A Novel Network-Based Computational Model for Prediction of Potential LncRNA–Disease Association


Abstract
:1. Introduction
2. Results
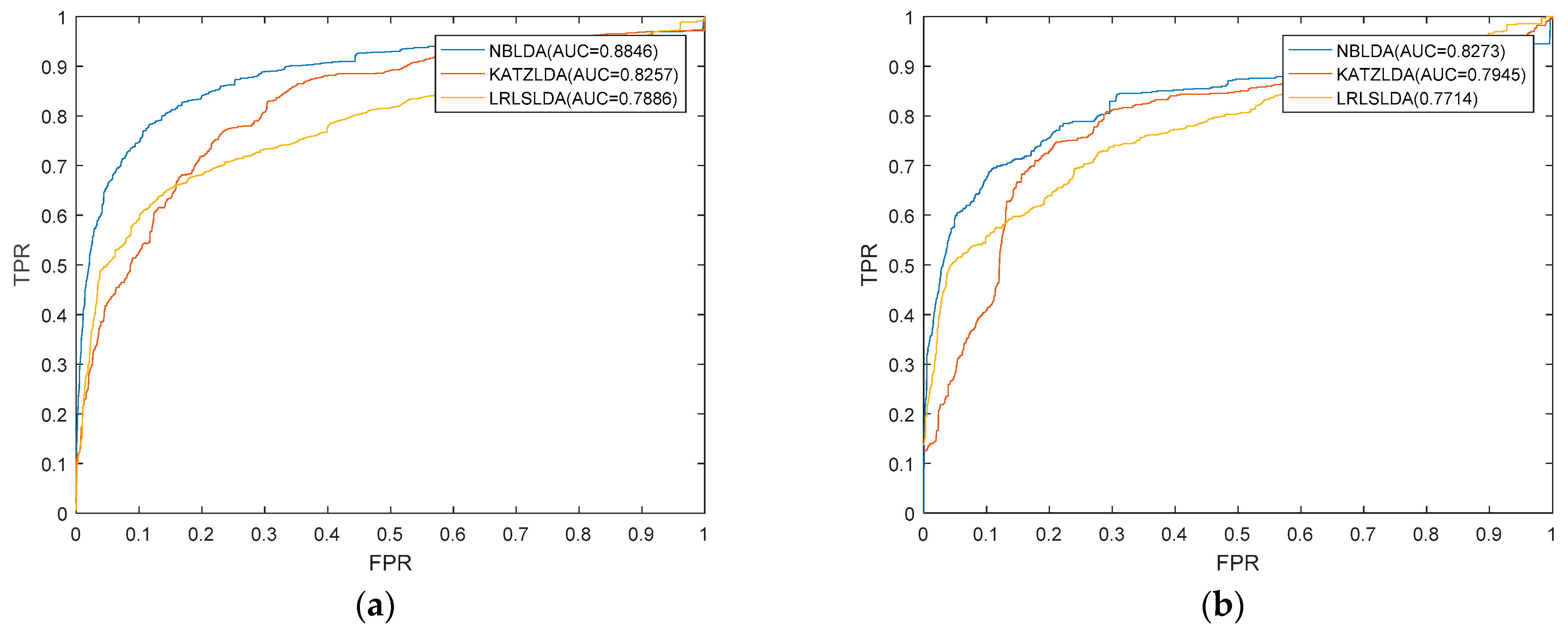
2.1. Performance Evaluation
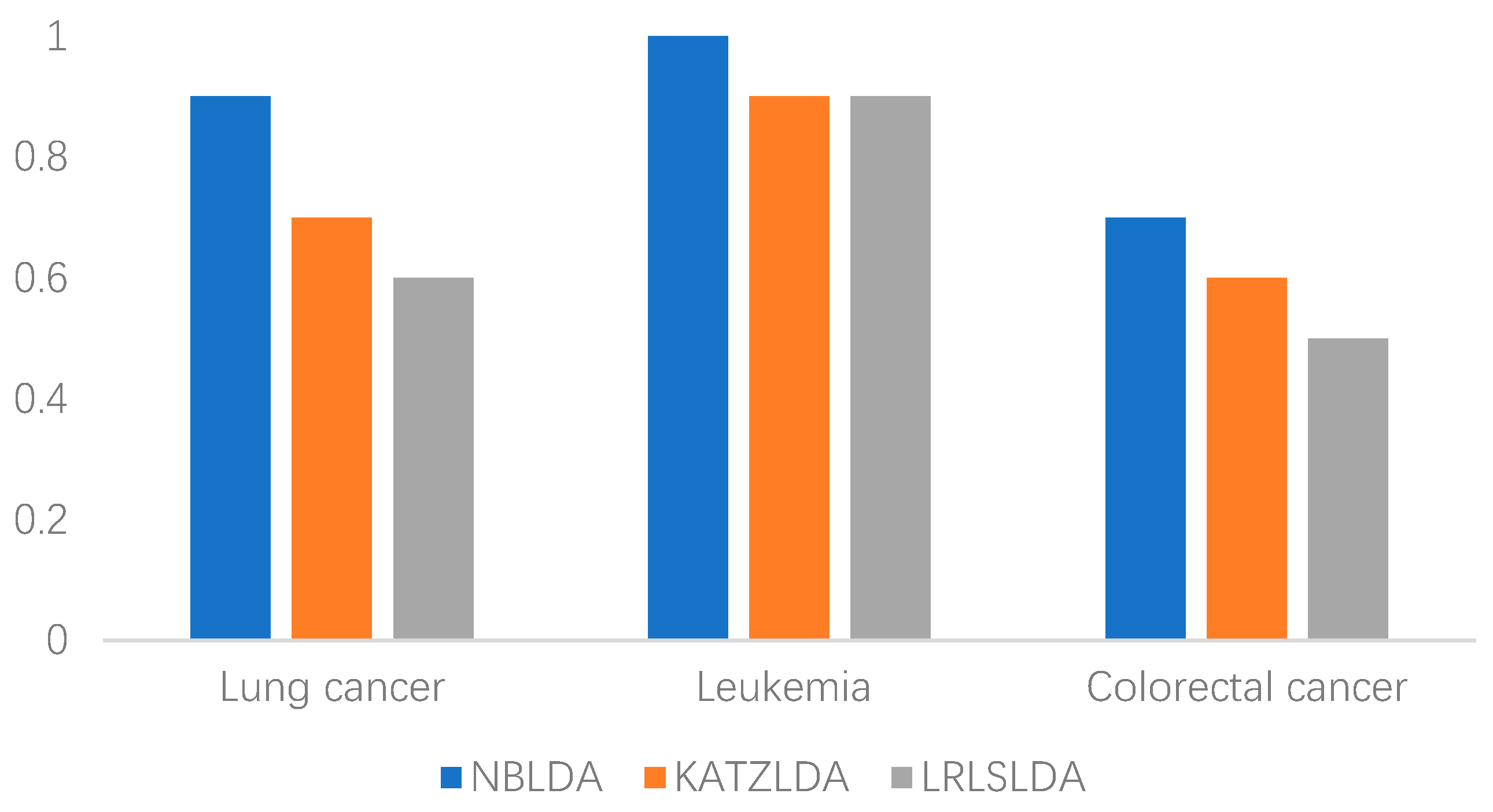
2.2. Case Studies
3. Discussion
4. Materials and Methods
4.1. Human lncRNA–Disease Associations
4.2. Gaussian Interaction Profile Kernel Similarity for lncRNAs and Diseases
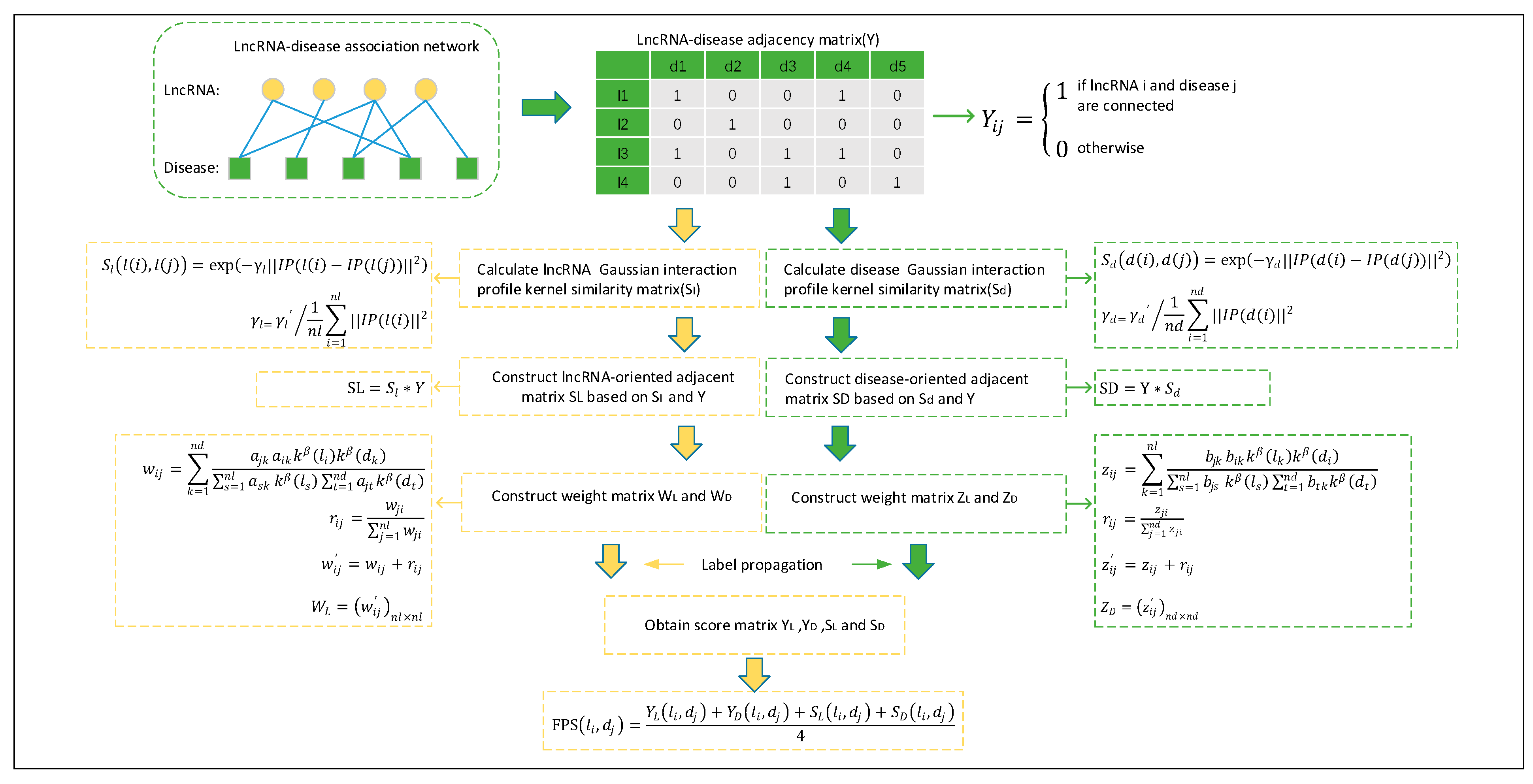
4.3. Prediction Model of NBLDA
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LOOCV | leave-one-out cross validation |
| FPS | final lncRNA–disease association score matrix |
| ROC | receiver operating characteristic |
| TPR | true positive rate |
| FPR | false positive rate |
| AUC | area under the ROC curve |
References
- Lv, J.; Huang, Z.; Liu, H.; Liu, H.; Cui, W.; Li, B.; He, H.; Guo, J.; Liu, Q.; Zhang, Y.; et al. Identification and characterization of long intergenic non-coding RNAs related to mouse liver development. Mol. Genet. Genom. 2014, 289, 1225–1235. [Google Scholar] [CrossRef]
- Yanofsky, C. Establishing the Triplet Nature of the Genetic Code. Cell 2007, 128, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Core, L.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Guttman, M.; Russell, P.; Ingolia, N.T.; Weissman, J.S.; Lander, E.S. Ribosome Profiling Provides Evidence that Large Noncoding RNAs Do Not Encode Proteins. Cell 2013, 154, 240–251. [Google Scholar] [CrossRef]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Wang, H.M.; Lu, J.H.; Chen, W.Y.; Gu, A.Q. Upregulated lncRNA-UCA1 contributes to progression of lung cancer and is closely related to clinical diagnosis as a predictive biomarker in plasma. Int. J. Clin. Exp. Med. 2015, 8, 11824–11830. [Google Scholar] [PubMed]
- Li, H.; An, J.; Wu, M.; Zheng, Q.; Gui, X.; Li, T.; Pu, H.; Lu, D. LncRNA HOTAIR promotes human liver cancer stem cell malignant growth through downregulation of SETD2. Oncotarget 2015, 6, 27847–27864. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar] [CrossRef]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: A manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016, 44, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Liao, B.; Li, X.; Cai, L.; Li, Z.; Li, K.; Yang, J. Global network random walk for predicting potential human lncRNA-disease associations. Sci. Rep. 2017, 7, 12442. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef]
- Yu, J.; Ping, P.; Wang, L.; Kuang, L.; Li, X.; Wu, Z. A Novel Probability Model for LncRNA–Disease Association Prediction Based on the Naïve Bayesian Classifier. Genes 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, H.; Wang, Z.; Zhang, C.; Liu, L.; Wang, L.; He, W.; Hao, D.; Liu, S.; Zhou, M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014, 10, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, L.; Li, J.; Yang, L.G.; Sun, Y.; Li, Z.; He, S.; Feng, F.; Li, H.; Li, Y. Global Prioritizing Disease Candidate lncRNAs via a Multi-level Composite Network. Sci. Rep. 2017, 7, 39516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 2015, 5, 13186. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Kuang, L.; Wang, L.; Ping, P.; Xuan, Z.; Pei, T.; Wu, Z. Prediction of microRNA-disease associations based on distance correlation set. BMC Bioinform. 2018, 19, 141. [Google Scholar] [CrossRef]
- Chen, X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 2014, 5, 16840. [Google Scholar] [CrossRef]
- Liu, R.R.; Liu, J.G.; Jia, C.X.; Wang, B.H. Personal recommendation via unequal resource allocation on bipartite networks. Phys. A Stat. Mech. Its Appl. 2010, 389, 3282–3289. [Google Scholar] [CrossRef]
- Spiess, P.E.; Dhillon, J.; Baumgarten, A.S.; Johnstone, P.A.; Giuliano, A.R. Pathophysiological basis of human papillomavirus in penile cancer: Key to prevention and delivery of more effective therapies. CA Cancer J. Clin. 2016, 66, 481–495. [Google Scholar] [CrossRef]
- Ruprecht, B.; Zaal, E.A.; Zecha, J.; Wu, W.; Berkers, C.R.; Kuster, B.; Lemeer, S. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res. 2017, 77, 1842–1853. [Google Scholar] [CrossRef]
- Barton, M.K. Local consolidative therapy may be beneficial in patients with oligometastatic non-small cell lung cancer. CA Cancer J. Clin. 2017, 67, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, R.T.; Murray, T.; Bolden, S.; Wingo, P.A. Cancer statistics, 2000. CA Cancer J. Clin. 2000, 50, 7–33. [Google Scholar] [CrossRef] [Green Version]
- White, N.M.; Cabanski, C.R.; Silva-Fisher, J.M.; Dang, H.X.; Govindan, R.; Maher, C.A. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014, 15, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lee, W.; Jiang, Z.; Jhunjhunwala, S.; Haverty, P.M.; Gnad, F.; Guan, Y.; Gilbert, H.N.; Stinson, J.; Klijn, C.; et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012, 22, 2315–2327. [Google Scholar] [CrossRef] [Green Version]
- Beasley, M.B.; Brambilla, E.; Travis, W.D. The 2004 World Health Organization classification of lung tumors. Semin. Roentgenol. 2005, 40, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. The Emergence of lncRNAs in Cancer Biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Yang, Y.R.; Zang, S.Z.; Zhong, C.L.; Li, Y.X.; Zhao, S.S.; Feng, X.J. Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6929–6935. [Google Scholar] [PubMed]
- Sun, C.; Li, S.; Zhang, F.; Xi, Y.; Wang, L.; Bi, Y.; Li, D. Long non-coding RNA NEAT1 promotes non-small cell lung cancer progression through regulation of miR-377-3p-E2F3 pathway. Oncotarget 2016, 7, 51784–51814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Ma, F.; Huang, W.; Fang, S.; Li, M.; Wei, T.; Guo, L. Long non-coding RNA TUG1 is involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2. Mol. Cancer 2017, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Franca, R.; Favretto, D.; Granzotto, M.; Decorti, G.; Rabusin, M.; Stocco, G. Epratuzumab and Blinatumomab as Therapeutic Antibodies for Treatment of Pediatric Acute Lymphoblastic Leukemia: Current Status and Future Perspectives. Curr. Med. Chem. 2017, 24, 1050–1065. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, X.; Zhang, X.; Xu, Y. The progress and current status of immunotherapy in acute myeloid leukemia. Ann. Hematol. 2017, 96, 1965–1982. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.F.; Jia, H.Z.; Zhang, Z.Z.; Zhao, X.S.; Zou, Y.F.; Zhang, W.; Wan, J.; Chen, X.F. LncRNA H19 regulates ID2 expression through competitive binding to hsa-miR-19a/b in acute myelocytic leukemia. Mol. Med. Rep. 2017, 16, 3687–3693. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, J.; Kaviani Gebelli, S.; Atashi, A. Evaluation of MALAT1 gene expression in AML and ALL cell lines. Koomesh 2015, 17, 179–186. [Google Scholar]
- Wu, S.; Zheng, C.; Chen, S.; Cai, X.; Shi, Y.; Lin, B.; Chen, Y. Overexpression of long non?coding RNA HOTAIR predicts a poor prognosis in patients with acute myeloid leukemia. Oncol. Lett. 2015, 10, 2410–2414. [Google Scholar] [CrossRef]
- Sahin, I.H.; Garrett, C.R. Current State-of-the-Science Adjuvant and Neoadjuvant Therapy in Surgically Resected Colorectal Cancer; IntechOpen Limited: London, UK, 2014. [Google Scholar]
- Bond, J.H. Colorectal cancer update. Prevention, screening, treatment, and surveillance for high-risk groups. Med. Clin. N. Am. 2000, 84, 1163–1182. [Google Scholar] [CrossRef]
- Xin, Y.; Li, Z.; Zheng, H.; Chan, M.T.V.; Ka, K.; Wu, W. CCAT2: A novel oncogenic long non-coding RNA in human cancers. Cell Prolif. 2017, 50, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Shaker, O.G.; Senousy, M.A.; Elbaz, E.M. Association of rs6983267 at 8q24, HULC rs7763881 polymorphisms and serum lncRNAs CCAT2 and HULC with colorectal cancer in Egyptian patients. Sci. Rep. 2017, 7, 16246. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; He, P.; Shao, T.; Li, Y.; Li, J.; Zhang, Y. Long non-coding RNA XIST functions as an oncogene in human colorectal cancer by targeting miR-132-3p. J. Buon Off. J. Balk. Union Oncol. 2017, 22, 696–703. [Google Scholar]
- Gu, L.; Lu, L.; Zhou, D.; Liu, Z. Long Noncoding RNA BCYRN1 Promotes the Proliferation of Colorectal Cancer Cells via Up-Regulating NPR3 Expression. Cell. Physiol. Biochem. 2018, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An Analysis of Human MicroRNA and Disease Associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Tian, H.; Zhang, P.; Hu, Z.; Zhou, T. Personalized recommendation based on unbiased consistence. EPL 2015, 111, 48007. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | LncRNA | Evidence (PMID) | Rank |
|---|---|---|---|
| Lung cancer | PVT1 | 26493997,28731781,28972861,27904703,29133127 | 1 |
| Lung cancer | NEAT1 | 25818739,29152741,28295289,28615056,29095526 | 2 |
| Lung cancer | TUG1 | 28069000,24853421,29277771,28121347,27485439 | 3 |
| Lung cancer | XIST | 29130102,29339211,26339353,29337100,28248928 | 4 |
| Lung cancer | HULC | 30575912 | 5 |
| Lung cancer | LINC-ROR | 28459375,28516515,29028092 | 6 |
| Lung cancer | PANDAR | 28121347,25719249 | 7 |
| Lung cancer | MIAT | 29487526,28843520,29228680,29795987,27981551 | 8 |
| Lung cancer | HNF1A-AS1 | 27981551,29289833 | 9 |
| Leukemia | H19 | 15645136,29703210,24685695,28765931,29643943 | 1 |
| Leukemia | MALAT1 | 28713913 | 2 |
| Leukemia | HOTAIR | 27748863,26622861,27875938,25979172,26261618 | 3 |
| Leukemia | MEG3 | 28407691,28190319,19595458,14602737,29029424 | 4 |
| Leukemia | PVT1 | 29510227,26545364 | 5 |
| Leukemia | GAS5 | 27951730 | 6 |
| Leukemia | UCA1 | 27854515,29762824,26053097,29663500 | 7 |
| Leukemia | TUG1 | 29654398 | 8 |
| Leukemia | XIST | 7981627 | 9 |
| Leukemia | SNHG5 | 28861326,29917184 | 10 |
| Colorectal cancer | CCAT2 | 29181105,27875818,28838211,26853146,23796952 | 1 |
| Colorectal cancer | XIST | 29495975,29137332,17143621,28730777,29484395 | 2 |
| Colorectal cancer | BCYRN1 | 30114690 | 3 |
| Colorectal cancer | HNF1A-AS1 | 28791380,29145164 | 4 |
| Colorectal cancer | MIAT | 29686537 | 5 |
| Colorectal cancer | ATB | 25750289 | 6 |
| Colorectal cancer | TUSC7 | 27683121,28214867,23680400,28979678 | 10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Feng, X.; Zhao, H.; Xuan, Z.; Wang, L. A Novel Network-Based Computational Model for Prediction of Potential LncRNA–Disease Association. Int. J. Mol. Sci. 2019, 20, 1549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071549
Liu Y, Feng X, Zhao H, Xuan Z, Wang L. A Novel Network-Based Computational Model for Prediction of Potential LncRNA–Disease Association. International Journal of Molecular Sciences. 2019; 20(7):1549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071549
Chicago/Turabian StyleLiu, Yang, Xiang Feng, Haochen Zhao, Zhanwei Xuan, and Lei Wang. 2019. "A Novel Network-Based Computational Model for Prediction of Potential LncRNA–Disease Association" International Journal of Molecular Sciences 20, no. 7: 1549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071549