Deep Transcriptome Analysis Reveals Reactive Oxygen Species (ROS) Network Evolution, Response to Abiotic Stress, and Regulation of Fiber Development in Cotton

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
2.1. ROS Network Genes Identification in Gossypium Species
2.2. Chromosome Mapping of the ROS Network Gene Family
2.3. Phylogenetic Tree, Gene Duplication, and Synteny Analysis of the ROS Network Gene Family
2.4. Orthologous ROS Gene Analysis and Evolution Prediction through Synonymous (Ks) and Non-Synonymous (Ka) Rate of Substituion
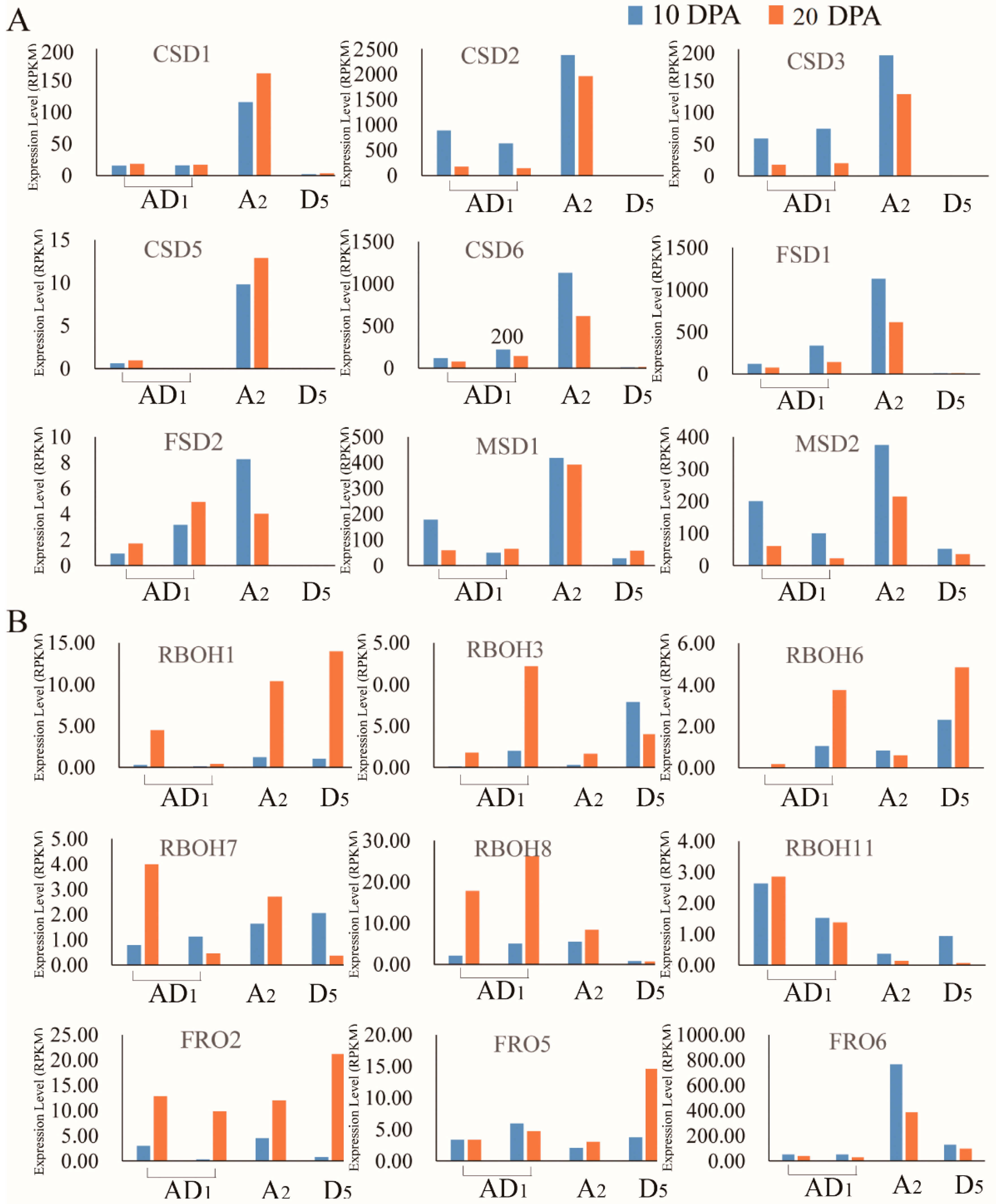
2.5. Analysis of the Expression Levels of Homeolog and Ortholog Genes ROS Network Genes at 10 DPA and 20 DPA of Cotton Fiber Development
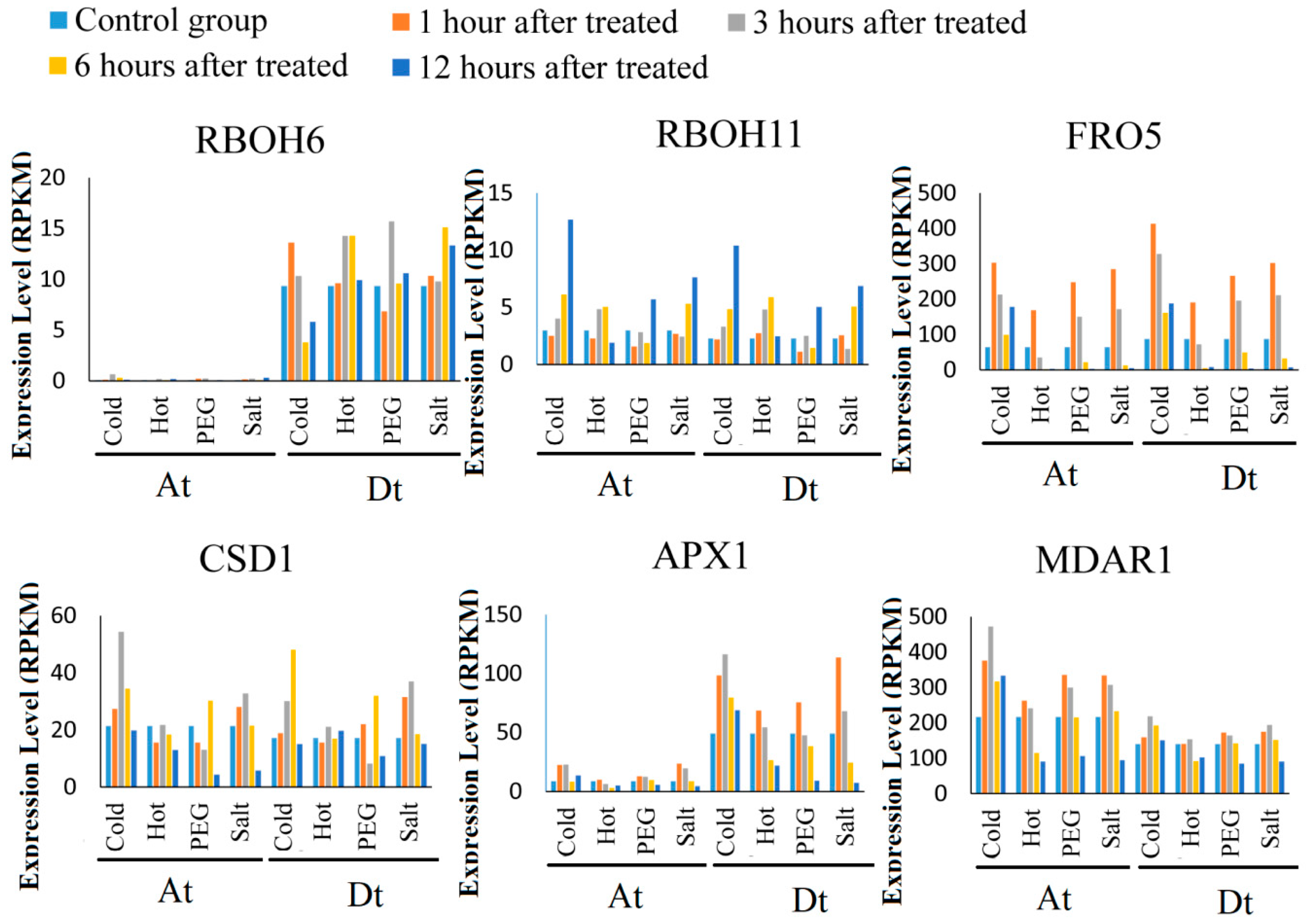
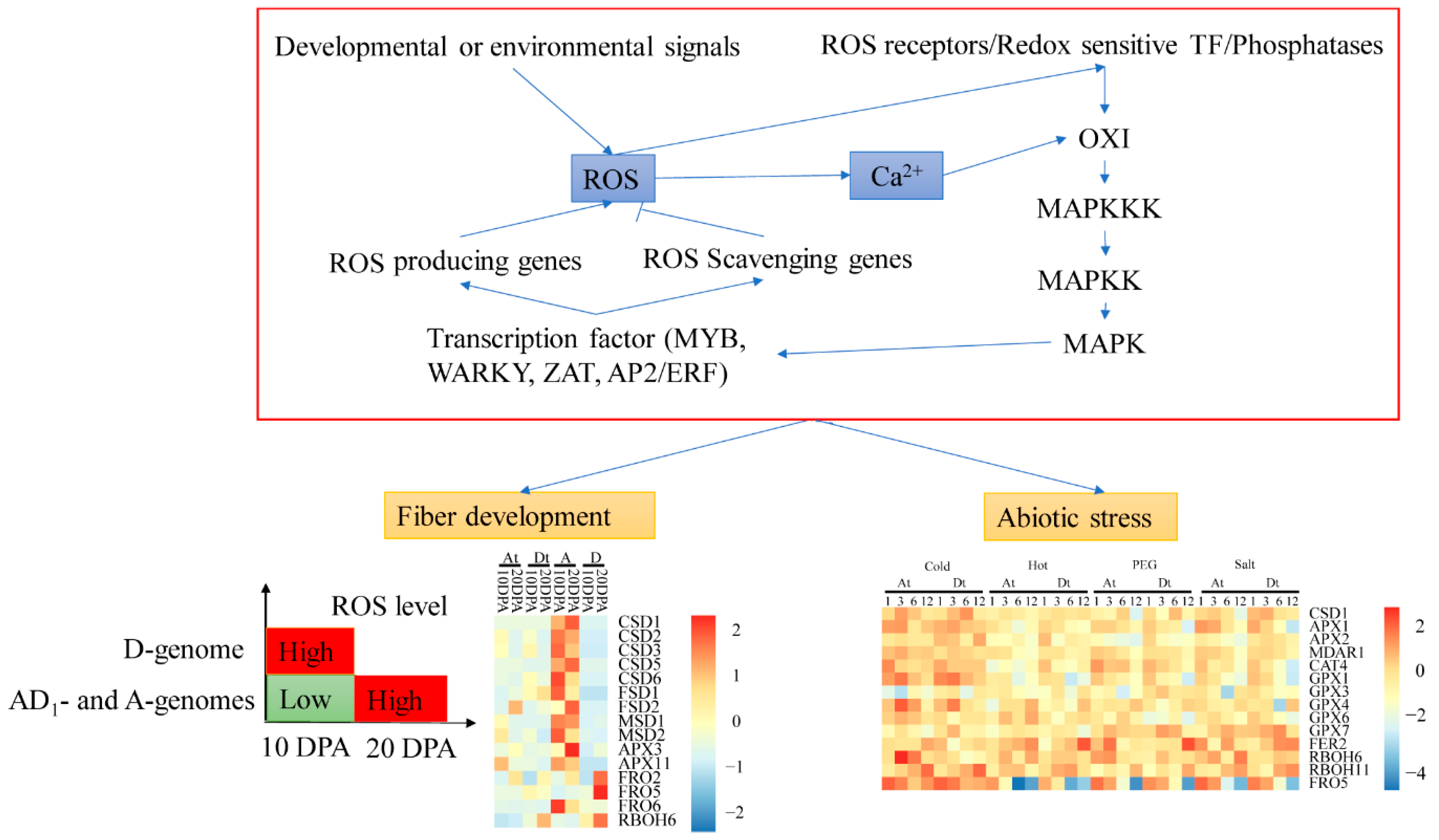
2.6. ROS Network Genes Response to Abiotic Stress
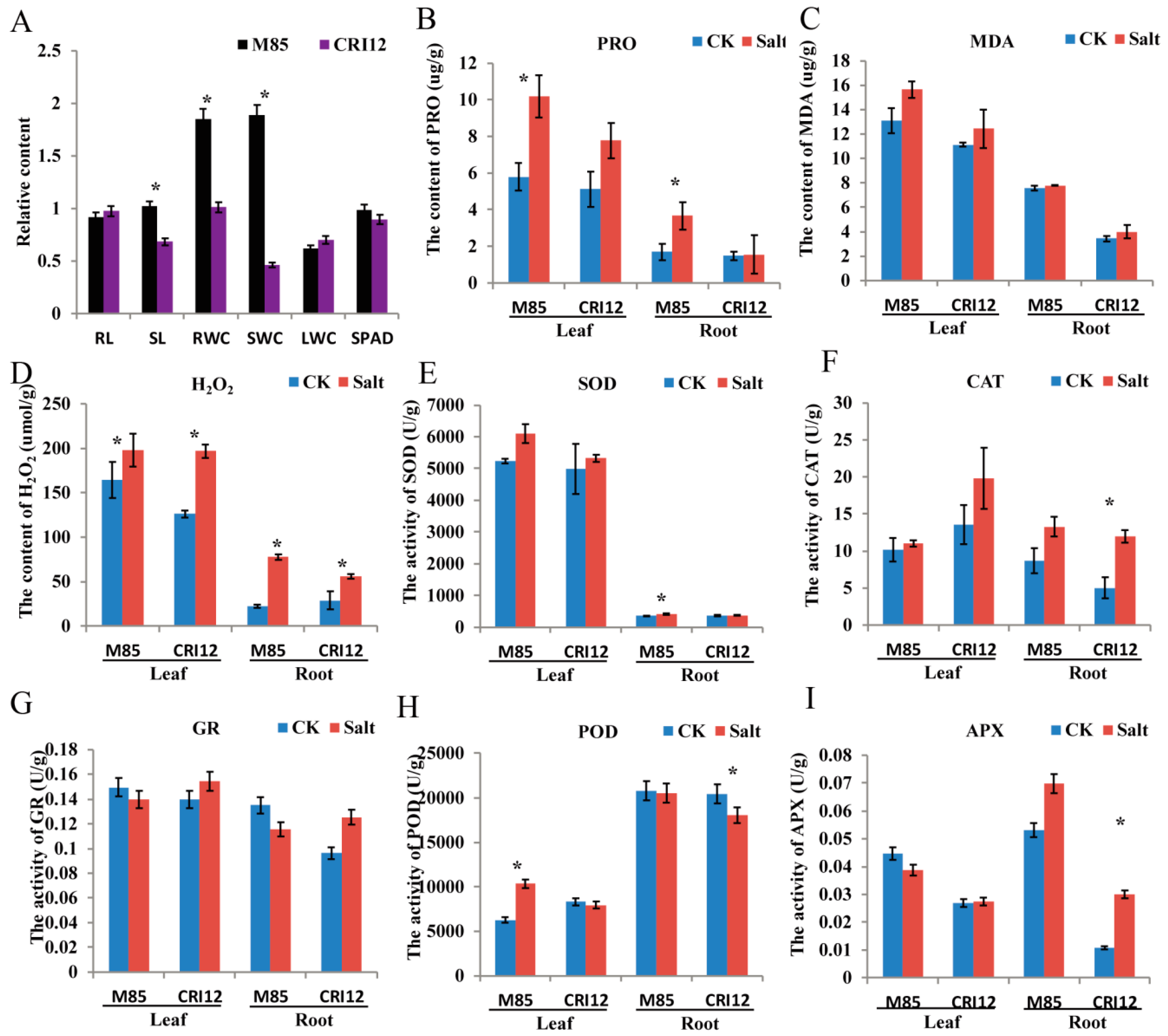
2.7. Analysis of Morphological and Physiological Changes under Salt Stress
2.8. RNA-Seq Analysis and ROS Network Response to Alkali-Salt Stress
3. Discussion
4. Materials and Methods
4.1. Identification of the ROS Genes in Cotton
4.2. Phylogenetic Tree, Orthologous Gene Pairs and Values of Ka, Ks and Ka/Ks
4.3. Chromosomal Locations, Conserved Synteny Blocks and Gene Duplication
4.4. Plant Materials and Salt Stress Treatment
4.5. Measurement of Morphological, Physiological and Biochemical Traits
4.6. RNA Extraction, cDNA Library Construction, and RNA-Seq
4.7. Expression Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| Ka | Synonymous |
| MDAR | Monodehydroascorbate reductase |
| Ks | Nonsynonymous |
| DHAR | Dehydroascorbate reductase |
| MYA | Million years ago |
| GR | Glutathione reductase |
| RBOH | Respiratory burst oxidase homologue |
| CAT | Catalase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| GPX | Glutathione peroxidase |
| APX | Ascorbate peroxidase |
| FER | Ferritin |
| AO | Ascorbate oxidase |
| FRO | NADPH-like oxidase |
| AOX | Ubiquinol oxidase |
| PrxR | Peroxiredoxin |
| GLR | Glutaredoxin |
| Trx | Thioredoxin reductase |
| PER | Peroxidase |
| SNP | Single nucleotide polymorphism |
| DPA | Days post anthesis |
| GWAS | Genome-wide association study. |
References
- Wendel, J.F.; Flagel, L.E.; Adams, K.L. Jeans, genes, and genomes: Cotton as a model for studying polyploidy. In Polyploidy and Genome Evolution; Springer: Berlin/Heidelberg, Germany, 2012; pp. 181–207. ISBN 9783642314421. [Google Scholar]
- Bradshaw, J.E. Genome Evolution and Polyploidy. In Plant Breeding: Past, Present and Future; Springer: Dordrecht, The Netherlands, 2016; pp. 233–269. [Google Scholar]
- Flagel, L.E.; Wendel, J.F.; Udall, J.A. Duplicate gene evolution, homoeologous recombination, and transcriptome characterization in allopolyploid cotton. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Shan, X.; Liu, Z.; Dong, Z.; Wang, Y.; Chen, Y.; Lin, X.; Long, L.; Han, F.; Dong, Y.; Liu, B. Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol. Biol. Evol. 2005, 22, 976–990. [Google Scholar] [CrossRef]
- Dos Soares, L.A.A.; Fernandes, P.D.; de Lima, G.S.; Brito, M.E.B.; do Nascimento, R.; Arriel, N.H.C. Physiology and production of naturally-colored cotton under irrigation strategies using salinized water. Pesqui. Agropecu. Bras. 2018, 53, 746–755. [Google Scholar] [CrossRef]
- Xu, Z.; Yu, J.; Kohel, R.J.; Percy, R.G.; Beavis, W.D.; Main, D.; Yu, J.Z. Distribution and evolution of cotton fiber development genes in the fibreless Gossypium raimondii genome. Genomics 2015, 106, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux, P.; Karpinski, S. Signal transduction in response to excess light: Getting out of the chloroplast. Curr. Opin. Plant Biol. 2002, 5, 43–48. [Google Scholar] [CrossRef]
- Ba, Q.S.; Liu, H.Z.; Wang, J.W.; Che, H.X.; Niu, N.; Wang, J.S.; Ma, S.C.; Zhang, G.S. Relationship between metabolism of reactive oxygen species and chemically induced male sterility in wheat (Triticum aestivum L.). Can. J. Plant Sci. 2013, 93, 675–681. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Perata, P.; Pucciariello, C.; Novi, G.; Parlanti, S.; Banti, V. Reactive Oxygen Species-Driven Transcription in Arabidopsis under Oxygen Deprivation. Plant Physiol. 2012, 159, 184–196. [Google Scholar] [CrossRef]
- Leymarie, J.; Vitkauskaité, G.; Hoang, H.H.; Gendreau, E.; Chazoule, V.; Meimoun, P.; Corbineau, F.; El-Maarouf-Bouteau, H.; Bailly, C. Role of reactive oxygen species in the regulation of arabidopsis seed dormancy. Plant Cell Physiol. 2012, 53, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Guo, C.; Wu, H.; Chen, C. Arginine decarboxylase ADC2 enhances salt tolerance through increasing ROS scavenging enzyme activity in Arabidopsis thaliana. Plant Growth Regul. 2017, 83, 253–263. [Google Scholar] [CrossRef]
- Waszczak, C.; Carmody, M.; Kangasjärvi, J. Reactive Oxygen Species in Plant Signaling. Annu. Rev. Plant Biol. 2018, 69. [Google Scholar] [CrossRef] [PubMed]
- Hovav, R.; Udall, J.A.; Chaudhary, B.; Hovav, E.; Flagel, L.; Hu, G.; Wendel, J.F. The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Tang, W.; Tu, L.; Yang, X.; Tan, J.; Deng, F.; Hao, J.; Guo, K.; Lindsey, K.; Zhang, X. The calcium sensor GhCaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol. 2014, 202, 509–520. [Google Scholar] [CrossRef]
- Damiano, S.; Fusco, R.; Morano, A.; de Mizio, M.; Paternò, R.; de Rosa, A.; Spinelli, R.; Amente, S.; Frunzio, R.; Mondola, P.; et al. Reactive oxygen species regulate the levels of dual oxidase (duox1-2) in human neuroblastoma cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Kwak, H.J.; Liu, P.; Bajrami, B.; Xu, Y.; Park, S.Y.; Nombela-Arrieta, C.; Mondal, S.; Sun, Y.; Zhu, H.; Chai, L.; et al. Myeloid Cell-Derived Reactive Oxygen Species Externally Regulate the Proliferation of Myeloid Progenitors in Emergency Granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef]
- Guo, K.; Du, X.; Tu, L.; Tang, W.; Wang, P.; Wang, M.; Liu, Z.; Zhang, X. Fibre elongation requires normal redox homeostasis modulated by cytosolic ascorbate peroxidase in cotton (Gossypium hirsutum). J. Exp. Bot. 2016, 67, 3289–3301. [Google Scholar] [CrossRef]
- Li, R.; Xin, S.; Tao, C.; Jin, X.; Li, H. Cotton ascorbate oxidase promotes cell growth in cultured tobacco bright yellow-2 cells through generation of apoplast oxidation. Int. J. Mol. Sci. 2017, 18, 1346. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, G.Q.; Zou, D.; Yan, J.Q.; Li, Y.; Hu, S.; Li, X.B. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2018, 217, 625–640. [Google Scholar] [CrossRef]
- Watkins, J.M.; Chapman, J.M.; Muday, G.K. Abscisic Acid-Induced Reactive Oxygen Species Are Modulated by Flavonols to Control Stomata Aperture. Plant Physiol. 2017, 175, 1807–1825. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Lu, X.; Shu, N.; Wang, X.; Yang, X.; Wang, S.; Wang, J.; Guo, L.; Wang, D.; et al. Transcriptome Analysis of Gossypium hirsutum L. Reveals Different Mechanisms among NaCl, NaOH and Na2CO3 Stress Tolerance. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Sugimoto, M.; Oono, Y.; Gusev, O.; Matsumoto, T.; Yazawa, T.; Levinskikh, M.A.; Sychev, V.N.; Bingham, G.E.; Wheeler, R.; Hummerick, M. Genome-wide expression analysis of reactive oxygen species gene network in Mizuna plants grown in long-term spaceflight. BMC Plant Biol. 2014, 14. [Google Scholar] [CrossRef]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.; Wang, X.; Cai, X.; Zhou, Z.; Zhang, Z.; Salih, H.; Wang, K.; et al. Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet. 2018, 19. [Google Scholar] [CrossRef]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef]
- Soltis, P.S.; Burleigh, J.G.; Chanderbali, A.S.; Yoo, M.-J.; Soltis, D.E. Gene and Genome Duplications in Plants. In Evolution after Gene Duplication; Wiley: London, UK, 2011; pp. 269–298. [Google Scholar]
- Wang, Q.; Wang, S.; Zhu, X.; Fang, L.; Hu, Y.; Chen, X.; Huang, X.; Du, X.; Chen, S.; Wan, Q.; et al. Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 2017, 18. [Google Scholar] [CrossRef]
- Shoemaker, R.C.; Polzin, K.; Labate, J.; Specht, J.; Brummer, E.C.; Olson, T.; Young, N.; Concibido, V.; Wilcox, J.; Tamulonis, J.P.; et al. Genome duplication in soybean (Glycine subgenus soja). Genetics 1996, 144, 329–338. [Google Scholar] [CrossRef]
- Small, R.L.; Wendel, J.F. Phylogeny, duplication, and intraspecific variation of Adh sequences in new world diploid cottons (Gossypium L., Malvaceae). Mol. Phylogenet. Evol. 2000, 16, 73–84. [Google Scholar] [CrossRef]
- Miao, Q.; Deng, P.; Saha, S.; Jenkins, J.N.; Hsu, C.-Y.; Abdurakhmonov, I.Y.; Buriev, Z.T.; Pepper, A.; Ma, D.-P. Transcriptome Analysis of Ten-DPA Fiber in an Upland Cotton (Gossypium hirsutum) Line with Improved Fiber Traits from Phytochrome A1 RNAi Plants. Am. J. Plant Sci. 2017, 8, 2530–2553. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Zhang, L.; Zuo, K. A Cotton Annexin Protein AnxGb6 Regulates Fiber Elongation through Its Interaction with Actin 1. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Li, H.B.; Qin, Y.M.; Pang, Y.; Song, W.Q.; Mei, W.Q.; Zhu, Y.X. A cotton ascorbate peroxidase is involved in hydrogen peroxide homeostasis during fibre cell development. New Phytol. 2007, 175, 462–471. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Enzymes and Other Agents That Enhance Cell Wall Extensibility. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2002, 50, 391–417. [Google Scholar] [CrossRef]
- Tao, C.; Jin, X.; Zhu, L.; Xie, Q.; Wang, X.; Li, H. Genome-wide investigation and expression profiling of APX gene family in Gossypium hirsutum provide new insights in redox homeostasis maintenance during different fiber development stages. Mol. Genet. Genomics 2018, 293, 685–697. [Google Scholar] [CrossRef]
- Davletova, S.; Rizhsky, L.; Liang, H.; Shengqiang, Z.; Oliver, D.J.; Coutu, J.; Shulaev, V.; Schlauch, K.; Mittler, R. Cytosolic Ascorbate Peroxidase 1 Is a Central Component of the Reactive Oxygen Gene Network of Arabidopsis. Plant Cell 2005, 17, 268–281. [Google Scholar] [CrossRef]
- Orman-Ligeza, B.; Parizot, B.; de Rycke, R.; Fernandez, A.; Himschoot, E.; Van Breusegem, F.; Bennett, M.J.; Périlleux, C.; Beeckman, T.; Draye, X. RBOH-mediated ROS production facilitates lateral root emergence in Arabidopsis. Development 2016, 143, 3328–3339. [Google Scholar] [CrossRef]
- Sarwar, M.B.; Sadique, S.; Hassan, S.; Riaz, S.; Rashid, B.; Bahaeldeen Babiker Mohamed, T.H. Physio-Biochemical and Molecular Responses in Transgenic Cotton under Drought Stress. J. Agric. Sci. 2017, 23, 157–166. [Google Scholar]
- Wei, Y.; Xu, Y.; Lu, P.; Wang, X.; Li, Z.; Cai, X.; Zhou, Z.; Wang, Y.; Zhang, Z.; Lin, Z.; et al. Salt stress responsiveness of a wild cotton species (Gossypium klotzschianum) based on transcriptomic analysis. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Enomoto, A.; Kido, N.; Ito, M.; Takamatsu, N.; Miyagawa, K. Serine-Threonine Kinase 38 is regulated by Glycogen Synthase Kinase-3 and modulates oxidative stress-induced cell death. Free Radic. Biol. Med. 2012, 52, 507–515. [Google Scholar] [CrossRef]
- Vaahtera, L.; Brosché, M.; Wrzaczek, M.; Kangasjärvi, J. Specificity in ROS Signaling and Transcript Signatures. Antioxid. Redox Signal. 2013, 21, 1422–1441. [Google Scholar] [CrossRef]
- Jiang, P.; Zhang, X.; Zhu, Y.; Zhu, W.; Xie, H.; Wang, X. Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep. 2007, 26, 1627–1634. [Google Scholar] [CrossRef]
- Deng, M.H.; Wen, J.F.; Huo, J.L.; Zhu, H.S.; Dai, X.Z.; Zhang, Z.Q.; Zhou, H.; Zou, X.X. Relationship of metabolism of reactive oxygen species with cytoplasmic male sterility in pepper (Capsicum annuum L.). Sci. Hortic. 2012, 134, 232–236. [Google Scholar] [CrossRef]
- Senchina, D.S.; Alvarez, I.; Cronn, R.C.; Liu, B.; Rong, J.; Noyes, R.D.; Paterson, A.H.; Wing, R.A.; Wilkins, T.A.; Wendel, J.F. Rate variation among nuclear genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 2003, 20, 633–643. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Magwanga, R.; Lu, P.; Kirungu, J.; Diouf, L.; Dong, Q.; Hu, Y.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Z.; et al. GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation. Int. J. Mol. Sci. 2018, 19, 1614. [Google Scholar] [CrossRef]
- Simon, U.K.; Polanschütz, L.M.; Koffler, B.E.; Zechmann, B. High Resolution Imaging of Temporal and Spatial Changes of Subcellular Ascorbate, Glutathione and H2O2 Distribution during Botrytis cinerea Infection in Arabidopsis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Heino, P.; Kariola, T.; Pennanen, V.; Broberg, M.; Survila, M.; Palva, E.T.; Sipari, N.; Davidsson, P.R. Peroxidase-Generated Apoplastic ROS Impair Cuticle Integrity and Contribute to DAMP-Elicited Defenses. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Rossi, F.R.; Krapp, A.R.; Bisaro, F.; Maiale, S.J.; Pieckenstain, F.L.; Carrillo, N. Reactive oxygen species generated in chloroplasts contribute to tobacco leaf infection by the necrotrophic fungus Botrytis cinerea. Plant J. 2017, 92, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.J.; Wendel, J.F. Comparative Evolutionary and Developmental Dynamics of the Cotton (Gossypium hirsutum) Fiber Transcriptome. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Li, S.; Zhang, F.; Wu, S.; Jin, X.; Zhang, T.; Guo, W.; Wang, L.; Cheng, C. A Cotton Annexin Affects Fiber Elongation and Secondary Cell Wall Biosynthesis Associated with Ca2+ Influx, ROS Homeostasis, and Actin Filament Reorganization. Plant Physiol. 2016, 171, 1750–1770. [Google Scholar] [CrossRef]
- Van Montagu, M.; Van Camp, W.; Vranova, E.; Villarroel, R.; Atichartpongkul, S.; Inze, D. Comprehensive analysis of gene expression in Nicotiana tabacum leaves acclimated to oxidative stress. Proc. Natl. Acad. Sci. USA 2002, 99, 10870–10875. [Google Scholar] [CrossRef]
- Rentel, M.C.; Lecourieux, D.; Ouaked, F.; Usher, S.L.; Petersen, L.; Okamoto, H.; Knight, H.; Peck, S.C.; Grierson, C.S.; Hirt, H.; et al. OXI1 kinase is necessary for oxidative burst-mediated signalling in Arabidopsis. Nature 2004, 427, 858–861. [Google Scholar] [CrossRef]
- Jia, H.; Hao, L.; Guo, X.; Liu, S.; Yan, Y.; Guo, X. A Raf-like MAPKKK gene, GhRaf19, negatively regulates tolerance to drought and salt and positively regulates resistance to cold stress by modulating reactive oxygen species in cotton. Plant Sci. 2016, 252, 267–281. [Google Scholar] [CrossRef]
- Chu, X.; Wang, C.; Chen, X.; Lu, W.; Li, H.; Wang, X.; Hao, L.; Guo, X. The cotton WRKY gene GhWRKY41 positively regulates salt and drought stress tolerance in transgenic Nicotiana benthamiana. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Tang, C.; Kang, Z.; Chen, S.; Deng, L.; Chang, D.; Wang, X. TaADF3, an Actin-Depolymerizing Factor, Negatively Modulates Wheat Resistance Against Puccinia striiformis. Front. Plant Sci. 2016, 6. [Google Scholar] [CrossRef]
- Shang, H.; Wang, Z.; Zou, C.; Zhang, Z.; Li, W.; Li, J.; Shi, Y.; Gong, W.; Chen, T.; Liu, A.; et al. Comprehensive analysis of NAC transcription factors in diploid Gossypium: Sequence conservation and expression analysis uncover their roles during fiber development. Sci. China Life Sci. 2016, 59, 142–153. [Google Scholar] [CrossRef]
- Miao, Y.; Wang, P.; Du, Y.; Song, C.-P.; Zhao, X. The MPK6-ERF6-ROS-Responsive cis-Acting Element7/GCC Box Complex Modulates Oxidative Gene Transcription and the Oxidative Response in Arabidopsis. Plant Physiol. 2013, 161, 1392–1408. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.A. Reactive Oxygen Species in the Elongation Zone of Maize Leaves Are Necessary for Leaf Extension. Plant Physiol. 2002, 129, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.A.; Ramiro Lascano, H.; Bustos, D.; Taleisnik, E. Salinity-induced decrease in NADPH oxidase activity in the maize leaf blade elongation zone. J. Plant Physiol. 2007, 164, 223–230. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Sharp, R.E. Complexity and coordination of root growth at low water potentials: Recent advances from transcriptomic and proteomic analyses. Plant Cell Environ. 2010, 33, 590–603. [Google Scholar] [CrossRef]
- Shivashankara, K.S.; Pavithra, K.C.; Geetha, G.A. Antioxidant protection mechanism during abiotic stresses. In Abiotic Stress Physiology of Horticultural Crops; Springer: New Delhi, India, 2016; pp. 47–70. ISBN 9788132227250. [Google Scholar]
- Lee, S.; Seo, P.J.; Lee, H.J.; Park, C.M. A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 2012, 70, 831–844. [Google Scholar] [CrossRef]
- Zhou, T.; Yang, X.; Wang, L.; Xu, J.; Zhang, X. GhTZF1 regulates drought stress responses and delays leaf senescence by inhibiting reactive oxygen species accumulation in transgenic Arabidopsis. Plant Mol. Biol. 2014, 85, 163–177. [Google Scholar] [CrossRef]
- Mahmood, K.; El-Kereamy, A.; Kim, S.H.; Nambara, E.; Rothstein, S.J. ANAC032 positively regulates age-dependent and stress-induced senescence in arabidopsis thaliana. Plant Cell Physiol. 2016, 57, 2029–2046. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Qiu, Q.; Wang, K.; Yang, H.; Abbott, R.J.; Liu, J.; Yu, L.; Ma, H.; Lu, Y.; et al. Erratum: Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, D.; Li, Y.; Li, X. Soil salinity evolution and its relationship with dynamics of groundwater in the oasis of inland river basins: Case study from the Fubei Region of Xinjiang Province, China. Environ. Monit. Assess. 2008, 140, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Yanchao, X.; Yangyang, W.; Zhenqing, L.; Xiaoyan, C.; Yuhong, W.; Xingxing, W.; Zhenmei, Z.; Kunbo, W.; Fang, L.; Zhongli, Z. Integrated Evaluation and the Physiological and Biochemical Responses of Semi-Wild Cotton under Complex Salt-Alkali Stress. Cotton Sci. 2018, 231–241. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.D.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Dong, Q.; Hu, Y.; Zhou, Z.; Cai, X.; Wang, X.; Hou, Y.; Wang, K.; et al. Cotton Late Embryogenesis Abundant (LEA2) Genes Promote Root Growth and Confers Drought Stress Tolerance in Transgenic Arabidopsis thaliana. G3 Genes Genomes Genet. 2018, 8, 2781–2803. [Google Scholar] [CrossRef]
- Yoshino, M. Climate Change and Its Effects on Agriculture. J. Agric. Meteorol. 2011, 46, 251–254. [Google Scholar] [CrossRef]
- Diouf, L.; Pan, Z.; He, S.-P.; Gong, W.-F.; Jia, Y.H.; Magwanga, R.O.; Romy, K.R.E.; Rashid, H.; Kirungu, J.N.; Du, X. High-Density Linkage Map Construction and Mapping of Salt-Tolerant QTLs at Seedling Stage in Upland Cotton Using Genotyping by Sequencing (GBS). Int. J. Mol. Sci. 2017, 18, 2622. [Google Scholar] [CrossRef]
- Kohli, S.K.; Handa, N.; Gautam, V.; Bali, S.; Sharma, A.; Khanna, K.; Arora, S.; Thukral, A.K.; Ohri, P.; Karpets, Y.V.; et al. ROS signaling in plants under heavy metal stress. In Reactive Oxygen Species and Antioxidant Systems in Plants: Role and Regulation under Abiotic Stress; Springer: Singapore, 2017; pp. 185–214. ISBN 9789811052545. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Synteny Block | ||||
|---|---|---|---|---|
| NGEB | At vs. AA | At vs. DD | Dt vs. AA | Dt vs. DD |
| 1 | 136 * | 63 | 140 | 58 |
| 2 | 19 | 10 | 19 | 13 |
| 3 | 3 | 11 | 4 | 8 |
| 4 | 2 | 7 | 2 | 3 |
| 5 | 1 | 7 | 1 | 4 |
| 6 | 0 | 3 | 0 | 3 |
| 7 | 0 | 1 | 0 | 2 |
| 8 | 0 | 2 | 0 | 2 |
| 9 | 0 | 0 | 0 | 2 |
| 10 | 0 | 1 | 0 | 0 |
| 11 | 0 | 0 | 0 | 2 |
| 15 | 0 | 0 | 0 | 0 |
| 16 | 0 | 0 | 0 | 1 |
| 20 | 0 | 0 | 0 | 1 |
| Accession | RL | SL | Area of Damaged Blades | |||
|---|---|---|---|---|---|---|
| First Leaf * | Second Leaf * | Third Leaf * | Fourth Leaf * | |||
| Latifolium 130 | 0.90 a | 0.80 c | 0.97 a | 0.73 a | 0.65 a | 0.00 a |
| Latifolium 32 | 0.92 a | 0.97 a,b | 0.17 b | 0.03 b | 0.00 b | 0.00 a |
| Latifolium 40 | 0.87 a | 0.89 b,c | 0.40 b | 0.20 b | 0.23 a,b | 0.03 a |
| Marie-galante 85 | 0.92 a | 1.02 a | 0.32 b | 0.03 b | 0.00 b | 0.00 a |
| CRI 12 | 0.98 a | 0.68 d | 0.38 b | 0.08 b | 0.02 b | 0.00 a |
| CRI 16 | 1.12 a | 0.94 a,b | 0.38 b | 0.10 b | 0.05 b | 0.05 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Magwanga, R.O.; Cai, X.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Jin, D.; Guo, X.; Wei, Y.; et al. Deep Transcriptome Analysis Reveals Reactive Oxygen Species (ROS) Network Evolution, Response to Abiotic Stress, and Regulation of Fiber Development in Cotton. Int. J. Mol. Sci. 2019, 20, 1863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081863
Xu Y, Magwanga RO, Cai X, Zhou Z, Wang X, Wang Y, Zhang Z, Jin D, Guo X, Wei Y, et al. Deep Transcriptome Analysis Reveals Reactive Oxygen Species (ROS) Network Evolution, Response to Abiotic Stress, and Regulation of Fiber Development in Cotton. International Journal of Molecular Sciences. 2019; 20(8):1863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081863
Chicago/Turabian StyleXu, Yanchao, Richard Odongo Magwanga, Xiaoyan Cai, Zhongli Zhou, Xingxing Wang, Yuhong Wang, Zhenmei Zhang, Dingsha Jin, Xinlei Guo, Yangyang Wei, and et al. 2019. "Deep Transcriptome Analysis Reveals Reactive Oxygen Species (ROS) Network Evolution, Response to Abiotic Stress, and Regulation of Fiber Development in Cotton" International Journal of Molecular Sciences 20, no. 8: 1863. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081863