Genetic and Epigenetic Alterations in Lung Cancer Associated with Pulmonary Fibrosis
5.1. Genetic Alterations
Most pulmonary fibrosis patients who have a background of familial clustering of familial interstitial pneumonia show mutations in genes that encode surfactant-associated protein C (SFTPC) [178,179], surfactant-associated protein A2 (SFTPA2) [180], telomerase components (TERT and TERC) [181,182], and genes associated with telomere biology, such as poly (a)-specific ribonuclease deadenylation nuclease (PARN) and regulator of telomere elongation helicase 1 (RTEL1) [183]. Further, several common variants in TERT, genomic regions near TERC, oligonucleotide-binding fold containing 1 (OBFC1), RTEL1, PARN, and toll-interacting protein (TOLLIP), have all been associated with a sporadic risk of developing IPF [184,185,186]. Nevertheless, the most important genetic risk factor for sporadic IPF is a common variant (rs35705950) in the promoter region of the MUC5B gene, although it is also associated with familial pulmonary fibrosis [148] (Table 4).
LC development is the result of a stepwise accumulation of multiple acquired mutations of tumour suppressor genes or candidates, and the overexpression and mutation of oncogenes (Table 4). In this context, multiple P53 gene mutations have been found during the early stage of bronchial carcinoma [187,188]. Frequent P53 gene alterations have also been detected in epithelial lesions from IPF patients [189] and in squamous metaplasia, distributed in the peripheral zone of the fibrotic area in patients with IPF [190]. Similarly, aberration and loss of function of the candidate tumour suppressor gene fragile histidine triad (FHIT) has been reported in NSCLC [191], as well as in IPF lesions [192]. Additionally, FHIT gene allelic loss has been seen more frequently among the metaplasias and bronchiolar epithelia samples obtained from LC-IPF patients than from IPF patients without LC [192]. Otherwise, the frequency of expression of Ras protein in ATII cells has been observed as being significantly greater in lung tissues from LC-IPF patients compared with lung tissues from IPF patients who did not have lung carcinoma. Moreover, a specific point mutation in codon 12 of the KRAS gene has been detected in LC-IPF patients [189]. Interestingly, this mutation has not been identified between the numerous KRAS mutations observed in lung carcinoma tissue [193]. However, contrasting results regarding the prevalence of KRAS mutations in LC-IPF patients have been reported recently [194,195]. In addition, a significantly higher prevalence of BRAF mutations in IPF-LC than in LC has been observed, with an equal distribution between ADC and SQC subtypes. Moreover, some of these somatic mutations have not been shown to be significant in NSCLC patients [195]. As such, it is rational to suggest that these somatic mutations in tumour suppressor genes and oncogenes, as well as oncogene overexpression, predispose IPF patients to develop LC. However, it also raises two controversial questions: Why is LC not the cause, instead of a consequence, of IPF?; and why do LC and IPF not independently and synchronously develop as a result of common pathogenetic mechanisms? In answering these questions, Hwan et al. [195] revealed a predominance of C>T somatic transitions in most of the somatic mutations detected in LC-IPF patients. By contrast, in the non-IPF SQC subtype, C>A transversions are the most frequent [196]. This suggests a potential association between APOBEC (cytidine deaminase, which converts cytosine to uracil)-related mutagenesis and the development of LC associated with pulmonary fibrosis.
Recently, germline mutations associated with familial NSCLC and predisposing to it are also being discovered [197] (Table 4). In this context, several findings suggest that some germline mutations that predispose patients to develop IPF also predispose them to developing lung carcinoma. Indeed, two heterozygous missense mutations, and a heterozygous missense mutation in SFTPA2 [198] and SFTPA1 [199], respectively, have been identified in LC-IPF families. All of these mutations are predicted to disrupt the structure of surfactant A protein (SP-A) and impair protein secretion [198,199], leading to protein instability and ER stress of resident ATII cells [179,200]. SP-A is produced by ATII and club cells [201], which have both been proposed as possible initiators of lung ADC [202]. Although the role of ER-stress in tumourigenesis is controversial [69,70], recent evidence showing that ER stress may attenuate senescence and promote tumorigenesis might explain the co-occurrence of LC (histological subtype ADC) and pulmonary fibrosis in families with an SFTPA1/2 mutation [71]. Two further germline mutations in TERT (rs2736100) and CDKN1A (rs2395655), which were previously reported to confer IPF risk [203], have also been identified in several LC-IPF patients [195]. These mutations affect telomerase function and impair the cellular response to DNA damage, respectively [204]. Accordingly, they might also explain the co-occurrence of both diseases. Furthermore, the germline variant, rs2736100, has been reported to be associated with lung ADC risk [205], and other TERT, TERC, OFBC1, and RTEL1 polymorphisms have also been revealed as risk factors of LC [206,207]. However, telomere functionality and its contribution to LC development is controversial. In fact, a gain at chromosomal region 5p15.33 in TERT is the most frequent genetic event in the early stages of NSCLC [208]. However, short telomeres in peripheral blood leukocytes have been related to an increased risk of lung carcinoma [209], probably via an increased mutation rate and the genomic instability induced by telomere dysfunction [210]. Therefore, it might be hypothesised that mutations associated with telomere biology in IPF lesions, which correlate with shortened telomeres in leukocytes and ATII cells [211], could drive the development of LC via an increased mutation rate and genomic instability.
Finally, the germline or somatic variant (rs35705950) in the MUC5B promoter region that consists of TT and GT genotypes (risk genotypes for IPF) has been reported to confer a survival advantage among patients with IPF [212]. However, these genotypes are associated with poorer overall survival in NSCLC patients [213]. Furthermore, significant associations between the MUC5B promoter polymorphism and the incidence of radiation pneumonitis in patients with NSCLC have not been identified [213]. This supports the idea that IPF underlies the development of LC and is not a consequence of it.
5.2. Epigenetic Alterations
Due to similar pathogenic mechanisms between IPF and LC, their global methylation patterns are also somewhat similar (Table 4). However, there are also differences, which may be explained partly by IPF or cancer-specific changes [214]. For example, it was found that as a consequence of promoter hypermethylation, the relative expression of the SMAD4 gene was significantly lower in the tumours of LC-IPF patients compared to those who had LC without IPF [215]. This was a surprising finding because SMAD4 has been identified as a tumour-suppressor gene [216], but TGFβ1 signalling is the main effector in pulmonary fibrosis. Thus, SMAD4 over-expression would be expected in this disease. Another epigenetic alteration involved in IPF is THY-1 promoter hypermethylation and the absence of fibroblast Thy-1 expression, which is linked to the transformation of fibroblasts into myofibroblasts [217]. Loss of this molecule has also been documented in cancer and is associated with a more invasive disease [218]. By contrast, promoter hypermethylation of the O-6-methylguanine DNA methyltransferase (MGMT) gene is one of the early epigenetic marks in LC [219], while in IPF fibroblasts, MGMT is one of the most hypomethylated genes [220].
Otherwise, ~10% of miRNAs are abnormally expressed in IPF [95]. These variations are all capable of influencing EMT and inducing the regulation of apoptosis or ECM [95]. Some of these variations are also found in LC. For example, common to IPF, mir-21 is overexpressed in LC [95], which is an independent negative prognostic factor for overall survival in NSCLC patients [221]. By contrast, Let-7d expression is found to be mostly down-regulated in IPF and LC and acts as an oncogene [95,219,222].
6. Therapeutic Management in Lung Cancer Associated with Pulmonary Fibrosis Patients
The focus of IPF treatment in previous decades has been to use anti-inflammatory/immunomodulatory drugs in combination with antioxidants. However, their therapeutic usefulness was recently questioned given the unfavourable outcome when N-acetyl L-cysteine (NAC) was used in combination with prednisolone/azathioprine [224]. Following this, NAC monotherapy results were also negative [225], although a subgroup of IPF patients with the rs3750920 (TOLLIP) TT genotype showed a favourable response [226]. Numerous cellular and preclinical studies hold that antioxidants protect against cancer [227,228]. However, it has been shown that NAC increases tumour progression and reduces survival in LC preclinical models [229], which contraindicates NAC treatment for LC-IPF.
In line with the antioxidant treatments tested in IPF, pirfenidone was initially considered as an antioxidant therapy since it demonstrated O2− scavenging activity [230,231]. Oral NAC has been used in conjunction with pirfenidone to treat IPF, but it does not substantially alter the tolerability profile of pirfenidone and is unlikely to be beneficial in IPF [232]. Beyond its antioxidant activity, pirfenidone is a pleiotropic molecule that inhibits TGF−β, collagen synthesis, and fibroblast proliferation, and also mediates tissue repair [233,234,235,236]. It is currently approved as an IPF therapy and Miuri et al. [237] observed that the incidence of LC in IPF patients treated with pirfenidone was significantly lower than in a non-pirfenidone IPF patient group. Furthermore, recent publications have shown that pirfenidone confers anti-fibrotic effects by interfering with the shh pathway [238], which can partly explain the observed lower LC incidence in IPF patients treated with pirfenidone. It has also been observed that perioperative pirfenidone treatment reduces the incidence of postoperative acute exacerbation of IPF in LC-IPF patients [239]. Moreover, experimental data have shown that the combination of pirfenidone and cisplatin may lead to an increase of CAF and tumour cell mortality in NSCLC preclinical models [240].
Advances in the understanding of IPF pathogenesis have resulted in further preclinical and clinical trials of drugs with antiproliferative and antifibrotic effects. For instance, tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib, gefitinib, erlotinib, nintedanib, SU5918, and SU11657, are being investigated [241,242,243,244,245,246,247]. The important role of these inhibitors in cancer treatment was previously shown [248]. Indeed, gefitinib and erlotinib are important oral treatments for NSCLC patients with mutations that activate the epidermal growth factor receptor (EGFR). In IPF, imatinib was tested in fibrotic patients, but failed to show any benefit on survival or lung function [249]. In contrast, the VEGF, FGF, and PDGF receptor inhibitor, nintedanib, has been approved for IPF treatment. Additionally, this drug is also approved for use in combination with docetaxel as an effective second-line option for patients with advanced ADC-NSCLC who have been previously treated with one course of platinum-based therapy [250].
Another class of antifibrotic drugs are the mTOR kinase inhibitors, including everolimus, which failed as an IPF treatment [251]. However, everolimus has shown modest beneficial effects in patients with advanced NSCLC who were previously treated with chemotherapy alone, or with chemotherapy and EGFR inhibitors [252]. It is also approved as a second-line treatment in renal and breast cancer. Currently, there are efforts towards assessing the efficacy of a new mTOR kinase inhibitor (GSK-2126458) for IPF and advanced solid tumour treatment.
In addition to the previously mentioned therapeutic strategies, a broad range of IPF therapies are currently being tested in clinical trials (Table 5). Some of these therapies target molecules and mechanisms mentioned in this review, and which are hallmarks of the progression of both diseases. These include anti-IL-13 antibodies (QAX576 and Lebrikizumab), anti-CCL2 antibodies (Carlumab and CNTO-888), anti-TGFβ1 antibodies (Fresolimumab and GC1008), anti-integrin αvβ6 antibodies (BG0011 and STX-100), integrin αvβ6 antagonist drugs (GSK3008348), LPAR1 antagonist drugs (BMS-986020), ATX-inhibiting drugs (GLPG1690), angiostatic drugs (Tetrathiomolybdate), shh pathway-inhibiting drugs (vismodegib), and galectin-3-inhibiting drugs (TD139). Only two of these drugs are being clinically developed for NSCLC patients. Fresolimumab, which was tested in combination with stereotactic ablative radiotherapy in a phase I study, and tetrathiomolybdate in combination with carboplatin and pemetrexed, which is currently being tested in a phase I study. There are also preclinical studies for NSCLC that include some of these target molecules. For example, CCR2 antagonism was demonstrated to supress CCL2-mediated viability, motility, and invasion of the NSCLC cell line, A549, in vitro [253]. Likewise, galectin-3 knockdown in NSCLC cell line-derived sphere resulted in attenuation of lung carcinogenesis by inhibiting stem-like properties [254]. In the same way, genetic deletion of ATX and LPAR1 was shown to attenuate lung carcinogenesis development in animal models [132]. Moreover, vismodegib is approved for the treatment of metastatic, local, or recurrent advanced basal cell carcinoma (BCC), although it has not been tested in NSCLC. Nevertheless, blockade of shh signaling synergistically has shown to increase sensitivity to EGFR-TKIs in primary and secondary resistant NSCLC cells [255].
Given the mechanistic similarities between LC and IPF diseases, and the concurrence of predominantly NSCLC and IPF, it is rational to consider the usefulness of the large number of approved NSCLC treatments for the management of pulmonary fibrosis. For example, Nivolumab is a new immunomodulatory agent that acts as a programmed death receptor-1-blocking antibody. One case study of an ADC patient with IPF showed a beneficial and sustained response in the lung, without any sign of IPF exacerbation after Nivolumab treatment [256]. This could be explained by the higher expression of programmed cell death ligand 1 reported in cancer cells from UIP-associated SQC versus non-UIP SQC patients [257]. Other examples of feasible IPF and LC-IPF treatment candidates include vantictumab, which interferes with Wnt signalling and has undergone Phase I trials for NSCLC (preclinical studies of Wnt pathway inhibition have also been performed in pulmonary fibrosis [258,259]), and Muc1-based therapeutic strategies. Indeed, four Muc1-based Phase III trials exploring cancer treatment have been completed, one of which used a Muc1 tandem repeat peptide as an immunogen (L-BLP25) in patients with stage III unresectable NSCLC after chemoradiation [260].
Finally, rovalpituzumab treatment, although not tested for NSCLC, is currently approved for SCLC, and it could also have potential in the treatment of IPF and LC-IPF, since it interferes with the Notch signalling pathway. In fact, it has been observed that artesunate ameliorates lung fibrosis via inhibiting the Notch signaling pathway in a rat bleomycin model [261].
7. Conclusions
The course of IPF disease and its resulting mortality are altered by the frequent co-occurrence of LC. This review supports the view of LC as a consequence of a genetic predisposition in IPF patients and, common cellular processes and molecular pathways between both diseases. Currently, there is no consensus regarding the treatment of patients with both diseases. However, pirfenidone and nintedanib are two novel drugs approved for IPF that have potential for treating patients with fibrosis, possibly extending the survival time and lowering the incidence of LC. However, we are some distance from realising effective therapeutic approaches that are capable of stopping the disease process, where disease progression still occurs in most IPF patients despite treatment. Nevertheless, we now have a great deal of knowledge about cancer biology and its similarities with IPF. Therefore, it seems reasonable to investigate whether specific cancer drugs may exert beneficial anti-fibrotic effects that are effective to treat LC-IPF patients. Furthermore, clinical trials that prospectively investigate the efficacy of currently approved anti-fibrotic agents (or agents under study) as treatments for LC-IPF patients are sorely needed.
Author Contributions
Conception and design: B.B., J.M. and J.C. Data acquisition: B.B., J.M., and J.C. Analysis and interpretation: B.B., J.M. and J.C. Writing and Original Draft Preparation, B.B., J.M. and J.C.; Writing Review & Editing, B.B., J.M. and J.C.; Visualization, B.B., J.M. and J.C.; Supervision, B.B., J.M. and J.C.; Project Administration, B.B., J.M. and J.C.; Funding Acquisition, B.B., J.M. and J.C.
Funding
This work was supported by the grants SAF2017–82913-R (J.C.), FIS PI17/02158 (J.M.), CIBERES (CB06/06/0027), TRACE (TRA2009–0311; Spanish Government), and by research grants from the Regional Government Prometeo 2017/023/UV (J.C.), ACIF/2016/341 (B.B.), from “Generalitat Valenciana”. Funding entities did not contribute to the study design or data collection, analysis and interpretation nor to the writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| IPF | Idiopathic pulmonary fibrosis |
| UIP | Usual interstitial pneumonia |
| LC | Lung cancer |
| LC-IPF | Lung cancer associated with idiopathic pulmonary fibrosis |
| NSCLC | Non-small cell-lung cancer |
| ADC | Adenocarcinoma |
| SQC | Squamous cell carcinoma |
| SCLC | Small cell lung cancer |
| ATII | Alveolar type II cells |
| ECM | Extracellular matrix |
| EMT | Epithelial to mesenchymal transition |
| EndMT | Endothelial to mesenchymal transition |
| α-SMA | α-smooth muscle actin |
| CAF | Cancer-associated fibroblast |
| ER | Endoplasmic reticulum stress |
| TGFβ | Transforming growth factor β |
| FGF | Fibroblast growth factor |
| VEGF | Vascular endothelial growth factor |
| PDGF | Platelet derived growth factor |
| IL-13 | Interleukin-13 |
| CCL2 | Chemokine ligand 2 |
| CCR2 | Chemokine receptor 2 |
| BALF | Bronchoalveolar lavage fluid |
| LPA | Lysophosphatidic acid |
| LPAR1 | Lysophosphatidic acid receptor 1 |
| ATX | Autotaxin |
| Shh | Sonic hedhehog |
| TKI | Tyrosine kinase inhibitor |
| EGFR | Epidermal growth factor |
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Meyer, E.C.; Liebow, A.A. Relationship of Interstitial Pneumonia Honeycombing and Atypical Epithelial Proliferation to Cancer of the Lung. Cancer 1965, 18, 322–351. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef]
- Wells, C.; Mannino, D.M. Pulmonary fibrosis and lung cancer in the United States: Analysis of the multiple cause of death mortality data, 1979 through 1991. South. Med J. 1996, 89, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Artinian, V.; Kvale, P.A. Cancer and interstitial lung disease. Curr. Opin. Pulm. Med. 2004, 10, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Kato, E.; Takayanagi, N.; Takaku, Y.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Sugita, Y. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2018, 4, 00111-2016. [Google Scholar] [CrossRef]
- Nagai, A.; Chiyotani, A.; Nakadate, T.; Konno, K. Lung cancer in patients with idiopathic pulmonary fibrosis. Tohoku J. Exp. Med. 1992, 167, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Aubry, M.C.; Myers, J.L.; Douglas, W.W.; Tazelaar, H.D.; Washington Stephens, T.L.; Hartman, T.E.; Deschamps, C.; Pankratz, V.S. Primary pulmonary carcinoma in patients with idiopathic pulmonary fibrosis. Mayo Clin. Proc. 2002, 77, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Tanai, C.; Tanaka, Y.; Noda, H.; Ishihara, T. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology 2011, 16, 326–331. [Google Scholar] [CrossRef] [PubMed]
- JafariNezhad, A.; YektaKooshali, M.H. Lung cancer in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0202360. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Tanaka, S.; Saiki, Y.; Hara, M.; Nakata, K.; Tanimura, S.; Banba, J. Lung cancer associated with usual interstitial pneumonia. Pathol. Int. 1995, 45, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.S.; Shim, T.S.; Lim, C.M.; Koh, Y.; Lee, S.D.; Kim, W.S.; Kim, W.D.; Lee, J.S.; Song, K.S. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Chung, M.P.; Kim, Y.W.; Lee, J.H.; Kim, K.S.; Ryu, J.S.; Lee, H.L.; Park, S.W.; Park, C.S.; Uh, S.T.; et al. Prevalence, risk factors and survival of lung cancer in the idiopathic pulmonary fibrosis. Thorac. Cancer 2012, 3, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuter, M.; Ehlers-Tenenbaum, S.; Schaaf, M.; Oltmanns, U.; Palmowski, K.; Hoffmann, H.; Schnabel, P.A.; Heussel, C.P.; Puderbach, M.; Herth, F.J.; et al. Treatment and outcome of lung cancer in idiopathic interstitial pneumonias. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2015, 31, 266–274. [Google Scholar]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef]
- Yoon, J.H.; Nouraie, M.; Chen, X.; Zou, R.H.; Sellares, J.; Veraldi, K.L.; Chiarchiaro, J.; Lindell, K.; Wilson, D.O.; Kaminski, N.; et al. Characteristics of lung cancer among patients with idiopathic pulmonary fibrosis and interstitial lung disease—Analysis of institutional and population data. Respir. Res. 2018, 19, 195. [Google Scholar] [CrossRef]
- Le Jeune, I.; Gribbin, J.; West, J.; Smith, C.; Cullinan, P.; Hubbard, R. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir. Med. 2007, 101, 2534–2540. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Hyldgaard, C.; Hilberg, O.; Bendstrup, E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir. Med. 2014, 108, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Yakumaru, K.; Suzuki, M.; Kageyama, K. Diffuse interstitial pulmonary fibrosis and lung cancer. Acta Pathol. Jpn. 1987, 37, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Nagai, K.; Yokose, T.; Yoshida, J.; Nishimura, M.; Takahashi, K.; Suzuki, K.; Kakinuma, R.; Nishiwaki, Y. Clinicopathological characteristics of surgically resected lung cancer associated with idiopathic pulmonary fibrosis. J. Surg. Oncol. 2001, 76, 53–57. [Google Scholar] [CrossRef]
- Saito, Y.; Kawai, Y.; Takahashi, N.; Ikeya, T.; Murai, K.; Kawabata, Y.; Hoshi, E. Survival after surgery for pathologic stage IA non-small cell lung cancer associated with idiopathic pulmonary fibrosis. Ann. Thorac. Surg. 2011, 92, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Park, J.Y.; Lee, H.Y.; Cho, Y.J.; Yoon, H.I.; Lee, J.H.; Jheon, S.; Lee, C.T.; Park, J.S. Lung cancer in patients with idiopathic pulmonary fibrosis: Clinical characteristics and impact on survival. Respir. Med. 2014, 108, 1549–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.A.; Kennedy, M.P.; Moore, E.; Crush, L.; Prendeville, S.; Maher, M.M.; Burke, L.; Henry, M.T. Radiological characteristics, histological features and clinical outcomes of lung cancer patients with coexistent idiopathic pulmonary fibrosis. Lung 2015, 193, 71–77. [Google Scholar] [CrossRef]
- Guyard, A.; Danel, C.; Theou-Anton, N.; Debray, M.P.; Gibault, L.; Mordant, P.; Castier, Y.; Crestani, B.; Zalcman, G.; Blons, H.; et al. Morphologic and molecular study of lung cancers associated with idiopathic pulmonary fibrosis and other pulmonary fibroses. Respir. Res. 2017, 18, 120. [Google Scholar] [CrossRef] [PubMed]
- Fraire, A.E.; Greenberg, S.D. Carcinoma and diffuse interstitial fibrosis of lung. Cancer 1973, 31, 1078–1086. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H. Pathological study of lung cancer associated with idiopathic interstitial pneumonia—With special reference to relationship between the primary site of lung cancer and honeycombing. Nihon Kyobu Shikkan Gakkai Zasshi 1985, 23, 873–881. [Google Scholar] [PubMed]
- Mizushima, Y.; Kobayashi, M. Clinical characteristics of synchronous multiple lung cancer associated with idiopathic pulmonary fibrosis. A review of Japanese cases. Chest 1995, 108, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Lever, V.; Rossi, A.; Gilioli, E.; Brunelli, M.; Dubini, A.; Tomassetti, S.; Piciucchi, S.; Nottegar, A.; Rossi, G.; et al. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology 2017, 71, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Strock, S.B.; Alder, J.K.; Kass, D.J. From bad to worse: When lung cancer complicates idiopathic pulmonary fibrosis. J. Pathol. 2018, 244, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Bonti, V.; Ferrari, K.; Rosi, E.; Bindi, A.; Bartolucci, M.; Chiara, M.; Voltolini, L. Lung Cancer in Patients with Severe Idiopathic Pulmonary Fibrosis: Critical Aspects. In Vivo 2017, 31, 773–777. [Google Scholar] [PubMed] [Green Version]
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol. Int. 1999, 49, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Singh, S.R.; Hall, I.P. Airway myofibroblasts and their relationship with airway myocytes and fibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 127–132. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., 3rd; Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- Evans, J.N.; Kelley, J.; Krill, J.; Low, R.B.; Adler, K.B. The myofibroblast in pulmonary fibrosis. Chest 1983, 83, 97S–98S. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.W.; Xie, Q.M.; Chen, J.Q.; Deng, Y.M.; Tang, H.F. TGF-beta1 induces alveolar epithelial to mesenchymal transition in vitro. Life Sci. 2004, 76, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrad, B.; Strieter, R.M. Fibrocytes and the pathogenesis of diffuse parenchymal lung disease. Fibrogenesis Tissue Repair 2012, 5 (Suppl. 1), S22. [Google Scholar] [CrossRef]
- Strieter, R.M. Pathogenesis and natural history of usual interstitial pneumonia: The whole story or the last chapter of a long novel. Chest 2005, 128, 526S–532S. [Google Scholar] [CrossRef] [PubMed]
- Metz, C.N. Fibrocytes: A unique cell population implicated in wound healing. Cell. Mol. Life Sci. CMLS 2003, 60, 1342–1350. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Sun, G.; Stacey, M.A.; Mori, L.; Mattoli, S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 2003, 171, 380–389. [Google Scholar] [CrossRef]
- Almudever, P.; Milara, J.; De Diego, A.; Serrano-Mollar, A.; Xaubet, A.; Perez-Vizcaino, F.; Cogolludo, A.; Cortijo, J. Role of tetrahydrobiopterin in pulmonary vascular remodelling associated with pulmonary fibrosis. Thorax 2013, 68, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Nataraj, D.; Ernst, A.; Kalluri, R. Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition. Am. J. Respir. Cell Mol. Biol. 2010, 43, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.M.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Nasreen, N.; Mohammed, K.A.; Mubarak, K.K.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; Antony, V.B. Pleural mesothelial cell transformation into myofibroblasts and haptotactic migration in response to TGF-beta1 in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L115–L124. [Google Scholar] [CrossRef] [PubMed]
- Mubarak, K.K.; Montes-Worboys, A.; Regev, D.; Nasreen, N.; Mohammed, K.A.; Faruqi, I.; Hensel, E.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 133–140. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Osterholzer, J.J.; Marazioti, A.; Stathopoulos, G.T. “Scar-cinoma”: Viewing the fibrotic lung mesenchymal cell in the context of cancer biology. Eur. Respir. J. 2016, 47, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Konigshoff, M. Lung cancer in pulmonary fibrosis: Tales of epithelial cell plasticity. Respir. Int. Rev. Thorac. Dis. 2011, 81, 353–358. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Mori, T.; Sahoo, D.; Xu, P.X.; Bermingham, J.R., Jr.; Weissman, I.L. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat. Cell Biol. 2012, 14, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Zhu, B.; Ferry, C.H.; Markell, L.K.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. The nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) promotes oncogene-induced cellular senescence through repression of endoplasmic reticulum stress. J. Biol. Chem. 2014, 289, 20102–20119. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGFbeta1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, D.; Vona, R.; Giordani, L.; Straface, E.; Giammarioli, A.M. Defective autophagy in fibroblasts may contribute to fibrogenesis in autoimmune processes. Curr. Pharm. Des. 2011, 17, 3878–3887. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Sortino, M.A.; Tomaselli, V.; Mastruzzo, C.; Condorelli, F.; Bellistri, G.; Pistorio, M.P.; Canonico, P.L.; Crimi, N. Different expression of TNF-alpha receptors and prostaglandin E(2)Production in normal and fibrotic lung fibroblasts: Potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell Mol. Biol. 2000, 22, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Ballinger, M.N.; White, E.S.; Green, M.E.; Herrygers, A.B.; Wilke, C.A.; Toews, G.B.; Peters-Golden, M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J. Immunol. 2005, 174, 5644–5649. [Google Scholar] [CrossRef]
- Nho, R.S.; Hergert, P.; Kahm, J.; Jessurun, J.; Henke, C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am. J. Pathol. 2011, 179, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.; Montano, M.; Garcia-Alvarez, J.; Ruiz, V.; Uhal, B.D.; Selman, M.; Pardo, A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am. J. Respir. Cell Mol. Biol. 2001, 24, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Mio, T.; Nagai, S.; Kitaichi, M.; Kawatani, A.; Izumi, T. Proliferative characteristics of fibroblast lines derived from open lung biopsy specimens of patients with IPF (UIP). Chest 1992, 102, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Chen, Y.Y.; Rusch, V.; Rabinovitch, P.S. Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am. Rev. Respir. Dis. 1988, 138, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Chanson, M.; Crespin, S. Connexins as therapeutic targets in lung disease. Expert Opin. Ther. Targets 2011, 15, 989–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovato-Salinaro, A.; Trovato-Salinaro, E.; Failla, M.; Mastruzzo, C.; Tomaselli, V.; Gili, E.; Crimi, N.; Condorelli, D.F.; Vancheri, C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 122. [Google Scholar] [CrossRef]
- Cesen-Cummings, K.; Fernstrom, M.J.; Malkinson, A.M.; Ruch, R.J. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis 1998, 19, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.S.; Thannickal, V.J.; Carskadon, S.L.; Dickie, E.G.; Livant, D.L.; Markwart, S.; Toews, G.B.; Arenberg, D.A. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: A role for phosphatase and tensin homologue deleted on chromosome 10. Am. J. Respir. Crit. Care Med. 2003, 168, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Lovgren, A.K.; Kovacs, J.J.; Xie, T.; Potts, E.N.; Li, Y.; Foster, W.M.; Liang, J.; Meltzer, E.B.; Jiang, D.; Lefkowitz, R.J.; et al. beta-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci. Transl. Med. 2011, 3, 74ra23. [Google Scholar] [CrossRef]
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2013, 22, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Niki, T.; Yamada, T.; Matsuno, Y.; Kondo, H.; Hirohashi, S. Increased expression of laminin-5 and its prognostic significance in lung adenocarcinomas of small size. An immunohistochemical analysis of 102 cases. Cancer 2001, 91, 1129–1141. [Google Scholar] [CrossRef]
- Garrido, C.; Schmitt, E.; Cande, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. HSP27 and HSP70: Potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef]
- Pelosi, G.; Pastorino, U.; Pasini, F.; Maissoneuve, P.; Fraggetta, F.; Iannucci, A.; Sonzogni, A.; De Manzoni, G.; Terzi, A.; Durante, E.; et al. Independent prognostic value of fascin immunoreactivity in stage I nonsmall cell lung cancer. Br. J. Cancer 2003, 88, 537–547. [Google Scholar] [CrossRef]
- Chilosi, M.; Zamo, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballinger, M.N.; Newstead, M.W.; Zeng, X.; Bhan, U.; Mo, X.M.; Kunkel, S.L.; Moore, B.B.; Flavell, R.; Christman, J.W.; Standiford, T.J. IRAK-M promotes alternative macrophage activation and fibroproliferation in bleomycin-induced lung injury. J. Immunol. 2015, 194, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Osterholzer, J.J.; Olszewski, M.A.; Murdock, B.J.; Chen, G.H.; Erb-Downward, J.R.; Subbotina, N.; Browning, K.; Lin, Y.; Morey, R.E.; Dayrit, J.K.; et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J. Immunol. 2013, 190, 3447–3457. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Kolb, M.; Bonniaud, P.; Gauldie, J. Re-evaluation of fibrogenic cytokines in lung fibrosis. Curr. Pharm. Des. 2003, 9, 39–49. [Google Scholar] [CrossRef]
- O’Riordan, T.G.; Smith, V.; Raghu, G. Development of novel agents for idiopathic pulmonary fibrosis: Progress in target selection and clinical trial design. Chest 2015, 148, 1083–1092. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Hetzel, M.; Bachem, M.; Anders, D.; Trischler, G.; Faehling, M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 2005, 183, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Fact. Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Noskovicova, N.; Petrek, M.; Eickelberg, O.; Heinzelmann, K. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am. J. Respir. Cell Mol. Biol. 2015, 52, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Battegay, E.J.; Raines, E.W.; Seifert, R.A.; Bowen-Pope, D.F.; Ross, R. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990, 63, 515–524. [Google Scholar] [CrossRef]
- Battegay, E.J.; Raines, E.W.; Colbert, T.; Ross, R. TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J. Immunol. 1995, 154, 6040–6047. [Google Scholar] [PubMed]
- Wang, P.; Song, L.; Ge, H.; Jin, P.; Jiang, Y.; Hu, W.; Geng, N. Crenolanib, a PDGFR inhibitor, suppresses lung cancer cell proliferation and inhibits tumor growth in vivo. Oncotargets Ther. 2014, 7, 1761–1768. [Google Scholar] [CrossRef]
- Shikada, Y.; Yonemitsu, Y.; Koga, T.; Onimaru, M.; Nakano, T.; Okano, S.; Sata, S.; Nakagawa, K.; Yoshino, I.; Maehara, Y.; et al. Platelet-derived growth factor-AA is an essential and autocrine regulator of vascular endothelial growth factor expression in non-small cell lung carcinomas. Cancer Res. 2005, 65, 7241–7248. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Muller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 2007, 29, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Frezzetti, D.; Gallo, M.; Maiello, M.R.; D’Alessio, A.; Esposito, C.; Chicchinelli, N.; Normanno, N.; De Luca, A. VEGF as a potential target in lung cancer. Expert Opin. Ther. Targets 2017, 21, 959–966. [Google Scholar] [CrossRef]
- Zhang, S.; Smartt, H.; Holgate, S.T.; Roche, W.R. Growth factors secreted by bronchial epithelial cells control myofibroblast proliferation: An in vitro co-culture model of airway remodeling in asthma. Lab. Investig. A J. Tech. Methods Pathol. 1999, 79, 395–405. [Google Scholar]
- Khalil, N.; Xu, Y.D.; O’Connor, R.; Duronio, V. Proliferation of pulmonary interstitial fibroblasts is mediated by transforming growth factor-beta1-induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38 MAPK and JNK. J. Biol. Chem. 2005, 280, 43000–43009. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Li, L.; Smith, C.; Dorry, S.J.; Koo, H.Y.; Chen, L.; Ornitz, D.M. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J. Biol. Chem. 2017, 292, 10364–10378. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yasuda, H.; Funaishi, K.; Arai, D.; Ishioka, K.; Ohgino, K.; Tani, T.; Hamamoto, J.; Ohashi, A.; Naoki, K.; et al. Multiple roles of extracellular fibroblast growth factors in lung cancer cells. Int. J. Oncol. 2015, 46, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Knight, R.A.; Bloor, C.A.; Spiteri, M.A. Enhanced insulin-like growth factor binding protein-related protein 2 (Connective tissue growth factor) expression in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am. J. Respir. Cell Mol. Biol. 1999, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Shih, J.Y.; Jeng, Y.M.; Su, J.L.; Lin, B.Z.; Chen, S.T.; Chau, Y.P.; Yang, P.C.; Kuo, M.L. Connective tissue growth factor and its role in lung adenocarcinoma invasion and metastasis. J. Natl. Cancer Inst. 2004, 96, 364–375. [Google Scholar] [CrossRef]
- Chien, W.; Yin, D.; Gui, D.; Mori, A.; Frank, J.M.; Said, J.; Kusuanco, D.; Marchevsky, A.; McKenna, R.; Koeffler, H.P. Suppression of cell proliferation and signaling transduction by connective tissue growth factor in non-small cell lung cancer cells. Mol. Cancer Res. MCR 2006, 4, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Spiteri, M.A. Growth factors in idiopathic pulmonary fibrosis: Relative roles. Respir. Res. 2002, 3, 13. [Google Scholar] [CrossRef]
- Kelly, K.; Kane, M.A.; Bunn, P.A., Jr. Growth factors in lung cancer: Possible etiologic role and clinical target. Med. Pediatr. Oncol. 1991, 19, 450–458. [Google Scholar] [CrossRef]
- Hodkinson, P.S.; Mackinnon, A.; Sethi, T. Targeting growth factors in lung cancer. Chest 2008, 133, 1209–1216. [Google Scholar] [CrossRef]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Magkrioti, C.; Oikonomou, N.; Kaffe, E.; Mouratis, M.A.; Xylourgidis, N.; Barbayianni, I.; Megadoukas, P.; Harokopos, V.; Valavanis, C.; Chun, J.; et al. The Autotaxin-Lysophosphatidic Acid Axis Promotes Lung Carcinogenesis. Cancer Res. 2018, 78, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Hancock, A.; Armstrong, L.; Gama, R.; Millar, A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am. J. Respir. Cell Mol. Biol. 1998, 18, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.L.; Rice, A.B.; Geisenhoffer, K.; Madtes, D.K.; Bonner, J.C. IL-13 and IL-1beta promote lung fibroblast growth through coordinated up-regulation of PDGF-AA and PDGF-Ralpha. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1132–1134. [Google Scholar]
- Kaviratne, M.; Hesse, M.; Leusink, M.; Cheever, A.W.; Davies, S.J.; McKerrow, J.H.; Wakefield, L.M.; Letterio, J.J.; Wynn, T.A. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J. Immunol. 2004, 173, 4020–4029. [Google Scholar] [CrossRef]
- Lee, C.G.; Kang, H.R.; Homer, R.J.; Chupp, G.; Elias, J.A. Transgenic modeling of transforming growth factor-beta(1): Role of apoptosis in fibrosis and alveolar remodeling. Proc. Am. Thorac. Soc. 2006, 3, 418–423. [Google Scholar] [CrossRef]
- Huang, M.; Wang, J.; Lee, P.; Sharma, S.; Mao, J.T.; Meissner, H.; Uyemura, K.; Modlin, R.; Wollman, J.; Dubinett, S.M. Human non-small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res. 1995, 55, 3847–3853. [Google Scholar]
- Pastuszak-Lewandoska, D.; Domanska-Senderowska, D.; Antczak, A.; Kordiak, J.; Gorski, P.; Czarnecka, K.H.; Migdalska-Sek, M.; Nawrot, E.; Kiszalkiewicz, J.M.; Brzezianska-Lasota, E. The Expression Levels of IL-4/IL-13/STAT6 Signaling Pathway Genes and SOCS3 Could Help to Differentiate the Histopathological Subtypes of Non-Small Cell Lung Carcinoma. Mol. Diagn. Ther. 2018, 22, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wu, X.J.; Zhang, J.J.; He, C.S. IL-13 receptor alpha2 is a negative prognostic factor in human lung cancer and stimulates lung cancer growth in mice. Oncotarget 2015, 6, 32902–32913. [Google Scholar] [CrossRef]
- Chakraborty, S.; Chopra, P.; Ambi, S.V.; Dastidar, S.G.; Ray, A. Emerging therapeutic interventions for idiopathic pulmonary fibrosis. Expert Opin. Investig. Drugs 2014, 23, 893–910. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Tseng, S.; Horner, R.M.; Tam, C.; Loda, M.; Rollins, B.J. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 2000, 404, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Qin, X.; Qin, C.Y.; Yin, Y.L.; Chen, Y.; Zhu, H.L. Expression of monocyte chemoattractant protein-1 and CC chemokine receptor 2 in non-small cell lung cancer and its significance. Cancer Immunol. Immunother. CII 2013, 62, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lan, T.; Hou, J.; Li, J.; Fang, R.; Yang, Z.; Zhang, M.; Liu, J.; Liu, B. NOX4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget 2014, 5, 4392–4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamy, S.M.; Rajasekaran, N.S.; Thannickal, V.J. Nuclear Factor-Erythroid-2-Related Factor 2 in Aging and Lung Fibrosis. Am. J. Pathol. 2016, 186, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.H.; Zhang, B.; Fan, Y.; Lin, N.M. Keap1-Nrf2 pathway: A promising target towards lung cancer prevention and therapeutics. Chronic Dis. Transl. Med. 2015, 1, 175–186. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Yu, C.J.; Yang, P.C.; Shun, C.T.; Lee, Y.C.; Kuo, S.H.; Luh, K.T. Overexpression of MUC5 genes is associated with early post-operative metastasis in non-small-cell lung cancer. Int. J. Cancer 1996, 69, 457–465. [Google Scholar] [CrossRef]
- Nagashio, R.; Ueda, J.; Ryuge, S.; Nakashima, H.; Jiang, S.X.; Kobayashi, M.; Yanagita, K.; Katono, K.; Satoh, Y.; Masuda, N.; et al. Diagnostic and prognostic significances of MUC5B and TTF-1 expressions in resected non-small cell lung cancer. Sci. Rep. 2015, 5, 8649. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Sanz, C.; González, S.; Guijarro, R.; Martínez, C.; Cortijo, J. Role of MUC1 in idiopathic pulmonary fibrosis. In Proceedings of the European Respiratory Society Congress, London, UK, 3–7 September 2016. [Google Scholar]
- Ballester, B.; Milara, J.; Guijarro, R.; Morcillo, E.; Cortijo, J. Role of MUC4 in idiopathic pulmonary fibrosis. In Proceedings of the European Respiratory Society Congress, Paris, France, 15–19 September 2018. [Google Scholar]
- Ballester, B.; Roger, I.; Contreras, S.; Montero, P.; Milara, J. Role of MUC1 in idiopathic pulmonary fibrosis: Mechanistic insights. In Proceedings of the European Respiratory Society Congress, Milan, Italy, 9–13 September 2017. [Google Scholar]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Bafna, S.; Kaur, S.; Batra, S.K. Membrane-bound mucins: The mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904. [Google Scholar] [CrossRef]
- Stroopinsky, D.; Rajabi, H.; Nahas, M.; Rosenblatt, J.; Rahimian, M.; Pyzer, A.; Tagde, A.; Kharbanda, A.; Jain, S.; Kufe, T.; et al. MUC1-C drives myeloid leukaemogenesis and resistance to treatment by a survivin-mediated mechanism. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Ramasamy, S.; Duraisamy, S.; Barbashov, S.; Kawano, T.; Kharbanda, S.; Kufe, D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol. Cell 2007, 27, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Farhad, M.; Rolig, A.S.; Redmond, W.L. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology 2018, 7, e1434467. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; Kaminski, N. Idiopathic pulmonary fibrosis: Aberrant recapitulation of developmental programs? PLoS Med. 2008, 5, e62. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef]
- Coon, D.R.; Roberts, D.J.; Loscertales, M.; Kradin, R. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp. Mol. Pathol. 2006, 80, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Moshai, E.F.; Wemeau-Stervinou, L.; Cigna, N.; Brayer, S.; Somme, J.M.; Crestani, B.; Mailleux, A.A. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.Z.; Zhang, Z.H.; Fan, X.Y.; Xu, X.L.; Chen, M.L.; Chang, B.W.; Zhang, Y.B. Notch3 overexpression associates with poor prognosis in human non-small-cell lung cancer. Med. Oncol. 2013, 30, 595. [Google Scholar] [CrossRef]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [Green Version]
- Zou, B.; Zhou, X.L.; Lai, S.Q.; Liu, J.C. Notch signaling and non-small cell lung cancer. Oncol. Lett. 2018, 15, 3415–3421. [Google Scholar] [CrossRef]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110gamma overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. A J. Tech. Methods Pathol. 2013, 93, 566–576. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Lee, D.Y.; Waghray, M.; Keshamouni, V.G.; Thomas, P.E.; Zhang, H.; Cui, Z.; Thannickal, V.J. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem. 2004, 279, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, W.M.; Klippel, A.; Escobedo, J.A.; Williams, L.T. Modification of the 85-kilodalton subunit of phosphatidylinositol-3 kinase in platelet-derived growth factor-stimulated cells. Mol. Cell. Biol. 1992, 12, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6, e24663. [Google Scholar] [CrossRef] [PubMed]
- Fumarola, C.; Bonelli, M.A.; Petronini, P.G.; Alfieri, R.R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 2014, 90, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef]
- Van Moorsel, C.H.; Ten Klooster, L.; van Oosterhout, M.F.; de Jong, P.A.; Adams, H.; Wouter van Es, H.; Ruven, H.J.; van der Vis, J.J.; Grutters, J.C. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2015, 192, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Bennett, W.P.; Colby, T.V.; Travis, W.D.; Borkowski, A.; Jones, R.T.; Lane, D.P.; Metcalf, R.A.; Samet, J.M.; Takeshima, Y.; Gu, J.R.; et al. p53 protein accumulates frequently in early bronchial neoplasia. Cancer Res. 1993, 53, 4817–4822. [Google Scholar] [PubMed]
- Sozzi, G.; Miozzo, M.; Donghi, R.; Pilotti, S.; Cariani, C.T.; Pastorino, U.; Della Porta, G.; Pierotti, M.A. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res. 1992, 52, 6079–6082. [Google Scholar] [PubMed]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, H.; Ogura, T.; Yokose, T.; Nagai, K.; Nishiwaki, Y.; Esumi, H. p53 gene alteration in atypical epithelial lesions and carcinoma in patients with idiopathic pulmonary fibrosis. Hum. Pathol. 2001, 32, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Veronese, M.L.; Negrini, M.; Baffa, R.; Cotticelli, M.G.; Inoue, H.; Tornielli, S.; Pilotti, S.; De Gregorio, L.; Pastorino, U.; et al. The FHIT gene 3p14.2 is abnormal in lung cancer. Cell 1996, 85, 17–26. [Google Scholar] [CrossRef]
- Uematsu, K.; Yoshimura, A.; Gemma, A.; Mochimaru, H.; Hosoya, Y.; Kunugi, S.; Matsuda, K.; Seike, M.; Kurimoto, F.; Takenaka, K.; et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001, 61, 8527–8533. [Google Scholar] [PubMed]
- Karachaliou, N.; Mayo, C.; Costa, C.; Magri, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS mutations in lung cancer. Clin. Lung Cancer 2013, 14, 205–214. [Google Scholar] [CrossRef]
- Masai, K.; Tsuta, K.; Motoi, N.; Shiraishi, K.; Furuta, K.; Suzuki, S.; Asakura, K.; Nakagawa, K.; Sakurai, H.; Watanabe, S.I.; et al. Clinicopathological, Immunohistochemical, and Genetic Features of Primary Lung Adenocarcinoma Occurring in the Setting of Usual Interstitial Pneumonia Pattern. J. Thorac. Oncol. Off. Publ. Int. Assoc. Stud. Lung Cancer 2016, 11, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, D.; Chun, S.M.; Bae, S.; Song, J.S.; Kim, M.Y.; Koo, H.J.; Song, J.W.; Kim, W.S.; Lee, J.C.; et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J. Pathol. 2018, 244, 25–35. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Clamon, G.H.; Bossler, A.D.; Abu Hejleh, T.; Furqan, M. Germline mutations predisposing to non-small cell lung cancer. Fam. Cancer 2015, 14, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N.; Giraud, V.; Picard, C.; Nunes, H.; Dastot-Le Moal, F.; Copin, B.; Galeron, L.; De Ligniville, A.; Kuziner, N.; Reynaud-Gaubert, M.; et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 2016, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef]
- Madsen, J.; Tornoe, I.; Nielsen, O.; Koch, C.; Steinhilber, W.; Holmskov, U. Expression and localization of lung surfactant protein A in human tissues. Am. J. Respir. Cell Mol. Biol. 2003, 29, 591–597. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Kazemier, K.M.; Ruven, H.J.; Grutters, J.C. Association between variations in cell cycle genes and idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e30442. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Chatterjee, N.; Yu, K.; Goldin, L.R.; Goldstein, A.M.; Rotunno, M.; Mirabello, L.; Jacobs, K.; Wheeler, W.; Yeager, M.; et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet. 2009, 85, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Tan, N.; Meng, C.; Li, J.; Jing, L.; Yan, M.; Jin, T.; Chen, F. Genetic variations in TERC and TERT genes are associated with lung cancer risk in a Chinese Han population. Oncotarget 2017, 8, 110145–110152. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet. Cytogenet. 2008, 182, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Amos, C.I.; Zhu, Y.; Zhao, H.; Grossman, B.H.; Shay, J.W.; Luo, S.; Hong, W.K.; Spitz, M.R. Telomere dysfunction: A potential cancer predisposition factor. J. Natl. Cancer Inst. 2003, 95, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Feldser, D.M.; Greider, C.W. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106, 275–286. [Google Scholar] [CrossRef]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef]
- Yang, J.; Xu, T.; Gomez, D.R.; Jeter, M.; Levy, L.B.; Song, Y.; Hahn, S.; Liao, Z.; Yuan, X. The Pulmonary Fibrosis Associated MUC5B Promoter Polymorphism Is Prognostic of the Overall Survival in Patients with Non-Small Cell Lung Cancer (NSCLC) Receiving Definitive Radiotherapy. Transl. Oncol. 2017, 10, 197–202. [Google Scholar] [CrossRef]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Gemma, A.; Yoshimura, A.; Hosoya, Y.; Nara, M.; Hosomi, Y.; Okano, T.; Kunugi, S.; Koizumi, K.; Fukuda, Y.; et al. Reduced transcription of the Smad4 gene during pulmonary carcinogenesis in idiopathic pulmonary fibrosis. Mol. Med. Rep. 2009, 2, 73–80. [Google Scholar] [PubMed]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.R.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar] [PubMed]
- Sanders, Y.Y.; Pardo, A.; Selman, M.; Nuovo, G.J.; Tollefsbol, T.O.; Siegal, G.P.; Hagood, J.S. Thy-1 promoter hypermethylation: A novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Lung, H.L.; Bangarusamy, D.K.; Xie, D.; Cheung, A.K.; Cheng, Y.; Kumaran, M.K.; Miller, L.; Liu, E.T.; Guan, X.Y.; Sham, J.S.; et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene 2005, 24, 6525–6532. [Google Scholar] [CrossRef]
- Langevin, S.M.; Kratzke, R.A.; Kelsey, K.T. Epigenetics of lung cancer. Transl. Res. J. Lab. Clin. Med. 2015, 165, 74–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef]
- Yang, M.; Shen, H.; Qiu, C.; Ni, Y.; Wang, L.; Dong, W.; Liao, Y.; Du, J. High expression of miR-21 and miR-155 predicts recurrence and unfavourable survival in non-small cell lung cancer. Eur. J. Cancer 2013, 49, 604–615. [Google Scholar] [CrossRef]
- Kolenda, T.; Przybyla, W.; Teresiak, A.; Mackiewicz, A.; Lamperska, K.M. The mystery of let-7d—A small RNA with great power. Contemp. Oncol. 2014, 18, 293–301. [Google Scholar] [CrossRef]
- Nguyen-Ngoc, T.; Bouchaab, H.; Adjei, A.A.; Peters, S. BRAF Alterations as Therapeutic Targets in Non-Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Stud. Lung Cancer 2015, 10, 1396–1403. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E., Jr.; Raghu, G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Sato, K.; Muramoto, Y.; Karakawa, T.; Kitamado, M.; Iwanaga, T.; Nabeshima, T.; Maruyama, K.; Nakagawa, K.; Ishida, K.; et al. Superoxide scavenging activity of pirfenidone-iron complex. Biochem. Biophys. Res. Commun. 2008, 372, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Montes, A.; Ruiz-Corro, L.; Lopez-Reyes, A.; Castrejon-Gomez, E.; Armendariz-Borunda, J. Potent antioxidant role of pirfenidone in experimental cirrhosis. Eur. J. Pharmacol. 2008, 595, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Bendstrup, E.; Crestani, B.; Gunther, A.; Olschewski, H.; Skold, C.M.; Wells, A.; Wuyts, W.; Koschel, D.; Kreuter, M.; et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2016, 4, 445–453. [Google Scholar] [CrossRef]
- Hisatomi, K.; Mukae, H.; Sakamoto, N.; Ishimatsu, Y.; Kakugawa, T.; Hara, S.; Fujita, H.; Nakamichi, S.; Oku, H.; Urata, Y.; et al. Pirfenidone inhibits TGF-beta1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm. Med. 2012, 12, 24. [Google Scholar] [CrossRef]
- Lin, X.; Yu, M.; Wu, K.; Yuan, H.; Zhong, H. Effects of pirfenidone on proliferation, migration, and collagen contraction of human Tenon’s fibroblasts in vitro. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Lee, K.; Ryu, S.W.; Im, M.; Kook, K.H.; Choi, C. Pirfenidone inhibits transforming growth factor-beta1-induced fibrogenesis by blocking nuclear translocation of Smads in human retinal pigment epithelial cell line ARPE-19. Mol. Vis. 2012, 18, 1010–1020. [Google Scholar] [PubMed]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, G.F.; Liao, X.P.; Li, X.J.; Zhang, J.; Lin, H.; Chen, Z.; Zhang, X. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int. J. Rheum. Dis. 2018, 21, 477–486. [Google Scholar] [CrossRef]
- Iwata, T.; Yoshino, I.; Yoshida, S.; Ikeda, N.; Tsuboi, M.; Asato, Y.; Katakami, N.; Sakamoto, K.; Yamashita, Y.; Okami, J.; et al. A phase II trial evaluating the efficacy and safety of perioperative pirfenidone for prevention of acute exacerbation of idiopathic pulmonary fibrosis in lung cancer patients undergoing pulmonary resection: West Japan Oncology Group 6711 L (PEOPLE Study). Respir. Res. 2016, 17, 90. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef]
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Grone, H.J.; Lipson, K.E.; et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Aono, Y.; Nishioka, Y.; Inayama, M.; Ugai, M.; Kishi, J.; Uehara, H.; Izumi, K.; Sone, S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 2005, 171, 1279–1285. [Google Scholar] [CrossRef]
- Ishii, Y.; Fujimoto, S.; Fukuda, T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 550–556. [Google Scholar] [CrossRef]
- Li, M.; Abdollahi, A.; Grone, H.J.; Lipson, K.E.; Belka, C.; Huber, P.E. Late treatment with imatinib mesylate ameliorates radiation-induced lung fibrosis in a mouse model. Radiat. Oncol. 2009, 4, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, C.K.; Lee, S.H.; Yoon, H.K.; Kim, S.C.; Lee, S.Y.; Kwon, S.S.; Kim, Y.K.; Kim, K.H.; Kim, T.J.; Kim, J.W. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respir. Int. Rev. Thorac. Dis. 2011, 82, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Mizoguchi, K.; Kawarada, S.; Miyoshi, A.; Suzuki, M.; Chiba, S.; Deki, T. Effects of erlotinib on lung injury induced by intratracheal administration of bleomycin (BLM) in rats. J. Toxicol. Sci. 2010, 35, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.E.; Lasky, J.A.; Limper, A.H.; Mieras, K.; Gabor, E.; Schroeder, D.R.; Imatinib, I.P.F.S.I. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 2010, 181, 604–610. [Google Scholar] [CrossRef]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Malouf, M.A.; Hopkins, P.; Snell, G.; Glanville, A.R.; Everolimus in, I.P.F.S.I. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 2011, 16, 776–783. [Google Scholar] [CrossRef]
- Soria, J.C.; Shepherd, F.A.; Douillard, J.Y.; Wolf, J.; Giaccone, G.; Crino, L.; Cappuzzo, F.; Sharma, S.; Gross, S.H.; Dimitrijevic, S.; et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 1674–1681. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Xue, Y.; Long, M.; Zhang, G.; Zhang, J.; Su, H. Targeting CCR2 with its antagonist suppresses viability, motility and invasion by downregulating MMP-9 expression in non-small cell lung cancer cells. Oncotarget 2017, 8, 39230–39240. [Google Scholar] [CrossRef]
- Chung, L.Y.; Tang, S.J.; Wu, Y.C.; Sun, G.H.; Liu, H.Y.; Sun, K.H. Galectin-3 augments tumor initiating property and tumorigenicity of lung cancer through interaction with beta-catenin. Oncotarget 2015, 6, 4936–4952. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.Y.; Zhang, X.C.; Yang, S.Q.; An, S.J.; Chen, Z.H.; Su, J.; Xie, Z.; Gou, L.Y.; Wu, Y.L. Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS ONE 2016, 11, e0149370. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Tanaka, K.; Sunami, S.; Asoh, T.; Maeyama, T.; Tsuruta, N.; Nakanishi, Y.; Okamoto, I. Durable response to nivolumab in a lung adenocarcinoma patient with idiopathic pulmonary fibrosis. Thorac. Cancer 2018, 9, 1519–1521. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Aokage, K.; Nishikawa, H.; Neri, S.; Nakamura, H.; Sugano, M.; Tane, K.; Miyoshi, T.; Kojima, M.; Fujii, S.; et al. Immunosuppressive tumor microenvironment of usual interstitial pneumonia-associated squamous cell carcinoma of the lung. J. Cancer Res. Clin. Oncol. 2018, 144, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; Meng, X.; Zhang, K.; Li, X.; Wang, C.; Xiang, Z.; Hu, K.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses bleomycin-induced pulmonary fibrosis by attenuating the expression of TGF-beta1 and FGF-2. Exp. Mol. Pathol. 2016, 101, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [PubMed]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.E.; Bosquee, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, G.; Mo, B.; Wang, C. Artesunate ameliorates lung fibrosis via inhibiting the Notch signaling pathway. Exp. Ther. Med. 2017, 14, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, D.; Zhu, S.; Gu, J.; Ding, F.; Bian, W.; Rong, Z.; Shen, C. Bleomycin-induced pulmonary fibrosis is attenuated by an antibody against KL-6. Exp. Lung Res. 2013, 39, 241–248. [Google Scholar] [CrossRef]
Figure 1.
Cell types and cellular processes involved in lung cancer (LC) and idiopathic pulmonary fibrosis (IPF). Lung resident fibroblasts, pericytes, pleural mesothelial cells (PMC), circulating fibrocytes, vascular endothelial cells, and epithelial cells (Alveolar type II cells (ATII) in IPF and cancer epithelial cells in LC) are transformed to IPF myofibroblast or mesenchymal phenotype and cancer-associated fibroblasts (CAFs). Myofibroblasts and cancer cells are characterized by altered cell-cell communication, migration properties, and tissue invasion through basement membranes and the extracellular matrix. IPF myofibroblasts and ATII cells acquire senescent identities, but the presence of this phenotype is controversial in LC. Otherwise, endoplasmic reticulum (ER) stress is induced in IPF, while autophagy is defective in IPF. However, the function of both processes is controversial in LC. Finally, apoptosis is induced in ATII cells, but IPF myofibroblasts and cancer cells evade apoptosis.
Figure 1.
Cell types and cellular processes involved in lung cancer (LC) and idiopathic pulmonary fibrosis (IPF). Lung resident fibroblasts, pericytes, pleural mesothelial cells (PMC), circulating fibrocytes, vascular endothelial cells, and epithelial cells (Alveolar type II cells (ATII) in IPF and cancer epithelial cells in LC) are transformed to IPF myofibroblast or mesenchymal phenotype and cancer-associated fibroblasts (CAFs). Myofibroblasts and cancer cells are characterized by altered cell-cell communication, migration properties, and tissue invasion through basement membranes and the extracellular matrix. IPF myofibroblasts and ATII cells acquire senescent identities, but the presence of this phenotype is controversial in LC. Otherwise, endoplasmic reticulum (ER) stress is induced in IPF, while autophagy is defective in IPF. However, the function of both processes is controversial in LC. Finally, apoptosis is induced in ATII cells, but IPF myofibroblasts and cancer cells evade apoptosis.


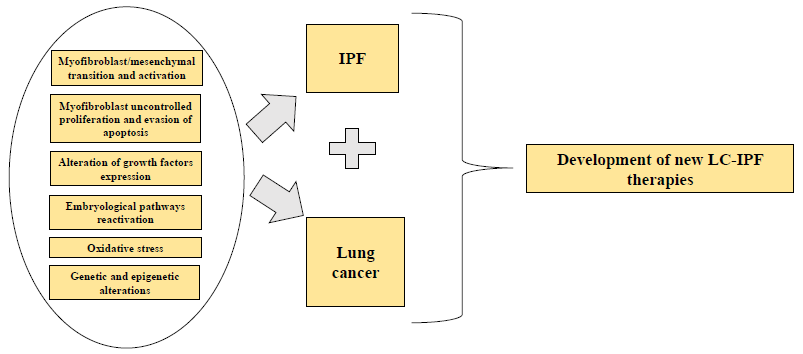{kind=link}
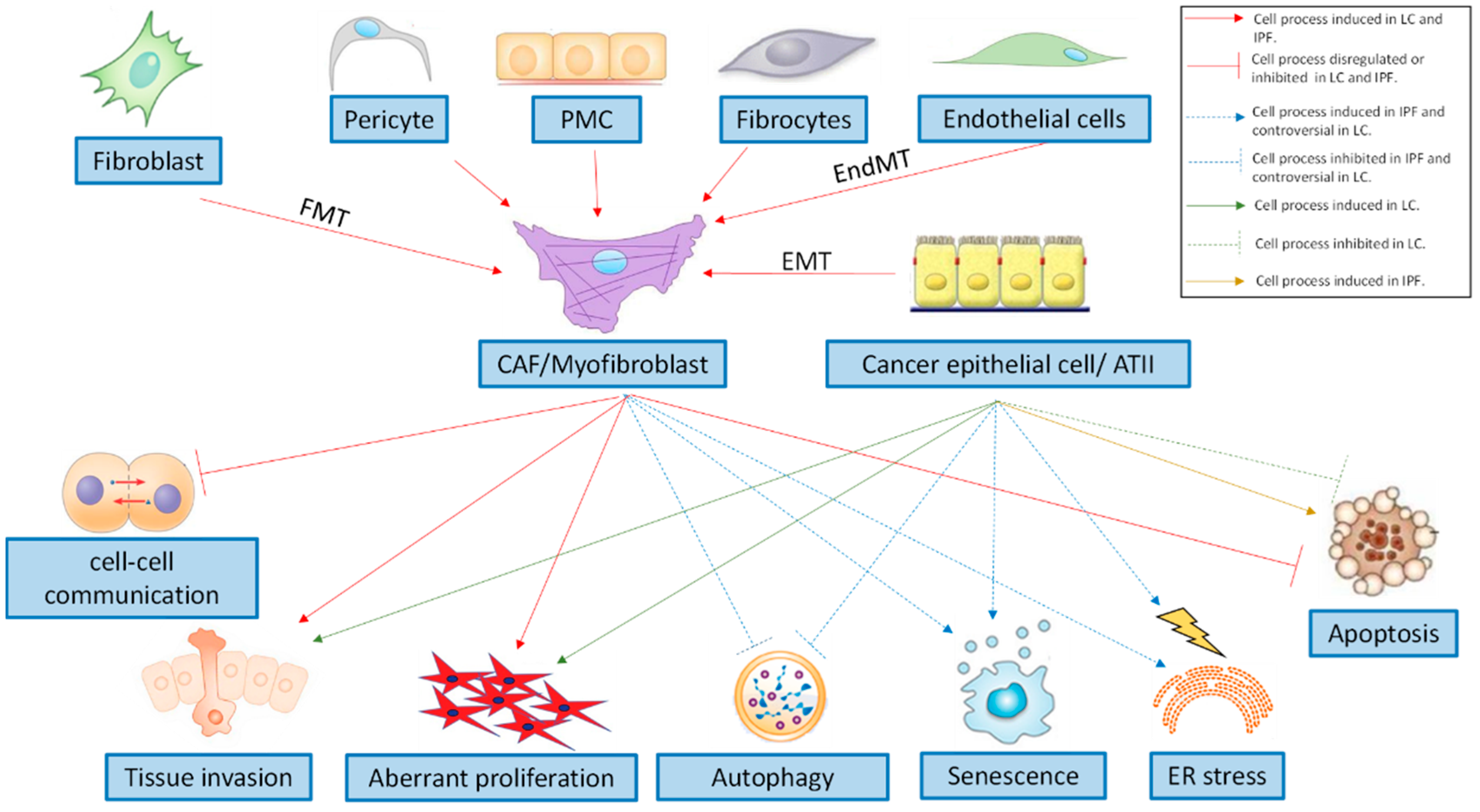{kind=link}
Table 1.
Prevalence of lung cancer (LC) in idiopathic pulmonary fibrosis (IPF) patients.
| Study | Number of Patients with IPF | Prevalence of LC (%) | LC-IPF Male (%) | LC-IPF Median Age | LC-IPF Smokers (%) | Reference |
|---|---|---|---|---|---|---|
| Nagai (1992) | 99 | 31.3% | 87.1% | 70.9 | 87.1% | [9] |
| Matsusitha (1995) | 20 | 48.2% | 90% | 66.4 | 74.3% | [13] |
| Park (2001) | 281 | 22.4% | 97% | 66.8 | 88.9% | [14] |
| Le Jeune (2007) | 1064 | 2.7% | ND | ND | ND | [20] |
| Ozawa (2009) | 103 | 20.4% | 95.2% | 65.5 | 66.7% | [15] |
| Lee (2012) | 1685 | 6.8% | 94.7% | 68.5 | 92.3% | [16] |
| Kreuter (2014) | 265 | 16% | ND | ND | ND | [17] |
| Tomasetti (2015) | 181 | 13% | 82.6% | 66.9 | 91.3% | [18] |
| Yoon (2018) | 1108 | 2.8% | 61% | 65 | 77% | [19] |
| Kato (2018) | 632 | 11.1% | 94.3% | 66.8 | 100% | [8] |
ND: not determined.
Table 2.
Lung cancer (LC) histological subtypes in patients with idiopathic pulmonary fibrosis (IPF).
Table 2.
Lung cancer (LC) histological subtypes in patients with idiopathic pulmonary fibrosis (IPF).
| Study | Number of Patients with LC-IPF | Squamous Cell Carcinoma | Adenocarcinoma | Other Histological Subtypes | Reference |
|---|---|---|---|---|---|
| Kawai (1987) | 8 | 12.5% | 75% | 12.5% | [24] |
| Nagai (1992) | 31 | 45.2% | 35.2% | 19.6% | [9] |
| Park (2001) | 63 | 35% | 30% | 35% | [14] |
| Kawasaki (2001) | 53 | 46% | 46% | 8% | [25] |
| Aubry (2002) | 24 | 67% | 29% | 4% | [10] |
| Ozawa (2009) | 21 | 38% | 29% | 33% | [15] |
| Saito (2011) | 28 | 67.9% | 25% | 7.1% | [26] |
| Lee (2014) | 70 | 40% | 30% | 30% | [27] |
| Kreuter (2015) | 42 | 36% | 31% | 33% | [17] |
| Tomasetti (2015) | 23 | 39% | 35% | 26% | [18] |
| Khan (2015) | 34 | 41% | 38% | 21% | [28] |
| Guyard (2017) | 18 | 44% | 33% | 23% | [29] |
| Yoon (2018) | 27 | 41% | 26% | 33% | [19] |
| Kato (2018) | 70 | 30% | 20% | 50% | [8] |
Table 3.
Principal fibrogenic molecules and signal transduction pathways participating in lung cancer (LC) and idiopathic pulmonary fibrosis (IPF).
Table 3.
Principal fibrogenic molecules and signal transduction pathways participating in lung cancer (LC) and idiopathic pulmonary fibrosis (IPF).
| IPF | LC | |
|---|---|---|
| Growth Factors | ||
| TGFβ1 | Overexpressed | Overexpressed |
| PDGF | Overexpressed | Overexpressed |
| VEGF | Overexpressed | Overexpressed |
| FGF | Overexpressed | Overexpressed |
| CTGF | Overexpressed | Downregulated |
| Profibrotic mediators | ||
| LPA | Overexpressed | Overexpressed |
| Galectin-3 | Overexpressed | Overexpressed |
| Cytokines | Overexpressed | Overexpressed |
| CCL2 | Overexpressed | Overexpressed |
| IL-13 | Overexpressed | Overexpressed |
| Mucins | ||
| Mucin 1 | Overexpressed | Overexpressed |
| Mucin 4 | Overexpressed | Overexpressed |
| Mucin 5B | Overexpressed | Overexpressed |
| Embryological pathways | ||
| Wnt pathway | Overexpressed | Overexpressed |
| Shh pathway | Overexpressed | Overexpressed |
| Notch pathway | Overexpressed | Overexpressed |
| Proliferation-related pathways | ||
| PI3K/AKT/mTOR pathway | Overexpressed | Overexpressed |
| Migration-related proteins | ||
| Laminin | Overexpressed | Overexpressed |
| Fascin | Overexpressed | Overexpressed |
| Hsp27 | Overexpressed | Overexpressed |
| Oxidative stress—related molecules | ||
| NOX4 | Overexpressed | Overexpressed |
| Nrf2 | Downregulated | Downregulated |
| Cell-cell communication—related proteins | ||
| Connexin 43 | Downregulated | Downregulated |
TGFβ1: transforming growth factor β1; PDGF: platelet derived growth factor; VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; CTGF: connective tissue growth factor; LPA: lysophosphatidic acid; CCL2: chemokine ligand 2; IL-13: interleukin 13; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; mTOR: mammalian Target of Rapamycin; Hsp27: heat shock protein 27; NOX4: NADPH oxidase 4.
Table 4.
Mutated genes, hypermethylated genes, and non-coding RNAs with altered expression in Idiopathic pulmonary fibrosis (IPF), lung cancer (LC), and LC-IPF patients.
Table 4.
Mutated genes, hypermethylated genes, and non-coding RNAs with altered expression in Idiopathic pulmonary fibrosis (IPF), lung cancer (LC), and LC-IPF patients.
| IPF | LC | LC-IPF | |
|---|---|---|---|
| Mutated Genes | |||
| SFTPA1 | Yes [199] | ND | Yes [199] |
| SFTPA2 | Yes [180] | ND | Yes [198] |
| TERT | Yes [181,184] | Yes [206,207] | Yes [195] |
| TERC | Yes [181,184] | Yes [206,207] | ND |
| PARN | Yes [183] | ND | ND |
| OBFC1 | Yes [184] | Yes [207] | ND |
| RTEL1 | Yes [183] | Yes [207] | ND |
| TOLLIP | Yes [186] | ND | ND |
| MUC5B | Yes [148] | Yes [213] | ND |
| P53 | Yes [189] | Yes [187,188] | Yes [190] |
| FHIT | Yes [192] | Yes [191] | Yes [192] |
| KRAS | ND | Yes [193] | Yes [194] |
| BRAF | ND | Yes [223] | Yes [195] |
| CDKN1A | Yes [203] | ND | Yes [195] |
| Hypermethylated Genes | |||
| SMAD4 | ND | ND | Yes [215] |
| THY-1 | Yes [217] | ND * | ND |
| MGMT | No [220] | Yes [219] | ND |
| Non-coding RNAs | |||
| Let-7d | Downregulated [95] | Downregulated [219] | ND |
| miR-21 | Upregulated [95] | Upregulated [221] | ND |
ND: Not determined. * Reported in metastatic nasopharyngeal carcinoma [218].
Table 5.
Development status of drugs targeting molecules and processes involved in lung cancer (LC) and Idiopathic pulmonary fibrosis (IPF).
Table 5.
Development status of drugs targeting molecules and processes involved in lung cancer (LC) and Idiopathic pulmonary fibrosis (IPF).
| Therapy | IPF | LC |
|---|---|---|
| Anti-PDGFR, VEGFR, FGFR (nintedanib) | Approved | Approved in combination with docetaxel (second-line treatment) for ADC-NSCLC |
| Anti-fibrotic drug (pirfenidone) | Approved | Preclinical studies for NSCLC [240] |
| Anti-IL13 | QAX576 (NCT00532233, NCT01266135: Phase II completed) | Not studied |
| Lebrikizumab (NCT01872689: Phase II completed) | ||
| Anti-CCL2 | Carlumab (CNTO-888) (NCT00786201: Phase II completed | Preclinical studies for NSCLC [253] |
| Galectin-3 inhibition | TD139 (NCT02257177: Phase I/II completed) | Preclinical studies for NSCLC [254] |
| Anti-TGFβ | Fresolimumab (GC1008) (NCT00125385: Phase I completed) | Fresolimumab (GC1008) (NCT02581787: Phase I/II suspended) (NSCLC patients) |
| Anti-αvβ6 integrin | BG0011 (STX-100) (NCT01371305: Phase II completed) | Not studied |
| αvβ6 antagonist | GSK3008348 (NCT02612051: Phase I completed) | Not studied |
| Anti-CTGF | Pamrevlumab (FG-3019) (NCT01262001: Phase II completed) | Not studied |
| LPAR1 antagonist | BMS-986020 (NCT01766817: Phase II completed) | Preclinical studies [132] |
| Autotaxin inhibition | GLPG1690 (NCT02738801: Phase II completed) | Preclinical studies [132] |
| Angiostatic agent | Tetrathiomolybdate (NCT00189176: Phase I/II completed) | Tetrathiomolybdate (NCT01837329: Phase I recruiting patients) (NSCLC patients) |
| mTOR inhibitor | GSK-2126458 (NCT01725139: Phase I completed) | Not studied * |
| Sirolimus (NCT01462006: Not applicable Phase) | ||
| TERT gene expression induction | Nandrolone decanoate (NCT02055456: Phase I/II (unknown recruitment status)) | Not studied |
| Shh pathway inhibitor | Vismodegib (NCT02648048: Phase Ib completed) | Preclinical studies for NSCLC [255] |
| Nivolumab | Not studied | Approved for NSCLC |
| Notch pathway inhibitor | Artesunate (preclinical studies [261]) | Rovalpituzumab (approved for SCLC) |
| Wnt pathway inhibitor | Preclinical studies [258,259] | Vantictumab (NCT01957007: Phase I completed) (NSCLC patients) |
| Muc1-based therapies | Anti-KL-6 (preclinical studies [262]) | Muc1 immunogen (L-BLP25 (Phase III completed [260])) (NSCLC patients) |
IPF: idiopathic pulmonary fibrosis; LC: lung cancer; NSCLC: non-small cell lung cancer; SCLC: small cell lung cancer; PDGFR: platelet derived growth factor receptor; VEGFR: vascular endothelial growth factor receptor; FGFR: fibroblast growth factor receptor; IL-13: interleukin 13; CCL2: chemokine ligand 2; TGFβ: transforming growth factor β; CTGF: connective tissue growth factor; LPAR1: lysophosphatidic acid receptor type 1; mTOR: mammalian Target of Rapamycin. * (NCT02581787: Phase I/II terminated for subjects with solid advanced tumors).
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030593
AMA Style
Ballester B, Milara J, Cortijo J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. International Journal of Molecular Sciences. 2019; 20(3):593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030593
Chicago/Turabian StyleBallester, Beatriz, Javier Milara, and Julio Cortijo. 2019. "Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets" International Journal of Molecular Sciences 20, no. 3: 593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030593
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.