Mechanisms of Chemotherapy-Induced Peripheral Neuropathy

 ,
,  ,
,  , , and
, , and
Abstract
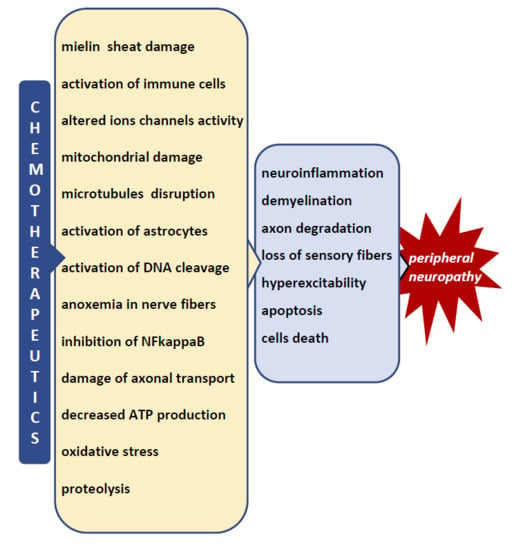
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
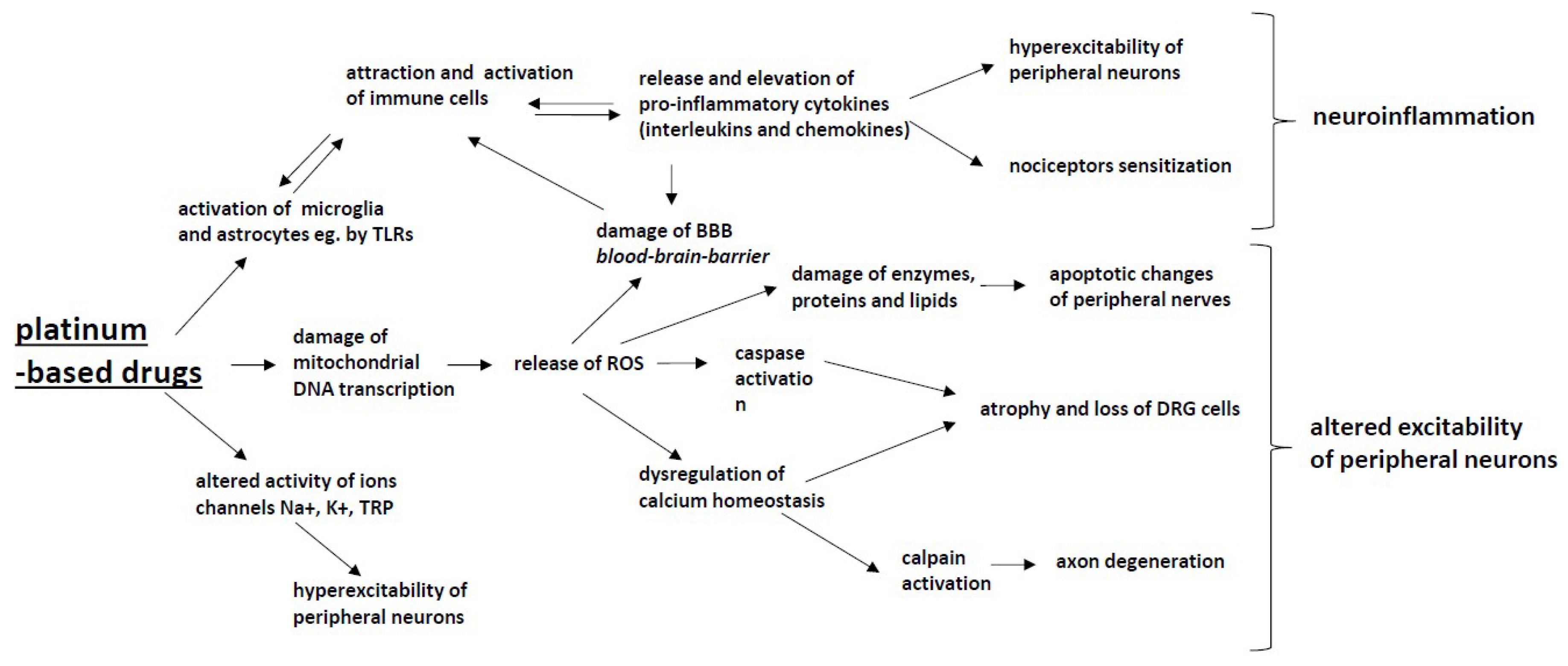
2. Platinum-Based Antineoplastics (Oxaliplatin, Cisplatin and Carboplatin)
- The binding to nuclear DNA (deoxyribonucleic acid) by cancer cells and the formation of DNA-platinum adducts, resulting in the inhibition of DNA replication and RNA (ribonucleic acid) transcription, followed by the arrest of cancer cell division, with the DNA adducts activating apoptotic pathways that induce cell death and tumor degradation;
- The alteration of mitochondrial function followed by the disruption of the respiratory chain function and an increased production of reactive oxygen species (ROS);
- The inhibition of mitochondrial DNA replication and transcription, leading to an altered mitochondrial function and the activation of apoptosis;
- The activation of the immune system (macrophages, T-cells and monocytes) followed by the release of pro-inflammatory cytokines and the activation of apoptosis;
- The influence on calcium signaling pathways and the function of protein kinase families (MAPK, mitogen activated protein kinases; JNK, c-Jun N-terminal kinase; PKC, protein kinase C; AKT, serine-threonine kinases), leading to tumor cell apoptosis.
2.1. Mitochondrial Dysfunction and Oxidative Stress
2.2. Intracellular Signaling
2.3. Ion Channels
2.4. Glial Cells
2.5. Inflammatory Mediators—Cytokines and Chemokines
2.6. Central Mechanisms
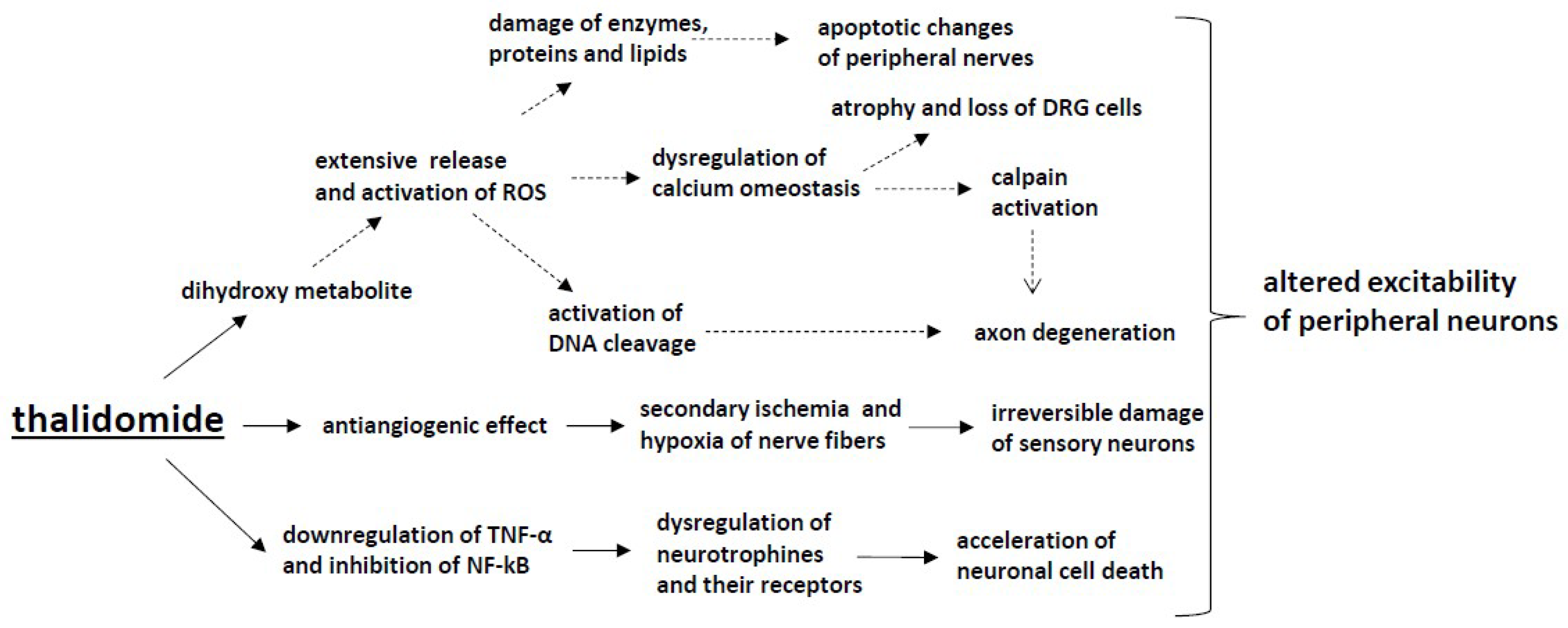
3. Immunomodulatory Drugs (Thalidomide)
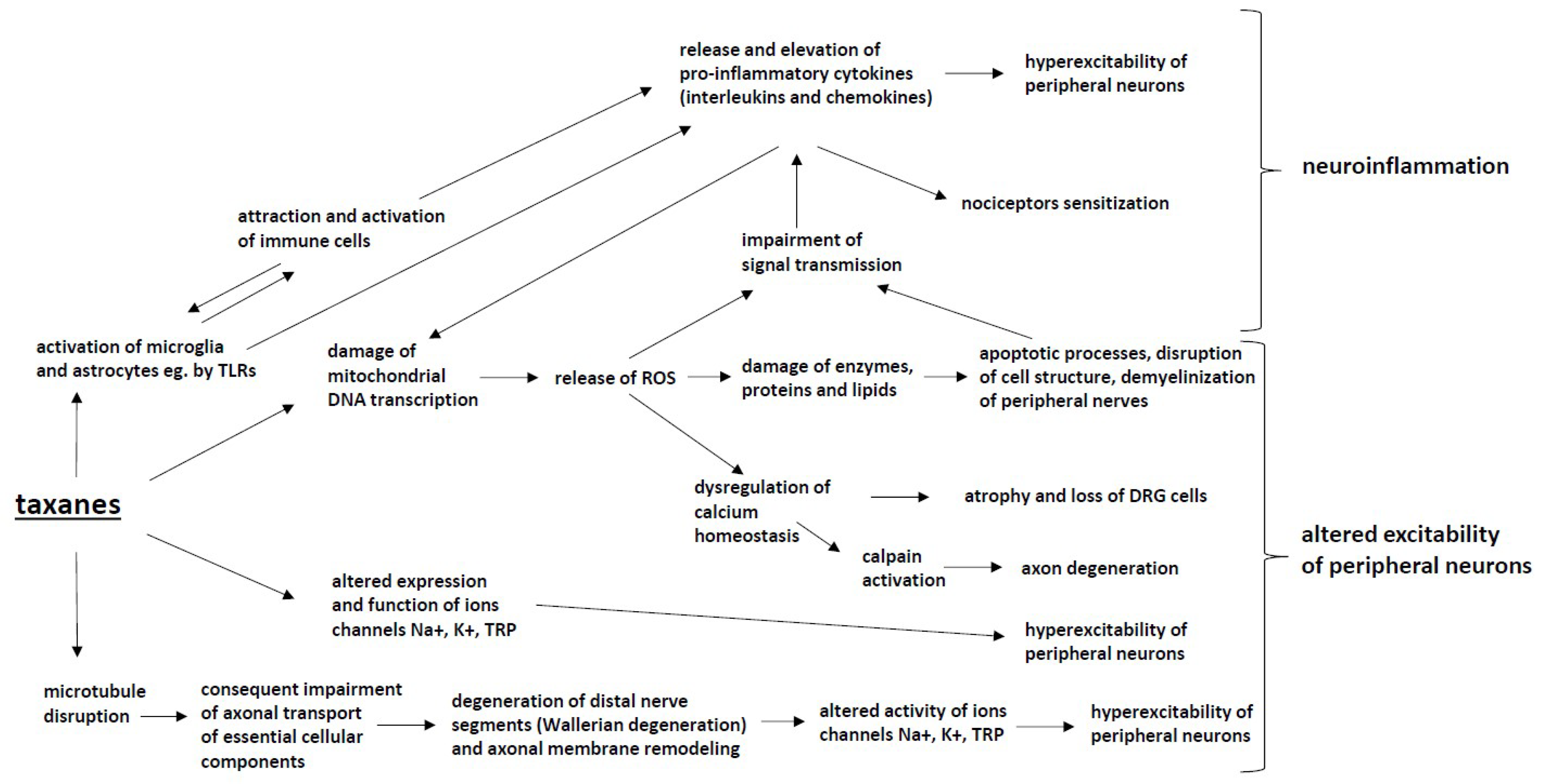
4. Taxanes
4.1. Microtubule Disruption
4.2. Mitochondrial Dysfunction
4.3. Axon Degeneration
4.4. Altered Calcium Homeostasis
4.5. Changes in Peripheral Nerve Excitability
4.6. Immune Processes and Neuroinflammation
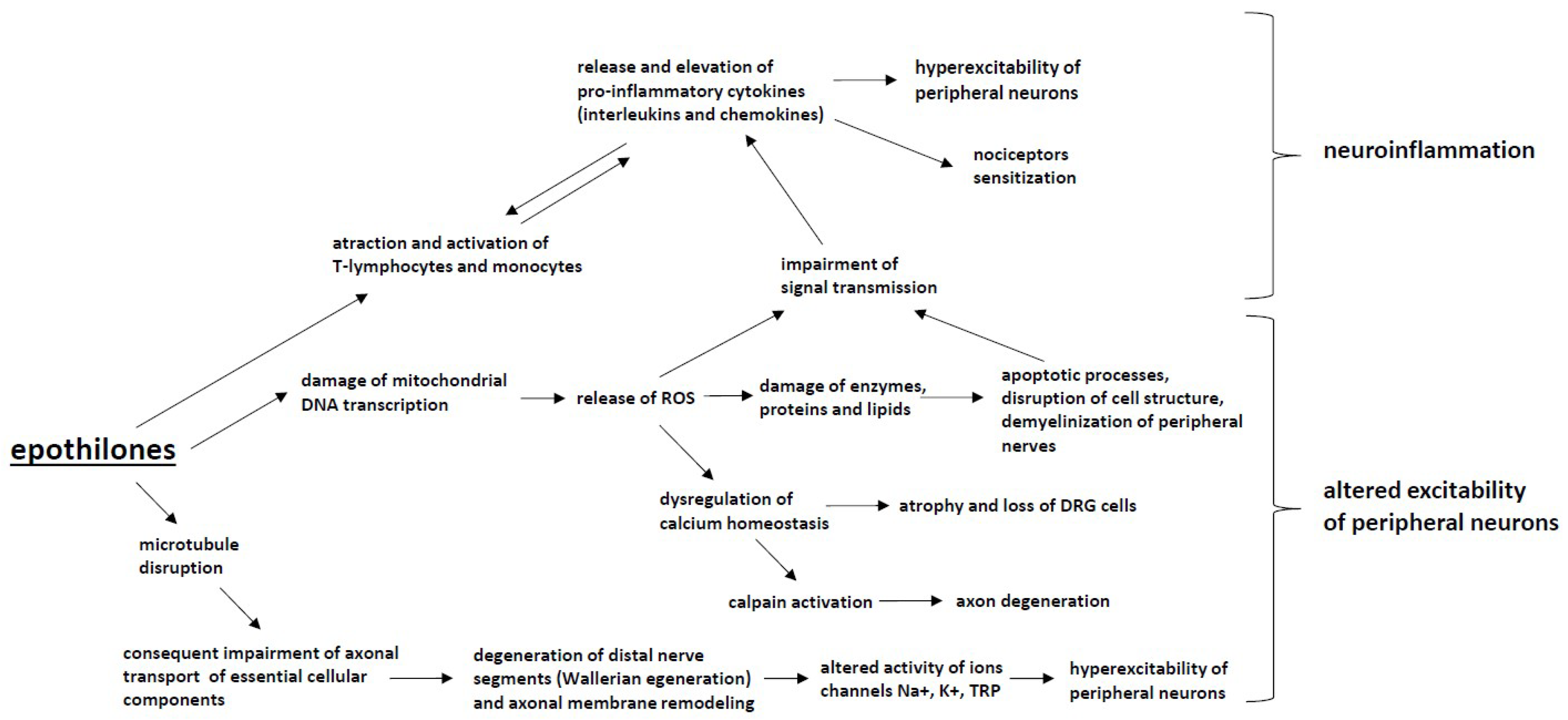
5. Epothilones (Ixabepilone)
6. Vinca Alkaloids
7. Protease Inhibitors: Bortezomib
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- American Society of Clinical Oncology. The state of cancer care in America, 2014: A report by the American Society of Clinical Oncology. J. Oncol. Pract. 2014, 10, 119–142. [Google Scholar] [CrossRef] [PubMed]
- Kent, E.E.; Forsythe, L.; Scoppa, S.; Hachey, M.; Rowland, J.H. Cancer survivors in the United States: Prevalence across the survivorship trajectory and implications for care. Cancer Epidemiol. Biomark. Prev. 2013, 22, 561–570. [Google Scholar]
- Glare, P.A.; Davies, P.S.; Finlay, E.; Gulati, A.; Lemanne, D.; Moryl, N.; Oeffinger, K.C.; Paice, J.A.; Stubblefield, M.D.; Syrjala, K.L. Pain in Cancer Survivors. J. Clin. Oncol. 2014, 32, 1739–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute: Chemotherapy Side Effects Sheets. Available online: http://www.cancer.gov/cancertopics/coping/physicaleffects/chemo-side-effects (accessed on 2 May 2014).
- Cioroiu, C.; Weimer, L.H. Update on Chemotherapy-Induced Peripheral Neuropathy. Curr. Neurol. Neurosci. Rep. 2017, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Juranek, J.K.; Zygulska, A.L. Chemotherapy-induced neuropathies—A growing problem for patients and health care providers. Brain Behav. 2016, 7, e00558. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Lacchetti, C.; Dworkin, R.H.; Lavoie-Smith, E.M.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Gavin, P.; Lavino, A.; Lustberg, M.B.; et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2014, 32, 1941–1967. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.T. Neuropathic pain in cancer. Br. J. Anaesth. 2013, 111, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, A.A.; Cavaletti, G.; Briani, C.; Velasco, R.; Bruna, J.; Campagnolo, M.; Alberti, P.; Bergamo, F.; Cortinovis, D.; Cazzaniga, M.; et al. Clinical pattern and associations of oxaliplatin acute neurotoxicity: A prospective study in 170 patients with colorectal cancer. Cancer 2013, 119, 438–444. [Google Scholar] [CrossRef]
- Maestri, A.; De Pasquale Ceratti, A.; Cundari, S.; Zanna, C.; Cortesi, E.; Crino, L. A pilot study on the effect of acetyl-L-carnitine in paclitaxel- andcisplatin-induced peripheral neuropathy. Tumori 2005, 91, 135–138. [Google Scholar]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174. [Google Scholar] [CrossRef]
- Cavaletti, G.; Alberti, P.; Frigeni, B.; Piatti, M.; Susani, E. Chemotherapy-induced neuropathy. Curr. Treat. Options Neurol. 2011, 13, 180–190. [Google Scholar] [CrossRef]
- Flatters, S.J.L.; Dougherty, P.M.; Colvin, L.A. Clinical and preclinical perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): A narrative review. Br. J. Anaesth. 2017, 119, 737–749. [Google Scholar] [CrossRef]
- Park, S.B.; Goldstein, D.; Krishnan, A.V.; Lin, C.S.; Friedlander, M.L.; Cassidy, J.; Koltzenburg, M.; Kiernan, M.C. Chemotherapy-induced peripheral neurotoxicity: A critical analysis. CA Cancer J. Clin. 2013, 63, 419–437. [Google Scholar] [CrossRef]
- Bernhardson, B.M.; Tishelman, C.; Rutqvist, L.E. Chemosensory changes experienced by patients undergoing cancer chemotherapy: A qualitative interview study. J. Pain Symptom Manag. 2007, 34, 403–412. [Google Scholar] [CrossRef]
- Kolb, N.A.; Smith, A.G.; Singleton, J.R.; Beck, S.L.; Stoddard, G.J.; Brown, S.; Mooney, K. The association of chemotherapy-induced peripheral neuropathy symptoms and the risk of falling. JAMA Neurol. 2016, 73, 860–866. [Google Scholar] [CrossRef]
- Mols, F.; van de Poll-Franse, L.V.; Vreugdenhil, G.; Beijers, A.J.; Kieffer, J.M.; Aaronson, N.K.; Husson, O. Reference data of the European Organisation for Research and Treatment of Cancer (EORTC) QLQ-CIPN20 Questionnaire in the general Dutch population. Eur. J. Cancer 2016, 69, 28–38. [Google Scholar] [CrossRef]
- Azhary, H.; Farooq, M.U.; Bhanushali, M.; Majid, A.; Kassab, M.Y. Peripheral neuropathy: Differential diagnosis and management. Am. Fam. Phys. 2010, 81, 887–892. [Google Scholar]
- Jones, D.; Zhao, F.; Brell, J.; Lewis, M.A.; Loprinzi, C.L.; Weiss, M.; Fisch, M.J. Neuropathic symptoms, quality of life, and clinician perception of patient care in medical oncology outpatients with colorectal, breast, lung, and prostate cancer. J. Cancer Surviv. 2015, 9, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.-W.; Huang, W.-J. Chemotherapy-induced peripheral neuropathy. Biomed. Rep. 2017, 6, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-TermEffects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharmacol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Areti, A.; Yerra, V.G.; Naidu, V.G.M.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Storey, D.J.; Sakala, M.; McLean, C.M.; Phillips, H.A.; Dawson, L.K.; Wall, L.R.; Fallon, M.T.; Clive, S. Capecitabine combined with oxaliplatin (CapOx) in clinical practice: How significant is peripheral neuropathy? Ann. Oncol. 2010, 21, 1657–1661. [Google Scholar] [CrossRef]
- Vanderhoop, R.G.; Vanderburg, M.E.L.; Huinink, W.W.T.; Vanhouwelingen, J.C.; Neijt, J.P. Incidence of neuropathy in 395 patients with ovarian cancer treated with or without cisplatin. Cancer 1990, 66, 1697–1702. [Google Scholar] [CrossRef] [Green Version]
- Krarup-Hansen, A.; Helweg-Larsen, S.; Schmalbruch, H.; Rorth, M.; Krarup, C. Neuronal involvement in cisplatin neuropathy: Prospective clinical and neurophysiological studies. Brain 2007, 130, 1076–1088. [Google Scholar] [CrossRef]
- Mollman, J.E.; Glover, D.J.; Hogan, W.M.; Furman, R.E. Cisplatin neuropathy. Risk factors, prognosis, and protection by WR-2721. Cancer 1988, 61, 2192–2195. [Google Scholar] [CrossRef] [Green Version]
- Gregg, R.W.; Molepo, J.M.; Monpetit, V.J.A.; Mikael, N.Z.; Redmond, D.; Gadia, M.; Stewart, D.J. Cisplatin neurotoxicity—The relationship between dosage, time, and platinum concentration in neurologic tissues, and morphological evidence of toxicity. J. Clin. Oncol. 1992, 10, 795–803. [Google Scholar] [CrossRef]
- Schmoll, H.J.; Kollmannsberger, C.; Metzner, B.; Hartmann, J.T.; Schleucher, N.; Schoffski, P.; Schleicher, J.; Rick, O.; Beyer, J.; Hossfeld, D.; et al. German Testicular Cancer Study. Long term results of first-line sequential high-dose etoposide, ifosfamide, and cisplatin chemotherapy plus autologous stem cell support for patients with advanced metastatic germ cell cancer: An extended phase I/II study of the German Testicular Cancer Study Group. J. Clin. Oncol. 2003, 21, 4083–4091. [Google Scholar]
- Hausheer, F.H.; Schilsky, R.L.; Bain, S.; Berghorn, E.J.; Lieberman, F. Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin. Oncol. 2006, 33, 15–49. [Google Scholar] [CrossRef]
- Leonard, G.D.; Wright, M.A.; Quinn, M.G.; Fioravanti, S.; Harold, N.; Schuler, B.; Thomas, R.R.; Grem, J.L. Survey of oxaliplatin-associated neurotoxicity using an interview-based questionnaire in patients with metastatic colorectal cancer. BMC Cancer 2005, 16, 116. [Google Scholar] [CrossRef]
- Gebremedhn, E.G.; Shortland, P.J.; Mahns, D.A. The incidence of acute oxaliplatin-induced neuropathy and its impact on treatment in the first cycle: A systematic review. BMC Cancer 2018, 18, 410. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Zolota, V.; Kyriakopoulou, O.; Kalofonos, H.P. Toxic peripheral neuropathy associated with commonly used chemotherapeutic agents. J BUON 2010, 15, 435–446. [Google Scholar]
- Deuis, J.R.; Zimmermann, K.; Romanovsky, A.A.; Possani, L.D.; Cabot, P.J.; Lewis, R.J.; Vetter, I. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for NaV1.6 in peripheral pain pathways. Pain 2013, 154, 1749–1757. [Google Scholar] [CrossRef] [Green Version]
- Attal, N.; Bouhassira, D.; Gautron, M.; Vaillant, J.N.; Mitry, E.; Lepere, C.; Rougier, P.; Guirimand, F. Thermal hyperalgesia as a marker of oxaliplatin neurotoxicity: A prospective quantified sensory assessment study. Pain 2009, 144, 245–252. [Google Scholar] [CrossRef]
- Arkenau, H.K. Capecitabine combined with oxaliplatin (CAPOX) in clinical practice: how significant is peripheral neuropathy? Ann Oncol. 2011, 22, 239–240. [Google Scholar] [CrossRef]
- Tofthagen, C.; McAllister, R.D.; McMillan, S.C. Peripheral neuropathy in patients with colorectal cancer receiving oxaliplatin. Clin. J. Oncol. Nurs. 2011, 15, 182–188. [Google Scholar] [CrossRef]
- Park, S.B.; Lin, C.S.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Long-term neuropathy after oxaliplatin treatment: Challenging the dictum of reversibility. Oncologist 2011, 16, 708–716. [Google Scholar] [CrossRef]
- Beijers, A.J.M.; Mols, F.; Vreugdenhil, G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support. Care Cancer 2014, 22, 1999–2007. [Google Scholar] [CrossRef] [Green Version]
- Briani, C.; Argyriou, A.A.; Izquierdo, C.; Velasco, R.; Campagnolo, M.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Bergamo, F.; Cortinovis, D.; et al. Long-term course of oxaliplatin-induced polyneuropathy: A prospective 2-year follow-up study. J. Peripher. Nerv. Syst. 2014, 19, 299–306. [Google Scholar] [CrossRef]
- Velasco, R.; Bruna, J.; Briani, C.; Argyriou, A.A.; Cavaletti, G.; Alberti, P.; Cacciavillani, M.; Frigeni, B.; Lonardi, S.; Cortinovis, D.; et al. Early predictors of oxaliplatin-induced cumulativeneuropathy in colorectal cancer patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 392–398. [Google Scholar] [CrossRef]
- Alejandro, L.M.; Behrendt, C.E.; Chen, K.; Openshaw, H.; Shibata, S. Predicting acute and persistent neuropathy associated with oxaliplatin. Am. J. Clin. Oncol. 2013, 36, 331–337. [Google Scholar] [CrossRef]
- Pulvers, J.N.; Marx, G. Factors associated with the development and severity of oxaliplatin-induced peripheral neuropathy: A systematic review. Asia Pac. J. Clin. Oncol. 2017, 13, 345–355. [Google Scholar] [CrossRef]
- Palugulla, S.; Thakkar, D.N.; Kayal, S.; Narayan, S.K.; Dkhar, S.A. Association of Voltage-Gated Sodium Channel Genetic Polymorphisms with Oxaliplatin-Induced Chronic Peripheral Neuropathy in South Indian Cancer Patients. Asian Pac. J. Cancer Prev. 2017, 18, 3157–3165. [Google Scholar]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Cortes-Funes, H.; Boni, C.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef]
- Ray, B.; Gupta, B.; Mehrotra, R. Binding of Platinum Derivative, Oxaliplatin to Deoxyribonucleic Acid: Structural Insight into Antitumor Action. J. Biomol. Struct. Dyn. 2018. [Google Scholar] [CrossRef]
- Riddell, I.A. Cisplatin and Oxaliplatin: Our Current Understanding of Their Actions. Met. Ions Life Sci. 2018. [Google Scholar] [CrossRef]
- McKeage, M.J.; Hsu, T.; Screnci, D.; Haddad, G.; Baguley, B.C. Nucleolar damage correlates with neurotoxicity induced by different platinum drugs. Br. J. Cancer 2001, 85, 1219–1225. [Google Scholar] [CrossRef]
- Pereira, A.F.; de Oliveira, F.F.B.; de Freitas Alves, B.W.; de Menezes, K.L.S.; de Mesquita, A.K.V.; Lisboa, M.R.P.; de Sousa, K.K.O.; Vale, M.L. Neurotoxic effect of oxaliplatin: Comparison with its oxalate-free analogue cis-[PtII(1R,2R-DACH)(3-acetoxy-1,1-cyclobutanedicarboxylato)] (LLC-1402) in mice. Toxicol. Appl. Pharmacol. 2018, 340, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kober, K.M.; Olshen, A.; Conley, Y.P.; Schumacher, M.; Topp, K.; Smoot, B.; Mazor, M.; Chesney, M.; Hammer, M.; Paul, S.M.; et al. Expression of mitochondrial dysfunction-related genes and pathways in paclitaxel-induced peripheral neuropathy in breast cancer survivors. Mol Pain. 2018, 14, 1744806918816462. [Google Scholar] [CrossRef]
- Jaggi, A.S.; Singh, N. Mechanisms in cancer chemotherapeutic drugs-induced peripheral neuropathy. Toxicology 2012, 291, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Viatchenko-Karpinski, V.; Ling, J.; Gu, J.G. Down-regulation of Kv4.3 channels and a-type K+ currents in V2 trigeminal ganglion neurons of rats following oxaliplatin treatment. Mol. Pain 2018, 14, 1–11. [Google Scholar] [CrossRef]
- Fujita, S.; Hirota, T.; Sakiyama, R.; Baba, M.; Ieiri, I. Identification of drug transporters contributing to oxaliplatin-induced peripheral neuropathy. J. Neurochem. 2019, 148, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, L.; Cai, X.; Fang, Y.; Wang, J.; Chen, G.; Yang, J.; Zhou, Q.; Sun, X.; Cheng, X.; et al. Nrf2 inhibits oxaliplatin-induced peripheral neuropathy via protection of mitochondrial function. Free Radic. Biol. Med. 2018, 120, 13–24. [Google Scholar] [CrossRef]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. PARP inhibition in platinum-based chemotherapy: Chemopotentiation and neuroprotection. Pharmacol. Res. 2018, 137, 104–113. [Google Scholar] [CrossRef]
- Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J. Pain 2008, 9, 463–472. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Zheng, H.; Xiao, W.H.; Bennett, G.J. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp. Neurol. 2011, 232, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced neuropathy: Oxidative stress as pathological mechanism. Protective effect of silibinin. J. Pain 2012, 13, 276–284. [Google Scholar] [CrossRef]
- Imai, S.; Koyanagi, M.; Azimi, Z.; Nakazato, Y.; Matsumoto, M.; Ogihara, T.; Yonezawa, A.; Omura, T.; Nakagawa, S.; Wakatsuki, S.; et al. Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Sci. Rep. 2017, 7, 5947. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced oxidative stress in nervous system-derived cellular models: Could it correlate with in vivo neuropathy? Free Radic Biol Med. 2013, 143–150. [Google Scholar] [CrossRef]
- Waseem, M.; Kaushik, P.; Tabassum, H.; Parvez, S. Role of Mitochondrial Mechanism in Chemotherapy-Induced Peripheral Neuropathy. Curr. Drug Metab. 2018, 19, 47–54. [Google Scholar] [CrossRef]
- Sharawy, N.; Rashed, L.; Youakim, M.F. Evaluation of multi neuroprotective effects of erythropoietin using cisplatin induced peripheral neurotoxicity model. Exp. Toxicol. Pathol. 2015, 67, 315–322. [Google Scholar] [CrossRef]
- Pan, L.; Song, K.; Hu, F.; Sun, W.; Lee, I. Nitric oxide induces apoptosis associated with TRPV1 channel-mediated Ca(2+) entryvia S-nitrosylation in osteoblasts. Eur. J. Pharmacol. 2013, 715, 280–285. [Google Scholar] [CrossRef]
- Jamieson, S.M.; Liu, J.; Connor, B.; McKeage, M.J. Oxaliplatin causes selective atrophy of a subpopulation of dorsal root ganglion neurons without inducing cell loss. Cancer Chemother. Pharmacol. 2005, 56, 391–399. [Google Scholar] [CrossRef]
- Apostolidis, L.; Schwarz, D.; Xia, A.; Weiler, M.; Heckel, A.; Godel, T.; Heiland, S.; Schlemmer, H.P.; Jäger, D.; Bendszus, M.; et al. Dorsal root ganglia hypertrophy as in vivo correlate of oxaliplatin-induced polyneuropathy. PLoS ONE 2017, 12, e0183845. [Google Scholar] [CrossRef]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, M.; Egashira, N.; Kawashiri, T.; Yano, T.; Ikesue, H.; Oishi, R. Oxaliplatin-induced neuropathy in the rat: Involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain 2009, 147, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Galimberti, A.; Maggioni, D.; Ravasi, M.; Pasini, S.; Nicolini, G.; Bossi, M.; Miloso, M.; Cavaletti, G.; Tredici, G. Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology 2009, 30, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmaker, J.G.J.; Faber, C.G.; Merkies, I.S.J.; Waxman, S.G. Painful peripheral neuropathy and sodium channel mutations. Neurosci. Lett. 2015, 596, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Sittl, R.; Lampert, A.; Huth, T.; Schuy, E.T.; Link, A.S.; Fleckenstein, J.; Alzheimer, C.; Grafe, P.; Carr, R.W. Anticancer drug oxaliplatin induces acute cooling aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc. Natl. Acad. Sci. USA 2012, 109, 6704–6709. [Google Scholar] [CrossRef] [PubMed]
- Lessans, S.; Lassiter, C.B.; Carozzi, V.; Heindel, P.; Semperboni, S.; Oggioni, N.; Chiorazzi, A.; Thompson, C.; Wagner, M.; Holden, J.; et al. Global transcriptomic profile of dorsal root ganglion and physiological correlates of cisplatin-induced peripheral neuropathy. Nurs Res. 2019, 68, 145–155. [Google Scholar] [CrossRef]
- Deuis, J.R.; Lim, Y.L.; de Sousa, S.R.; Lewis, R.J.; Alewood, P.F.; Cabot, P.J.; Vetter, I. Analgesic effects of clinically used compounds in novel mousemodels of polyneuropathy induced by oxaliplatin and cisplatin. Neuro Oncol. 2014, 16, 1324–1332. [Google Scholar] [CrossRef]
- Adelsberger, H.S.; Quasthoff, S.; Grosskreutz, J.; Lepier, A.; Eckel, F.; Lersch, C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur. J. Pharmacol. 2000, 406, 1–25. [Google Scholar] [CrossRef]
- Ghelardini, C.; Desaphy, J.F.; Muraglia, M.; Corbo, F.; Matucci, R.; Dipalma, A.; Bertucci, C.; Pistolozzi, M.; Nesi, M.; Norcini, M.; et al. Effects of a new potent analog of tocainide on hNaV1.7 sodium channels and in vivo neuropathic pain models. Neuroscience 2010, 169, 863–873. [Google Scholar] [CrossRef]
- Lolignier, S.; Bonnet, C.; Gaudioso, C.; Noël, J.; Ruel, J.; Amalem, M.; Ferrier, J.; Rodat-Despoix, L.; Bouvier, V.; Aissouni, Y.; et al. The NaV1.9 channel is a key determinant of cold pain sensation and cold allodynia. Cell Rep. 2015, 11, 1067–1078. [Google Scholar] [CrossRef]
- Heide, R.; Bostock, H.; Ventzel, L.; Grafe, P.; Bergmans, J.; Fuglsang-Frederiksen, A.; Finnerup, N.B.; Tankisi, H. Axonal excitability changes and acute symptoms of oxaliplatin treatment: In vivo evidence for slowed sodium channel inactivation. Clin. Neurophysiol. 2018, 129, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Antonacopoulou, A.; Genazzani, A.A.; Briani, C.; Bruna, J.; Terrazzino, S.; Velasco, R.; Alberti, P.; Campagnolo, M.; et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer 2013, 119, 3570–3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodeling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef]
- Poupon, L.; Lamoine, S.; Pereira, V.; Barriere, D.A.; Lolignier, S.; Giraudet, F.; Aissouni, Y.; Meleine, M.; Prival, L.; Richard, D.; et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology 2018, 140, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Thibault, K.; Calvino, B.; Dubacq, S.; Roualle-de-Rouville, M.; Sordoillet, V.; Rivals, I.; Pezet, S. Cortical effect of oxaliplatin associated with sustained neuropathic pain: Exacerbation of cortical activity and down-regulation of potassium channel expression in somatosensory cortex. Pain 2012, 153, 1636–1647. [Google Scholar] [CrossRef]
- Chukyo, A.; Chiba, T.; Kambe, T.; Yamamoto, K.; Kawakami, K.; Taguchi, K.; Abe, K. Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 2018, 67, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ta, L.E.; Bieber, A.J.; Carlton, S.M.; Loprinzi, C.L.; Low, P.A.; Windebank, A.J. Transient receptor potential vanilloid 1 is essential forcisplatin-induced heat hyperalgesia in mice. Mol. Pain 2010, 6, 15. [Google Scholar] [CrossRef]
- Yamamoto, K.; Chiba, N.; Chiba, T.; Kambe, T.; Abe, K.; Kawakami, K.; Utsunomiya, I.; Taguchi, K. Transient receptor potential ankyrin 1 that is induced in dorsal root ganglion neurons contributes to acute cold hypersensitivity after oxaliplatin administration. Mol. Pain 2015, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Isami, K.; Nakamura, S.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol. Pain 2012, 8, 55. [Google Scholar] [CrossRef]
- Nassini, R.; Gees, M.; Harrison, S.; De Siena, G.; Materazzi, S.; Moretto, N.; Failli, P.; Preti, D.; Marchetti, N.; Cavazzini, A.; et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Kaneko, S. Roles of Transient Receptor Potential Ankyrin 1 in Oxaliplatin-Induced Peripheral Neuropathy. Biol. Pharm. Bull. 2017, 40, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Nakamura, M.; Meng, Z.; Hamano, S.; Inoue, K.; Numata, T.; Takahashi, N.; Nagayasu, K.; Shirakawa, H.; Mori, Y.; et al. Distinct Mechanism of Cysteine Oxidation-Dependent Activation and Cold Sensitization of Human Transient Receptor Potential Ankyrin 1 Channel by High and Low Oxaliplatin. Front. Physiol. 2017, 8, 878. [Google Scholar] [CrossRef] [PubMed]
- Riva, B.; Dionisi, M.; Potenzieri, A.; Chiorazzi, A.; Cordero-Sanchez, C.; Rigolio, R.; Carozzi, V.A.; Lim, D.; Cavaletti, G.; Marmiroli, P.; et al. Oxaliplatin induces pH acidification in dorsal root ganglia neurons. Sci. Rep. 2018, 8, 15084. [Google Scholar] [CrossRef]
- Kawashiri, T.; Egashira, N.; Kurobe, K.; Tsutsumi, K.; Yamashita, Y.; Ushio, S.; Yano, T.; Oishi, R. L Type Ca2+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol. Pain 2012, 8, 7. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Z.F.; Liao, M.F.; Yao, W.L.; Wang, J.; Wang, X.R. Blocking PAR2 attenuates oxaliplatin-induced neuropathic pain via TRPV1 and releases of substance P and CGRP in superficial dorsal horn of spinal cord. J. Neurol. Sci. 2015, 352, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Robinson, C.R.; Zhang, H.; Dougherty, P.M. Spinal astrocyte gap junctions contribute to oxaliplatin-induced mechanical hypersensitivity. J. Pain 2013, 14, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare Mannelli, L.; Pacini, A.; Bonaccini, L.; Zanardelli, M.; Mello, T.; Ghelardini, C. Morphologic features and glial activation in rat oxaliplatin-dependent neuropathic pain. J. Pain 2013, 14, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Pacini, A.; Micheli, L.; Tani, A.; Zanardelli, M.; Ghelardini, C. Glial role in oxaliplatin induced neuropathic pain. Exp. Neurol. 2014, 261, 22–33. [Google Scholar] [CrossRef]
- Robinson, C.R.; Zhang, H.; Dougherty, P.M. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience 2014, 274, 308–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlman, C.; Doyle, T.M.; Little, J.W.; Luongo, L.; Janes, K.; Chen, Z.; Esposito, E.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 2018, 159, 1025–1034. [Google Scholar] [CrossRef]
- Makker, P.G.; Duffy, S.S.; Lees, J.G.; Perera, C.J.; Tonkin, R.S.; Butovsky, O.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Characterisation of immune and neuroinflammatory changes associated with chemotherapy-induced peripheral neuropathy. PLoS ONE 2017, 12, e0170814. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.Y.; Zhou, Y.; Cui, W.Q.; Hu, X.M.; Du, L.X.; Mi, W.L.; Chu, Y.X.; Wu, G.C.; Wang, Y.Q.; Mao-Ying, Q.L. Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain Behav. Immun. 2018, 68, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Uta, D.; Kato, M.; Toume, K.; Komatsu, K.; Kuraishi, Y. prophylactic administration of aucubin inhibits paclitaxel-induced mechanical allodynia via the inhibition of endoplasmic reticulum stress in peripheral Schwann Cells. Biol Pharm Bull. 2017, 40, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Makker, P.G.S.; Tonkin, R.S.; Abdulla, M.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 2017, 73, 22–29. [Google Scholar] [CrossRef]
- Warwick, R.A.; Hanani, M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur. J. Pain 2013, 17, 571–580. [Google Scholar] [CrossRef]
- Wang, X.M.; Lehky, T.J.; Brell, J.M.; Dorsey, S.G. Discover in cytokines as targets for chemotherapy-induced painful peripheral neuropathy. Cytokine 2012, 59, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.; Wahlman, C.; Little, J.W.; Doyle, T.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model ofoxaliplatin-induced peripheral neuropathy. Brain Behav. Immun. 2015, 44, 91–99. [Google Scholar] [CrossRef]
- Li, C.; Deng, T.; Shang, Z.; Wang, D.; Xiao, Y. Blocking TRPA1 and TNF-α Signal Improves Bortezomib-Induced Neuropathic Pain. Cell. Physiol. Biochem. 2018, 51, 2098–2110. [Google Scholar] [CrossRef]
- Park, H.J.; Stokes, J.A.; Corr, M.; Yaksh, T.L. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother. Pharmacol. 2014, 73, 25–34. [Google Scholar] [CrossRef]
- Jin, X.; Gereau, R.W.T. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensoryneurons by tumor necrosis factor-alpha. J. Neurosci. 2006, 26, 246–255. [Google Scholar] [CrossRef]
- Xu, D.; Hui Zhao, H.; Gao, H.; Zhao, H.; Liu, D.; Li, J. Participation of pro-inflammatorycytokines in neuropathic pain evoked bychemotherapeutic oxaliplatin via centralGABAergic pathway. Mol. Pain 2018, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Miller, R.J. Insights into the regulation of chemokine receptors by molecularsignaling pathways: Functional roles in neuropathic pain. Brain Behav. Immun. 2010, 24, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Li, Y.Y.; Cui, W.; Li, L.B.; Zhang, Z.C.; Tian, B.P.; Zhang, G.S. Melatonin attenuates pain hypersensitivity and decreases astrocyte-mediated spinal neuroinflammation ina rat model of oxaliplatin-induced pain. Inflammation 2017, 40, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Illias, A.M.; Gist, A.C.; Zhang, H.; Kosturakis, A.; Dougherty, P.M. Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain 2018, 159, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Yang, B.; Donnelly, D.F.; Ma, C.; La Motte, R.H. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J. Neurophysiol. 2006, 96, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Li, H.; Liu, Z.L.; Li, Q.; Qiu, H.W.; Zeng, L.J.; Yang, W.; Zhang, X.Z.; Li, Z.Y. Activation of STAT3-mediated CXCL12 up-regulation in the dorsal root ganglion contributes to oxaliplatin-induced chronic pain. Mol. Pain 2017, 13. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.Z.; Li, D.; Ou-Yang, H.D.; Liu, C.C.; Liu, X.G.; Ma, C.; Wei, J.Y.; Liu, Y.; Xin, W.J. Cerebrospinal fluid oxaliplatin contributes to the acute pain induced by systemicadministration of oxaliplatin. Anesthesiology 2016, 124, 1109–1121. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.S.; Tao, R.; Zhang, J.; Liu, L.; Jiang, Y.H.; Ma, S.H.; Song, L.X.; Xia, L.J. Upregulation of CX3CL1 mediated by NF-κB activation in dorsal root ganglion contributes to peripheral sensitization and chronic pain induced by oxaliplatin administration. Mol. Pain 2017, 13. [Google Scholar] [CrossRef]
- Dietrich, J.; Prust, M.; Kaiser, J. Chemotherapy, cognitive impairment and hippocampal toxicity. Neuroscience 2015, 309, 224–232. [Google Scholar] [CrossRef]
- Horky, L.L.; Gerbaudo, V.H.; Zaitsev, A.; Plesniak, W.; Hainer, J.; Govindarajulu, U.; Kikinis, R.; Dietrich, J. Systemic chemotherapy decreases brain glucose metabolism. Ann. Clin. Transl. Neurol. 2014, 1, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Janelsins, M.C.; Kohli, S.; Mohile, S.G.; Usuki, K.; Ahles, T.A.; Morrow, G.R. An update on cancer- and chemotherapy related cognitive dysfunction: Current status. Semin. Oncol. 2011, 38, 431–438. [Google Scholar] [CrossRef]
- Nudelman, K.N.; McDonald, B.C.; Wang, Y.; Smith, D.J.; West, J.D.; O’Neill, D.P.; Zanville, N.R.; Champion, V.L.; Schneider, B.P.; Saykin, A.J. Cerebral perfusion and gray matter changes associated with chemotherapy-induced peripheral neuropathy. J. Clin. Oncol. 2016, 34, 677–683. [Google Scholar] [CrossRef]
- Jacobs, S.S.; Fox, E.; Dennie, C.; Morgan, L.B.; McCully, C.L.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin. Cancer Res. 2005, 11, 1669–1674. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of blood–brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: Consequences for interendothelial adherens and tight junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1beta regulates blood–brain barrier permeability via reactivation of the hypoxia angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Becatti, M.; Paternostro, F.; Gulisano, M.; Ghelardini, C.; Salvemini, D.; Di Cesare Mannelli, L.; Pacini, A. Oxaliplatin-induced blood brain barrier loosening: A new point of view on chemotherapy-induced neurotoxicity. Oncotarget 2018, 9, 23426–23438. [Google Scholar] [CrossRef]
- Sanna, M.D.; Ghelardini, C.; Galeotti, N. Altered Expression of Cytoskeletal and Axonal Proteins in Oxaliplatin-Induced Neuropathy. Pharmacology 2016, 97, 146–150. [Google Scholar] [CrossRef]
- Richardson, P.; Hideshima, T.; Anderson, K. Thalidomide in multiple myeloma. Biomed. Pharmacother. 2002, 56, 115–128. [Google Scholar] [CrossRef]
- Fernyhough, P.; Smith, D.R.; Schapansky, J.; Van Der Ploeg, R.; Gardiner, N.J.; Tweed, C.W.; Kontos, A.; Freeman, L.; Purves-Tyson, T.D.; Glazner, G.W. Activation of nuclearfactor-(kappa) B via endogenous tumor necrosis factor (alpha) regulates survival of axotomized adult sensory neurons. J. Neurosci. 2005, 25, 1682–1690. [Google Scholar] [CrossRef]
- Mohty, B.; El-Cheikh, J.; Yakoub-Agha, I.; Moreau, P.; Harousseauj, L.; Mohty, M. Peripheral neuropathy and new treatments for multiple myeloma: Background and practical recommendations. Haematologica 2010, 95, 311–319. [Google Scholar] [CrossRef]
- Morawska, M.; Grzasko, N.; Kostyra, M.; Wojciechowicz, J.; Hus, M. Therapy-related peripheral neuropathy in multiple myeloma patients. Hematol. Oncol. 2015, 33, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Chen, C.I.; Sutton, D.; Reece, D.; Voralia, M.; Stewart, A.K. Intermediate dose thalidomide (200 mg daily) has comparable efficacy and less toxicity than higher doses in relapsed multiple myeloma. Leuk. Lymphoma 2003, 44, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Schenkel, B.; Mileshkin, L. An analysis of clinical trials assessing the efficacy and safety of single-agent thalidomide in patients with relapsed or refractory multiple myeloma. Leuk. Lymphoma 2007, 48, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Bramuzzo, M.; Stocco, G.; Montico, M.; Arrigo, S.; Calvi, A.; Lanteri, P.; Costa, S.; Pellegrino, S.; Magazzù, G.; Barp, J.; et al. Risk Factors and Outcomes of Thalidomide-induced Peripheral Neuropathy in a Pediatric Inflammatory Bowel Disease Cohort. Inflamm. Bowel Dis. 2017, 23, 1810–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sanz, R.; Corchete, L.A.; Alcoceba, M.; Chillon, M.C.; Jiménez, C.; Prieto, I.; García-Álvarez, M.; Puig, N.; Rapado, I.; Barrio, S.; et al. Prediction of peripheral neuropathy in multiple myeloma patients receiving bortezomib and thalidomide: A genetic study based on a single nucleotide polymorphism array. Hematol. Oncol. 2017, 35, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Corthals, S.L.; Walker, B.A.; Ross, F.M.; Gregory, W.M.; Dickens, N.J.; Lokhorst, H.M.; Goldschmidt, H.; Davies, F.E.; Durie, B.G.; et al. Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J. Clin. Oncol. 2011, 29, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Goloubeva, O.; Dalal, J.S.; Can, I.; Thompson, J.; Rapoport, A.P.; Heyman, M.; Akpek, G.; Fenton, R.G. Neurotoxicity of bortezomib therapy in multiple myeloma: A single-center experience and review of the literature. Cancer 2007, 110, 1042–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamilarasan, K.P.; Kolluru, G.K.; Rajaram, M.; Indhumathy, M.; Saranya, R.; Chatterjee, S. Thalidomide attenuates nitric oxide mediated angiogenesis by blocking migration of endothelial cells. BMC Cell Biol. 2006, 7, 17. [Google Scholar] [CrossRef]
- Jongen, J.L.M.; Broijl, A.; Sonneveld, P. Chemotherapy-induced peripheral neuropathies in hematological malignancies. J. Neurooncol. 2015, 121, 229–237. [Google Scholar] [CrossRef]
- Keifer, A.; Guttridge, D.C.; Ashburner, B.P.; Baldwin, A.S. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J. Biol. Chem. 2001, 276, 22382–22387. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Macedo-Júnior, S.J.; Borges, F.R.; Cremonese, R.P.; da Silva, M.D.; Luiz-Cerutti, M.; Martins, D.F.; Rodrigues, A.L.; Santos, A.R. Thalidomide reduces mechanical hyperalgesia and depressive-like behavior induced by peripheral nerve crush in mice. Neuroscience 2015, 303, 51–58. [Google Scholar] [CrossRef]
- Wani, T.H.; Chakrabarty, A.; Shibata, N.; Yamazaki, H.; Guengerich, F.P.; Chowdhury, G. The Dihydroxy Metabolite of the Teratogen Thalidomide Causes Oxidative DNA Damage. Chem. Res. Toxicol. 2017, 30, 1622–1628. [Google Scholar] [CrossRef]
- Yared, J.A.; Tkaczuk, K.H. Update on taxane development: New analogs and new formulations. Drug Des. Dev. Ther. 2012, 6, 371–384. [Google Scholar]
- Scripture, C.D.; Figg, W.D.; Sparreboom, A. Peripheral neuropathy induced by paclitaxel: Recent insights and future perspectives. Curr. Neuropharmacol. 2006, 4, 165–172. [Google Scholar] [CrossRef]
- De Iuliis, F.; Taglieri, L.; Salerno, G.; Lanza, R.; Scarpa, S. Taxane induced neuropathy in patients affected by breast cancer: Literature review. Crit. Rev. Oncol./Hematol. 2015, 96, 34–45. [Google Scholar] [CrossRef]
- Eckhoff, A.S.; Knoop, M.B.; Jensen, M.; Ewertz, M. Persistence of docetaxel-induced neuropathy and impact on quality of life among breast cancer survivors. Eur. J. Cancer 2015, 51, 292–300. [Google Scholar] [CrossRef]
- Peng, L.; Bu, Z.; Ye, X.; Zhou, Y.; Zhao, Q. Incidence and risk of peripheral neuropathy with nab-paclitaxel in patients with cancer: A meta-analysis. Eur. J. Cancer Care 2017, 26. [Google Scholar] [CrossRef]
- Gornstein, E.L.; Schwarz, T.L. Neurotoxic Mechanisms of Paclitaxel Are Local to the Distal Axon and Independent of Transport Defects. Exp. Neurol. 2017, 288, 153–166. [Google Scholar] [CrossRef]
- Bober, B.G.; Shah, S.B. Paclitaxel Alters Sensory Nerve Biomechanical Properties. J. Biomech. 2015, 48, 3559–3567. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Brady, S.T.; Feinstein, S.C.; Wilson, L.; Jordan, M.A. Effects of Eribulin, Vincristine, Paclitaxel and Ixabepilone on Fast Axonal Transport and Kinesin-1 Driven Microtubule Gliding: Implications for Chemotherapy-Induced Peripheral Neuropathy. Neurotoxicology 2013, 37, 231–239. [Google Scholar] [CrossRef]
- Shemesh, O.A.; Spira, M.E. Paclitaxel Induces Axonal Microtubules Polar Reconfiguration and Impaired Organelle Transport: Implications for the Pathogenesis of Paclitaxel-Induced Polyneuropathy. Acta Neuropathol. 2010, 119, 235–248. [Google Scholar] [CrossRef]
- Wozniak, K.M.; Vornov, J.J.; Wum, Y.; Liu, Y.; Carozzi, V.A.; Rodriguez-Menendez, V.; Ballarini, E.; Alberti, P.; Pozzi, E.; Semperboni, S.; et al. Peripheral neuropathy induced by microtubule-targeted chemotherapies: insights into acute injury and long-term recovery. Cancer Res. 2018, 78, 817–829. [Google Scholar] [CrossRef]
- Bobylev, I.; Joshi, A.R.; Barham, M.; Ritter, C.; Neiss, W.F.; Höke, A.; Lehmann, H.C. Paclitaxel Inhibits MRNA Transport in Axons. Neurobiol. Dis. 2015, 82, 321–331. [Google Scholar] [CrossRef]
- Doyle, T.; Chen, Z.; Muscoli, C.; Bryant, L.; Esposito, E.; Cuzzocrea, S.; Dagostino, C.; Ryerse, J.; Rausaria, S.; Kamadulski, A.; et al. Targeting the Overproduction of Peroxynitrite for the Prevention and Reversal of Paclitaxel-Induced Neuropathic Pain. J. Neurosci. 2012, 32, 6149–6160. [Google Scholar] [CrossRef] [Green Version]
- Duggett, N.A.; Griffiths, L.A.; McKenna, O.E.; de Santis, V.; Yongsanguanchai, N.; Mokori, E.B.; Flatters, S.J.L. Oxidative Stress in the Development, Maintenance and Resolution of Paclitaxel-Induced Painful Neuropathy. Neuroscience 2016, 333, 13–26. [Google Scholar] [CrossRef]
- Xiao, W.H.; Zheng, H.; Zheng, F.Y.; Nuydens, R.; Meert, T.F.; Bennett, G.J. Mitochondrial Abnormality in Sensory, but Not Motor, Axons in Paclitaxel-Evoked Painful Peripheral Neuropathy in the Rat. Neuroscience 2011, 199, 461–469. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Griffiths, L.A.; Flatters, S.J.L. Pharmacological Modulation of the Mitochondrial Electron Transport Chain in Paclitaxel-Induced Painful Peripheral Neuropathy. J. Pain 2015, 16, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Duggett, N.A.; Griffiths, L.A.; Flatters, S.J.L. Paclitaxel-Induced Painful Neuropathy Is Associated with Changes in Mitochondrial Bioenergetics, Glycolysis, and an Energy Deficit in Dorsal Root Ganglia Neurons. Pain 2017, 158, 1499–1508. [Google Scholar] [CrossRef]
- Flatters, S.J.L.; Bennett, G.J. Studies of Peripheral Sensory Nerves in Paclitaxel-Induced Painful Peripheral Neuropathy: Evidence for Mitochondrial Dysfunction. Pain 2006, 122, 245–257. [Google Scholar] [CrossRef]
- Xiao, W.H.; Bennett, G.J. Effects of Mitochondrial Poisons on the Neuropathic Pain Produced by the Chemotherapeutic Agents, Paclitaxel and Oxaliplatin. Pain 2012, 153, 704–709. [Google Scholar] [CrossRef]
- Sahenk, Z.; Barohn, R.; New, P.; Mendell, J.R. Taxol Neuropathy. Electrodiagnostic and Sural Nerve Biopsy Findings. Arch. Neurol. 1994, 51, 726–729. [Google Scholar] [CrossRef]
- Boehmerle, W.; Huehnchen, P.; Peruzzaro, S.; Balkaya, M.; Endres, M. Electrophysiological, Behavioral and Histological Characterization of Paclitaxel, Cisplatin, Vincristine and Bortezomib-Induced Neuropathy in C57Bl/6 Mice. Sci. Rep. 2015, 4, 6370. [Google Scholar] [CrossRef]
- Siau, C.; Xiao, W.; Bennett, G.J. Paclitaxel- and Vincristine-Evoked Painful Peripheral Neuropathies: Loss of Epidermal Innervation and Activation of Langerhans Cells. Exp. Neurol. 2006, 201, 507–514. [Google Scholar] [CrossRef]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology. 2014, 76, 175–183. [Google Scholar] [CrossRef]
- Boyette-Davis, J.; Xin, W.; Zhang, H.; Dougherty, P.M. Intraepidermal Nerve Fiber Loss Corresponds to the Development of Taxol-Induced Hyperalgesia and Can Be Prevented by Treatment with Minocycline. Pain 2011, 152, 308–313. [Google Scholar] [CrossRef]
- Ferrari, G.; Nallasamy, N.; Downs, H.; Dana, R.; Oaklander, A.L. Corneal Innervation as a Window to Peripheral Neuropathies. Exp. Eye Res. 2013, 113, 148–150. [Google Scholar] [CrossRef]
- Zhang, H.; Boyette-Davis, J.A.; Kosturakis, A.K.; Li, Y.; Yoon, S.-Y.; Walters, E.T.; Dougherty, P.M. Induction of Monocyte Chemoattractant Protein-1 (MCP-1) and Its Receptor CCR2 in Primary Sensory Neurons Contributes to Paclitaxel-Induced Peripheral Neuropathy. J. Pain 2013, 14, 1031–1044. [Google Scholar] [CrossRef]
- Siau, C.; Bennett, G.J. Dysregulation of Cellular Calcium Homeostasis in Chemotherapy-Evoked Painful Peripheral Neuropathy. Anesth. Analg. 2006, 102, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, E.; Watkins, S.C.; Gold, M.S. Paclitaxel-Induced Increase in Mitochondrial Volume Mediates Dysregulation of Intracellular Ca2+ in Putative Nociceptive Glabrous Skin Neurons from the Rat. Cell Calcium 2017, 62, 16–28. [Google Scholar] [CrossRef]
- Kidd, J.F.; Pilkington, M.F.; Schell, M.J.; Fogarty, K.E.; Skepper, J.N.; Taylor, C.W.; Thorn, P. Paclitaxel Affects Cytosolic Calcium Signals by Opening the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2002, 277, 6504–6510. [Google Scholar] [CrossRef]
- Mironov, S.L.; Ivannikov, M.V.; Johansson, M. [Ca 2+ ] i Signaling between Mitochondria and Endoplasmic Reticulum in Neurons Is Regulated by Microtubules. J. Biol. Chem. 2005, 280, 715–721. [Google Scholar] [CrossRef]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel Induces Calcium Oscillations via an Inositol 1,4,5-Trisphosphate Receptor and Neuronal Calcium Sensor 1-Dependent Mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal Root Ganglion Neurons Become Hyperexcitable and Increase Expression of Voltage-Gated T-Type Calcium Channels (Cav3.2) in Paclitaxel-Induced Peripheral Neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef]
- Okubo, K.; Takahashi, T.; Sekiguchi, F.; Kanaoka, D.; Matsunami, M.; Ohkubo, T.; Yamazaki, J.; Fukushima, N.; Yoshida, S.; Kawabata, A. Inhibition of T-Type Calcium Channels and Hydrogen Sulfide-Forming Enzyme Reverses Paclitaxel-Evoked Neuropathic Hyperalgesia in Rats. Neuroscience 2011, 188, 148–156. [Google Scholar] [CrossRef]
- Zhang, H.; Dougherty, P.M. Enhanced Excitability of Primary Sensory Neurons and Altered Gene Expression of Neuronal Ion Channels in Dorsal Root Ganglion in Paclitaxel-Induced Peripheral Neuropathy. Anesthesiology 2014, 120, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Chiba, T.; Abe, K.; Makabe, A.; Ikeno, S.; Kawakami, K.; Utsunomiya, I.; Hama, T.; Taguchi, K. Effect of Paclitaxel on Transient Receptor Potential Vanilloid 1 in Rat Dorsal Root Ganglion. Pain 2013, 154, 882–889. [Google Scholar] [CrossRef]
- Materazzi, S.; Fusi, C.; Benemei, S.; Pedretti, P.; Patacchini, R.; Nilius, B.; Prenen, J.; Creminon, C.; Geppetti, P.; Nassini, R. TRPA1 and TRPV4 Mediate Paclitaxel-Induced Peripheral Neuropathy in Mice via a Glutathione-Sensitive Mechanism. Pflugers Arch. 2012, 463, 561–569. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, C.; Wang, Z.J. Proteinase-Activated Receptor 2 Sensitizes Transient Receptor Potential Vanilloid 1, Transient Receptor Potential Vanilloid 4, and Transient Receptor Potential Ankyrin 1 in Paclitaxel-Induced Neuropathic Pain. Neuroscience 2011, 193, 440–451. [Google Scholar] [CrossRef]
- Li, Y.; North, R.Y.; Rhines, L.D.; Tatsui, C.E.; Rao, G.; Edwards, D.D.; Cassidy, R.M.; Harrison, D.S.; Johansson, C.A.; Zhang, H.; et al. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. J. Neurosci. 2018, 38, 1124–1136. [Google Scholar] [CrossRef]
- Aromolaran, K.A.; Goldstein, P.A. Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy; cause and effect? Mol Pain. 2017, 13, 1744806917714693. [Google Scholar] [CrossRef] [PubMed]
- Zaks-Zilberman, M.; Zaks, T.Z.; Vogel, S.N. Induction of proinflammatory and chemokine genes by lipopolysaccharide and paclitaxel (TaxolTM) in murine and human breast cancer cell lines. Cytokine 2001, 15, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Eijkelkamp, N.; Laumet, G.; Hack, C.E.; Li, Y.; Dougherty, P.M.; Heijnen, C.J.; Kavelaars, A. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J. Neurosci. 2016, 36, 11074–11083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yoon, S.-Y.; Zhang, H.; Dougherty, P.M. Evidence That Spinal Astrocytes but Not Microglia Contribute to the Pathogenesis of Paclitaxel-Induced Painful Neuropathy. J. Pain 2012, 13, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Medina, J.; Baulies, A.; Bura, S.A.; Valverde, O. Paclitaxel-Induced Neuropathic Pain Is Age Dependent and Devolves on Glial Response. Eur. J. Pain 2013, 17, 75–85. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; de Carvalho-Barbosa, M.; Kavelaars, A.; Heijnen, C.J.; Albrecht, P.J.; Dougherty, P.M. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J. Pain 2016, 17, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lu, N.; Cui, Y.; Yang, T.; Zhao, Z.-Q.; Xin, W.-J.; Liu, X.-G. Prevention of Paclitaxel-Induced Allodynia by Minocycline: Effect on Loss of Peripheral Nerve Fibers and Infiltration of Macrophages in Rats. Mol. Pain 2010, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Zhang, H.; Kosturakis, A.K.; Jawad, A.B.; Dougherty, P.M. Toll-Like Receptor 4 Signaling Contributes to Paclitaxel-Induced Peripheral Neuropathy. J. Pain 2014, 15, 712–725. [Google Scholar] [CrossRef]
- Lopus, M.; Smiyun, G.; Miller, H.; Oroudjev, E.; Wilson, L.; Jordan, M.A. Mechanism of action of ixabepilone and its interactions with the βIII-tubulin isotype. Cancer Chemother. Pharmacol. 2015, 76, 1013–1024. [Google Scholar] [CrossRef]
- Heigener, D.F.; von Pawel, J.; Eschbach, C.; Brune, A.; Schmittel, A.; Schmelter, T.; Reck, M.; Fischer, J.R. Prospective, multicenter, randomized, independent-group, open-label phase II study to investigate the efficacy and safety of three regimens with two doses of sagopilone as second-line therapy in patients with stage IIIB or IV non-small-cell lung cancer. Lung Cancer 2013, 80, 319–325. [Google Scholar] [CrossRef]
- Vahdat, L.T.; Thomas, E.S.; Roché, H.H.; Hortobagyi, G.N.; Sparano, J.A.; Yelle, L.; Fornier, M.N.; Martín, M.; Bunnell, C.A.; Mukhopadhyay, P.; et al. Ixabepilone-associated peripheral neuropathy: Data from across the phase II and III clinical trials. Support. Care Cancer 2012, 20, 2661–2668. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, G.J.; Carlson, K.; Donovan, D.; Cobham, M.; Chuang, E.; Moore, A.; Cigler, T.; Ward, M.; Lane, M.E.; Ramnarain, A.; et al. Ixabepilone-Induced Mitochondria and Sensory Axon Loss in Breast Cancer Patients. Ann. Clin. Transl. Neurol. 2014, 1, 639–649. [Google Scholar] [CrossRef]
- Raffa, R.B.; Pergolizzi, J.V., Jr. Cancer Chemotherapy–Induced Neuropathic Pain. The Underlying Peripheral Neuropathy. In Chemotherapy—Induced Neuropathic Pain; Raffa, R.B., Langford, R., Pergolizzi, J.V., Jr., Porecca, F., Tallarida, R.J., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2013; pp. 113–135. [Google Scholar]
- Boyette–Davis, J.A.; Hou, S.; Abdi, S.; Dougherty, P.M. An updated understanding of the mechanisms involved in chemotherapy-induced neuropathy. Pain Manag. 2018, 8, 363–375. [Google Scholar] [CrossRef]
- Topp, K.S.; Tanner, K.D.; Levine, J.D. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine induced painful peripheral neuropathy in the rat. J. Comp. Neurol. 2000, 424, 563–576. [Google Scholar] [CrossRef]
- Casey, E.B.; Jellife, A.M.; Le Quesne, P.M.; Millett, Y.L. Vincristine neuropathy. Clinical and electrophysiological observations. Brain 1973, 96, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Sandler, S.G.; Tobin, W.; Henderson, E.S. Vincristine-induced neuropathy. A clinical study of fifty leukemic patients. Neurology 1969, 19, 367–374. [Google Scholar] [CrossRef]
- Graf, W.D.; Chance, P.F.; Lensch, M.W.; Eng, L.J.; Lipe, H.P.; Bird, T.D. Severe vincristine neuropathy in Charcot-Marie-Tooth disease type 1A. Cancer 1996, 77, 1356–1362. [Google Scholar] [CrossRef]
- Nakamura, T.; Hashiguchi, A.; Suzuki, S.; Uozumi, K.; Tokunaga, S.; Takashima, H. Vincristine exacerbates asymptomatic Charcot–Marie–Tooth disease with a novel EGR2 mutation. Neurogenetics 2012, 13, 77–82. [Google Scholar] [CrossRef]
- Diouf, B.; Crews, K.R.; Lew, G.; Pei, D.; Cheng, C.; Bao, J.; Zheng, J.J.; Yang, W.; Fan, Y.; Wheeler, H.E.; et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 2015, 313, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Barzegar–Fallah, A.; Alimoradi, H.; Mehrzadi, S.; Barzegar–Fallah, N.; Zendedel, A.; Abbasi, A.; Dehpour, A.R. The neuroprotective effect of tropisetron on vincristine-induced neurotoxicity. Neurotoxicology 2014, 41, 1–8. [Google Scholar] [CrossRef]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; Di Antonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef] [Green Version]
- Saifee, T.A.; Elliott, K.J.; Rabin, N.; Yong, K.L.; D’Sa, S.; Brandner, S.; Lunn, M.P.; Blake, J.; Reilly, M.M. Bortezomib-induced inflammatory neuropathy. J. Peripher. Nerv. Syst. 2010, 15, 366–368. [Google Scholar] [CrossRef]
- Thawani, S.P.; Tanji, K.; De Sousa, E.A.; Weimer, L.H.; Brannagan, T.H., 3rd. Bortezomib-associated demyelinating neuropathy—Clinical and pathologic features. J. Clin. Neuromuscul. Dis. 2015, 16, 202–209. [Google Scholar] [CrossRef]
- Peng, L.; Ye, X.; Zhou, Y.; Zhang, J.; Zhao, Q. Meta-analysis of incidence and risk of peripheral neuropathy associated with intravenous bortezomib. Support. Care Cancer 2015, 23, 2813–2824. [Google Scholar] [CrossRef]
- Farquhar-Smith, P. Chemotherapy-induced neuropathic pain. Curr. Opin. Support. Palliat. Care 2011, 5, 1–7. [Google Scholar] [CrossRef]
- Hu, B.; Zhou, Q.; Wu, T.; Zhuang, L.; Yi, L.; Cao, J.; Yang, X.; Wang, J. Efficacy and safety of subcutaneous versus intravenous bortezomib in multiple myeloma: A meta-analysis. Int. J. Clin. Pharmacol. Ther. 2017, 55, 329–338. [Google Scholar] [CrossRef]
- Liu, H.; Xu, R.; Huang, H. Peripheral neuropathy outcomes and efficacy of subcutaneous bortezomib when combined with thalidomide and dexamethasone in the treatment of multiple myeloma. Exp. Ther. Med. 2016, 12, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Doyle, T.; Kress, M.; Nicol, G. Therapeutic targeting of the ceramide-to-sphingosine 1-phosphate pathway in pain. Trends Pharmacol. Sci. 2013, 34, 110–118. [Google Scholar] [CrossRef]
- Dawkins, J.L.; Hulme, D.J.; Brahmbhatt, S.B.; Auer-Grumbach, M.; Nicholson, G.A. Mutations in SPT LC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001, 27, 309–312. [Google Scholar] [CrossRef]
- Stockstill, K.; Doyle, T.M.; Yan, X.; Chen, Z.; Janes, K.; Little, J.W.; Braden, K.; Lauro, F.; Giancotti, L.A.; Harada, C.M.; et al. Dysregulation of sphingolipid Metabolism contributes to bortezomib-inducedneuropathicpain. J. Exp. Med. 2018, 215, 1301–1313. [Google Scholar] [CrossRef]
- Emery, E.C.; Wood, J.N. Gaining on Pain. N. Engl. J. Med. 2018, 379, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Broyl, A.; Corthals, S.L.; Jongen, J.L.; van der Holt, B.; Kuiper, R.; de Knegt, Y.; van Duin, M.; el Jarari, L.; Bertsch, U.; Lokhorst, H.M.; et al. Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: A prospective analysis of data from the HOVON-65/GMMG-HD4 trial. Lancet Oncol. 2010, 11, 1057–1065. [Google Scholar] [CrossRef]
- Magrangeas, F.; Kuiper, R.; Avet-Loiseau, H.; Gouraud, W.; Guerin-Charbonnel, C.; Ferrer, L.; Aussem, A.; Elghazel, H.; Suhard, J.; Sakissian, H.; et al. A genome-wide association study identifies a novel locus for bortezomib-induced peripheral neuropathy in European patients with multiple myeloma. Clin. Cancer Res. 2016, 22, 4350–4355. [Google Scholar] [CrossRef]
- Ale, A.; Bruna, J.; Calls, A.; Karamita, M.; Haralambous, S.; Probert, L.; NaVarro, X.; Udina, E. Inhibition of the neuronal NFkappaB pathway attenuates bortezomib induced neuropathy in a mouse model. Neurotoxicology 2016, 55, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Udd, K.A.; Vidisheva, A.; Swift, R.A.; Spektor, T.M.; Bravin, E.; Ibrahim, E.; Treisman, J.; Masri, M.; Berenson, J.R. Low serum vitamin D occurs commonly among multiple myeloma patients treated with bortezomib and/or thalidomide and is associated with severe neuropathy. Support. Care Cancer 2016, 24, 3105–3110. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xiao, W.H.; Bennett, G.J. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp. Neurol. 2012, 238, 225–234. [Google Scholar] [CrossRef]
- Hou, S.; Huh, B.; Kim, H.K.; Kim, K.H.; Abdi, S. Treatment of chemotherapy-induced peripheral neuropathy: systematic review and recommendations. Pain Physician. 2018, 21, 571–592. [Google Scholar] [PubMed]
- Chua, K.C.; Kroetz, D.L. Genetic Advances Uncover Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Clin. Pharmacol. Ther. 2017, 101, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Oveissi, V.; Ram, M.; Bahramsoltani, R.; Ebrahimi, F.; Rahimi, R.; Naseri, R.; Belwal, T.; Devkota, H.P.; Abbasabadi, Z.; Farzaei, M.H. Medicinal plants and their isolated phytochemicals for the management of chemotherapy-induced neuropathy: Therapeutic targets and clinical perspective. Daru 2019. [Google Scholar] [CrossRef]
- Wu, B.Y.; Liu, C.T.; Su, Y.L.; Chen, S.Y.; Chen, Y.H.; Tsai, M.Y. A review of complementary therapies with medicinal plants for chemotherapy-induced peripheral neuropathy. Complement. Ther. Med. 2019, 42, 226–232. [Google Scholar] [CrossRef]
- Masocha, W.; Thomas, A. Indomethacin plus minocycline coadministration relieves chemotherapy and antiretroviral drug-induced neuropathic pain in a cannabinoid receptors-dependent manner. J. Pharmacol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wrzosek, A.; Mika, J.; Wordliczek, J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2019, 20, 1451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061451
Zajączkowska R, Kocot-Kępska M, Leppert W, Wrzosek A, Mika J, Wordliczek J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. International Journal of Molecular Sciences. 2019; 20(6):1451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061451
Chicago/Turabian StyleZajączkowska, Renata, Magdalena Kocot-Kępska, Wojciech Leppert, Anna Wrzosek, Joanna Mika, and Jerzy Wordliczek. 2019. "Mechanisms of Chemotherapy-Induced Peripheral Neuropathy" International Journal of Molecular Sciences 20, no. 6: 1451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061451