Dissecting the Genetic Architecture of Seed Protein and Oil Content in Soybean from the Yangtze and Huaihe River Valleys Using Multi-Locus Genome-Wide Association Studies

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Variation of Seed Protein and Oil Content
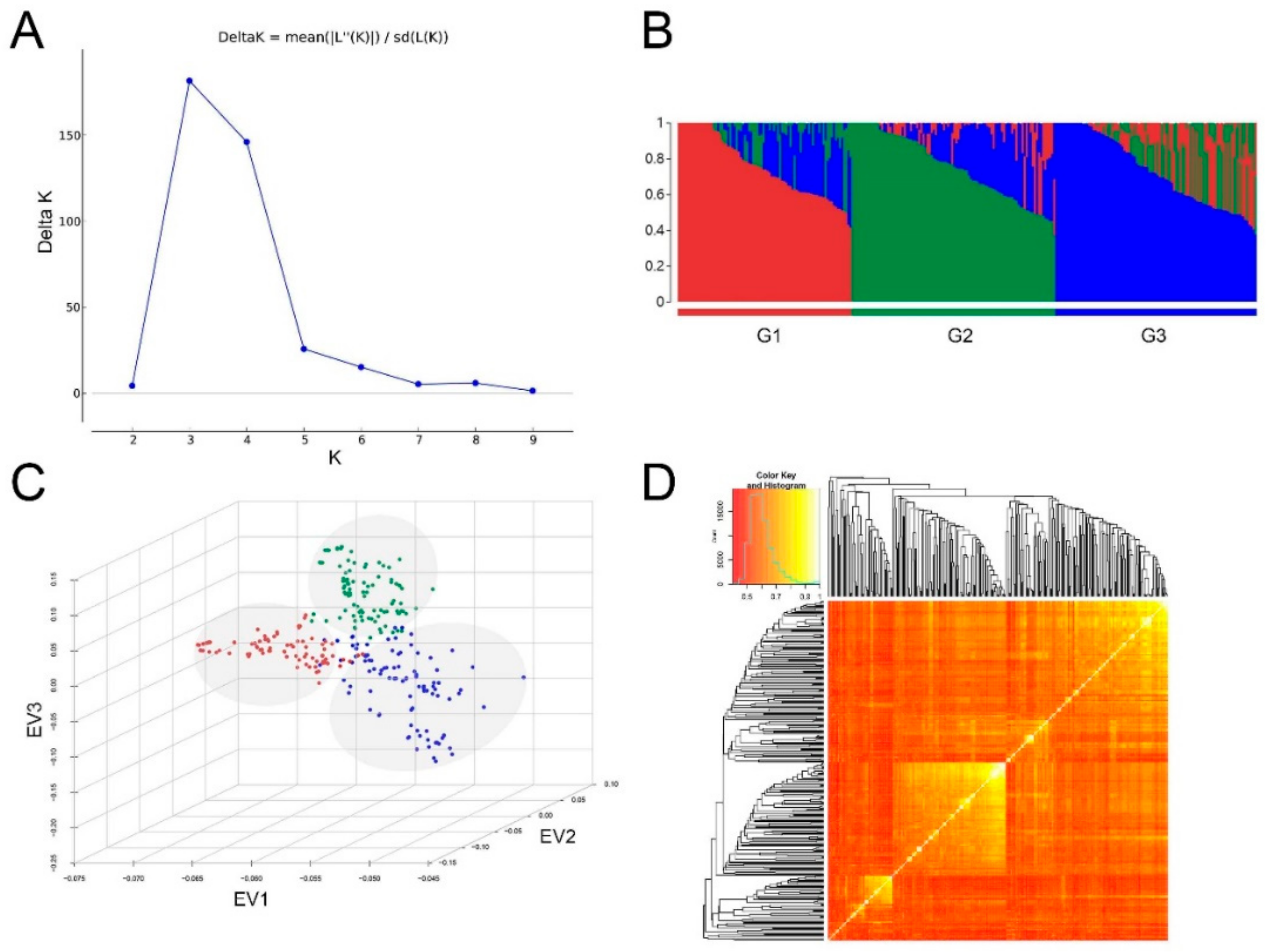
2.2. Genetic Diversity and Population Structure Analysis Based on SNPLDB Markers
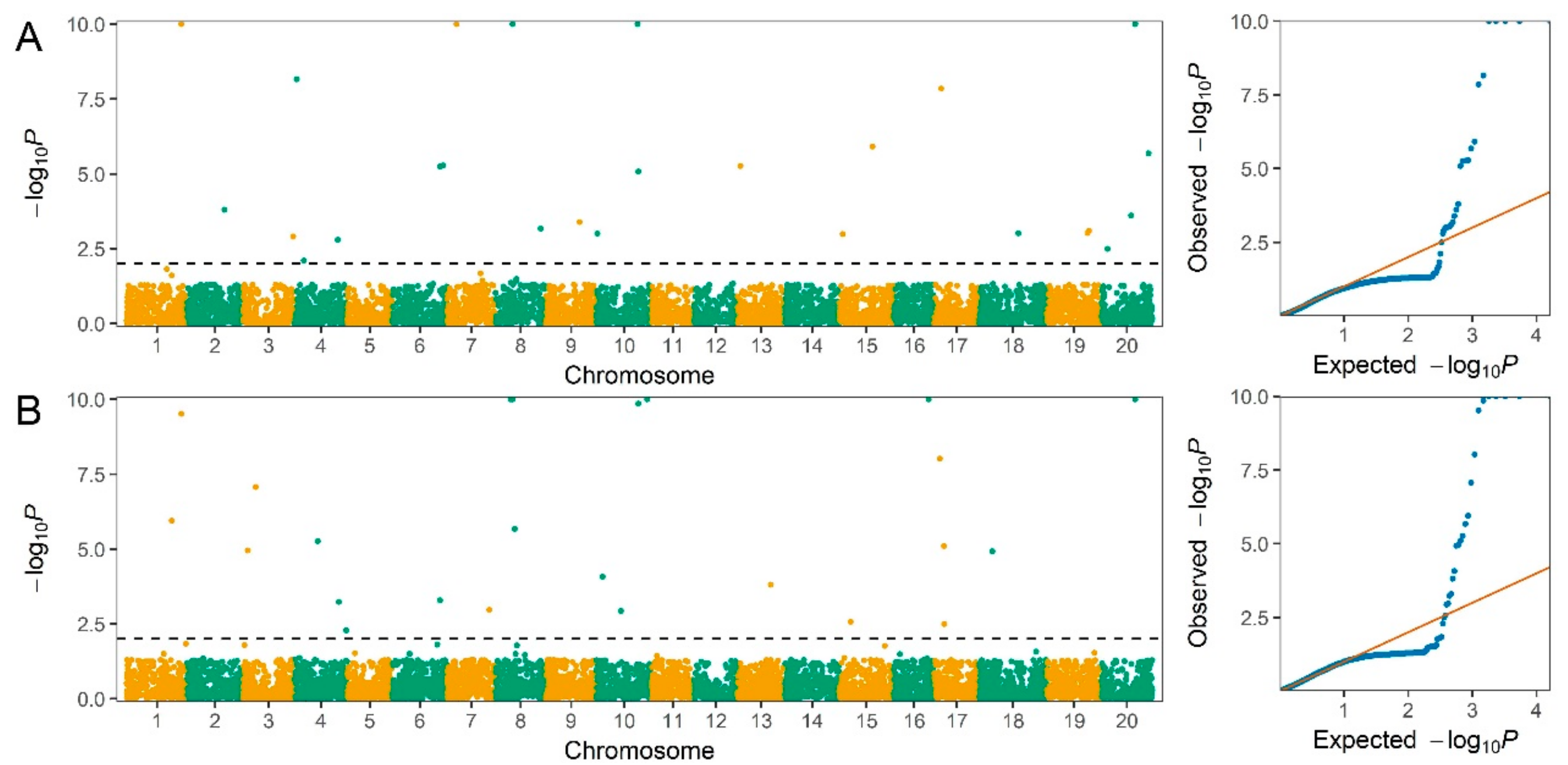
2.3. Genome-Wide Association Study for Seed Protein and Oil Content
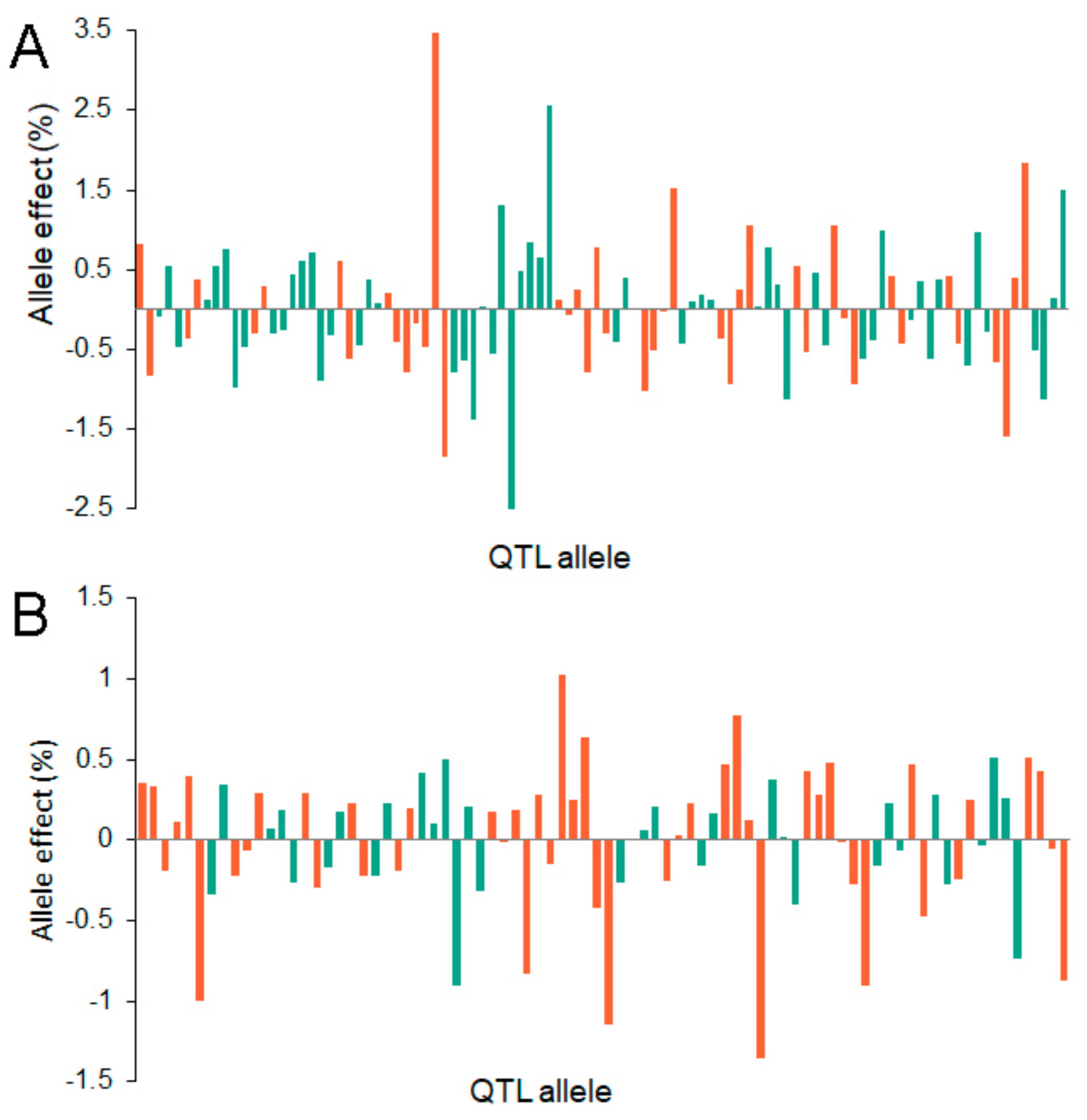
2.4. QTL-Allele Matrices of Seed Protein and Oil Content
2.5. The Common QTLs Associated with Seed Protein and Oil Content
2.6. Candidate Genes Controlling Seed Protein and Oil Content
3. Discussion
3.1. Efficient Multi-Locus GWAS Procedure for Dissecting the Genetic Architecture of Complex Traits
3.2. Previously Reported and Novel QTLs Detected with Multi-Locus GWAS Analysis
3.3. Candidate Genes for Seed Protein and Oil Content for Further Study
4. Materials and Methods
4.1. Plant Materials and Field Experiments
4.2. Phenotypic Evaluation and Statistical Analysis
4.3. SNP Genotyping and SNPLDB Marker Construction
4.4. Genetic Diversity and Population Structure Analysis Based on SNPLDB Markers
4.5. Multi-Locus Genome-Wide Association Study
4.6. Candidate Gene Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| GSC | genetic similarity coefficient |
| GWAS | genome-wide association study |
| MAF | minor allele frequency |
| NIR | near-infrared reflectance |
| PIC | polymorphic information content |
| QTL | quantitative trait loci |
| RAD-seq | restriction site-associated DNA sequencing |
| RIL | recombinant inbred line |
| RTM-GWAS | restricted two-stage multi-locus multi-allele (RTM) genome-wide association study (GWAS) |
| SNP | single nucleotide polymorphism |
| SNPLDB | SNP linkage disequilibrium block |
References
- American Soybean Association. SoyStats: A Reference Guide to Soybean Facts and Figures; American Soybean Association: St. Louis, MO, USA, 2018; Available online: http://soystats.com/ (accessed on 20 March 2019).
- Chaudhary, J.; Patil, G.B.; Sonah, H.; Deshmukh, R.K.; Vuong, T.D.; Valliyodan, B.; Nguyen, H.T. Expanding omics resources for improvement of soybean seed composition traits. Front. Plant Sci. 2015, 6, 1021. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.F. Seed composition. In Soybeans: Improvement, Production, and Uses, 3rd ed.; Boerma, R.H., Specht, J.E., Eds.; American Soybean Association: Madison, WI, USA, 2004; pp. 621–677. [Google Scholar]
- Hwang, E.Y.; Song, Q.; Jia, G.; Specht, J.E.; Hyten, D.L.; Costa, J.; Cregan, P.B. A genome-wide association study of seed protein and oil content in soybean. BMC Genom. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.; Cober, E.R.; Rajcan, I. Genetic control of soybean seed oil: II. QTL and genes that increase oil concentration without decreasing protein or with increased seed yield. Theor. Appl. Genet. 2013, 126, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Akond, M.; Liu, S.; Boney, M.; Kantartzi, S.K.; Meksem, K.; Bellaloui, N.; Lightfoot, D.A.; Kassem, M.A. Identification of quantitative trait loci (QTL) underlying protein, oil, and five major fatty acids’ contents in soybean. Am. J. Plant Sci. 2014, 5, 158–167. [Google Scholar] [CrossRef]
- Li, D.; Zhao, X.; Han, Y.; Li, W.; Xie, F. Genome-wide association mapping for seed protein and oil contents using a large panel of soybean accessions. Genomics 2019, 111, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.; Mian, R.; Vuong, T.; Pantalone, V.; Song, Q.; Chen, P.; Shannon, G.J.; Carter, T.C.; Nguyen, H.T. Molecular mapping and genomics of soybean seed protein: A review and perspective for the future. Theor. Appl. Genet. 2017, 130, 1975–1991. [Google Scholar] [CrossRef] [PubMed]
- Pathan, S.M.; Vuong, T.; Clark, K.; Lee, J.-D.; Shannon, J.G.; Roberts, C.A.; Ellersieck, M.R.; Burton, J.W.; Cregan, P.B.; Hyten, D.L.; et al. Genetic mapping and confirmation of quantitative trait loci for seed protein and oil contents and seed weight in soybean. Crop Sci. 2013, 53, 765–774. [Google Scholar] [CrossRef]
- Han, Y.; Teng, W.; Wang, Y.; Zhao, X.; Wu, L.; Li, D.; Li, W.; Morris, B. Unconditional and conditional QTL underlying the genetic interrelationships between soybean seed isoflavone, and protein or oil contents. Plant Breed. 2015, 134, 300–309. [Google Scholar] [CrossRef]
- Mao, T.; Jiang, Z.; Han, Y.; Teng, W.; Zhao, X.; Li, W. Identification of quantitative trait loci underlying seed protein and oil contents of soybean across multi-genetic backgrounds and environments. Plant Breed. 2013, 132, 630–641. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, S.; Li, W.; Liu, S.; Li, X.; Fang, Y.; Zhang, J.; Wang, Y.; Xu, S.; Zhang, J.; et al. Identification of QTNs controlling seed protein content in soybean using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 1690. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Van, K.; Sung, M.; Nelson, R.; LaMantia, J.; McHale, L.K.; Mian, M.A.R. Genome-wide association study of seed protein, oil and amino acid contents in soybean from maturity groups I to IV. Theor. Appl. Genet. 2019, 123, 1639–1659. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Guo, C.; Sun, F.; Zhang, J.; Wang, Z.; Kong, J.; He, Q.; Sharmin, R.A.; Zhao, T. Genome-wide association studies for dynamic plant height and number of nodes on the main stem in summer sowing soybeans. Front. Plant Sci. 2018, 9, 1184. [Google Scholar] [CrossRef] [PubMed]
- Bandillo, N.; Jarquin, D.; Song, Q.; Nelson, R.; Cregan, P.; Specht, J.; Lorenz, A. A Population Structure and Genome-wide association analysis on the USDA soybean germplasm collection. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; Meng, S.; Liu, M.; Xing, G.; Li, Y.; Yang, S.; Yang, J.; Zhao, T.; Gai, J. Identifying QTL–allele system of seed protein content in Chinese soybean landraces for population differentiation studies and optimal cross predictions. Euphytica 2018, 214, 157. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; Wang, H.; Meng, S.; Xing, G.; Li, Y.; Yang, S.; Zhao, J.; Zhao, T.; Gai, J. Detecting the QTL-Allele System of Seed Oil Traits Using Multi-Locus Genome-Wide Association Analysis for Population Characterization and Optimal Cross Prediction in Soybean. Front. Plant Sci. 2018, 9, 1793. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Feng, J.; Ren, W.; Huang, B.; Zhou, L.; Wen, Y.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Rakitsch, B.; Lippert, C.; Stegle, O.; Borgwardt, K. A Lasso multi-marker mixed model for association mapping with population structure correction. Bioinformatics 2013, 29, 206–214. [Google Scholar] [CrossRef]
- He, J.; Meng, S.; Zhao, T.; Xing, G.; Yang, S.; Li, Y.; Guan, R.; Lu, J.; Wang, Y.; Xia, Q.; et al. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theor. Appl. Genet. 2017, 130, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.; Wang, Y.; Xing, G.; Zhao, J.; Li, Y.; Yang, S.; Palmer, R.G.; Zhao, T.; Gai, J. Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. J. Exp. Bot. 2015, 66, 6311–6325. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; He, J.; Zhao, T.; Xing, G.; Li, Y.; Yang, S.; Lu, J.; Wang, Y.; Gai, J. Detecting the QTL-allele system of seed isoflavone content in Chinese soybean landrace population for optimal cross design and gene system exploration. Theor. Appl. Genet. 2016, 129, 1557–1576. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cao, Y.; He, J.; Zhao, T.; Gai, J. Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theor. Appl. Genet. 2017, 130, 2297–2314. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Tong, F.; Wang, W.; He, J.; Zhao, T.; Gai, J. Analysis of QTL–allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [Glycine max (L.) Merr.] using a novel GWAS procedure. Planta 2018, 248, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; He, J.; Zhao, T.; Xing, G.; Wang, Y.; Yu, D.; Chen, S.; Gai, J. Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. Theor. Appl. Genet. 2018, 131, 2581–2599. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, N.; Niu, J.; Wang, Z.; Cui, X.; Sun, J.; Zhao, T.; Xing, H. Loci and candidate gene identification for resistance to Phytophthora sojae via association analysis in soybean [Glycine max (L.) Merr]. Mol Genet. Genom. 2016, 291, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.N.; Nelson, R.L.; Song, Q.; Cregan, P.B.; Li, Z. The genetic architecture of seed composition in soybean is refined by genome-wide association scans across multiple populations. G3 Genes Genomes Genet. 2014, 4, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Kan, G.; Hu, Z.; Cheng, H.; Zhang, Y.; Wang, Q.; Wang, H.; Yang, Y.; Li, H.; Hao, D.; et al. Use of single nucleotide polymorphisms and haplotypes to identify genomic regions associated with protein content and water-soluble protein content in soybean. Theor. Appl. Genet. 2014, 127, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, X.; Liu, D.; Li, Y.; Lightfoot, D.A.; Yang, Z.; Zhao, L.; Zhou, G.; Wang, Z.; Huang, L.; et al. Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytol. 2016, 209, 871–884. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Lu, Y.; Bhusal, S.J.; Song, Q.; Cregan, P.B.; Yen, Y.; Brown, M.; Jiang, G.-L. Genome-wide scan for seed composition provides insights into soybean quality improvement and the impacts of domestication and breeding. Mol. Plant 2018, 11, 460–472. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, S.; Wang, Z.; Chang, F.; Kong, J.; Gai, J.; Zhao, T. Identification of Major Quantitative Trait Loci for Seed Oil Content in Soybeans by Combining Linkage and Genome-Wide Association Mapping. Front. Plant Sci. 2017, 8, 1222. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Warrington, C.V.; Abdel-Haleem, H.; Hyten, D.L.; Cregan, P.B.; Orf, J.H.; Killam, A.S.; Bajjalieh, N.; Li, Z.; Boerma, H.R. QTL for seed protein and amino acids in the Benning x Danbaekkong soybean population. Appl. Genet. 2015, 128, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wen, Z.; Li, H.; Yuan, D.; Li, J.; Zhang, H.; Huang, Z.; Cui, S.; Du, W. Identification of the quantitative trait loci (QTL) underlying water soluble protein content in soybean. Theor. Appl. Genet. 2013, 126, 425–433. [Google Scholar] [CrossRef]
- Pandurangan, S.; Pajak, A.; Molnar, S.J.; Cober, E.R.; Dhaubhadel, S.; Hernandezsebastia, C.; Kaiser, W.M.; Nelson, R.L.; Huber, S.C.; Marsolais, F. Relationship between asparagine metabolism and protein concentration in soybean seed. J. Exp. Bot. 2012, 63, 3173–3184. [Google Scholar] [CrossRef]
- Fasoula, V.A.; Harris, D.K.; Boerma, H.R. Validation and designation of quantitative trait loci for seed protein, seed oil, and seed weight from two soybean populations. Crop Sci. 2004, 44, 1218–1225. [Google Scholar] [CrossRef]
- Diers, B.W.; Keim, P.; Fehr, W.R.; Shoemaker, R.C. RFLP analysis of soybean seed protein and oil content. Theor. Appl. Genet. 1992, 83, 608–612. [Google Scholar] [CrossRef]
- Chung, J.; Babka, H.L.; Graef, G.L.; Staswick, P.E.; Lee, D.J.; Cregan, P.B.; Shoemaker, R.C.; Specht, J.E. The seed protein, oil, and yield QTL on soybean linkage group I. Crop Sci. 2003, 43, 1053–1067. [Google Scholar] [CrossRef]
- Hyten, D.L.; Pantalone, V.R.; Sams, C.E.; Saxton, A.M.; Landauellis, D.; Stefaniak, T.R.; Schmidt, M.E. Seed quality QTL in a prominent soybean population. Theor. Appl. Genet. 2004, 109, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Tajuddin, T.; Watanabe, S.; Yamanaka, N.; Harada, K. Analysis of quantitative trait loci for protein and lipid contents in soybean seeds using recombinant inbred lines. Breed. Sci. 2003, 53, 133–140. [Google Scholar] [CrossRef]
- Csanádi, G.; Vollmann, J.; Stift, G.; Lelley, T. Seed quality QTLs identified in a molecular map of early maturing soybean. Theor. Appl. Genet. 2001, 103, 912–919. [Google Scholar] [CrossRef]
- Kabelka, E.A.; Diers, B.W.; Fehr, W.R.; LeRoy, A.R.; Baianu, I.C.; You, T.; Neece, D.J.; Nelson, R.L. Putative Alleles for Increased Yield from Soybean Plant Introductions. Crop Sci. 2004, 44, 784–791. [Google Scholar] [CrossRef]
- Qi, Z.; Wu, Q.; Han, X.; Sun, Y.; Du, X.; Liu, C.; Jiang, H.; Hu, G.; Chen, Q. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 2011, 179, 499–514. [Google Scholar] [CrossRef]
- Qi, Z.; Han, X.; Sun, Y.; Wu, Q.; Shan, D.; Du, X.; Liu, C.; Jiang, H.; Hu, G.; Chen, Q. An Integrated Quantitative Trait Locus Map of Oil Content in Soybean, Glycine max (L.) Merr., Generated Using a Meta-Analysis Method for Mining Genes. Agric. Sci. China 2011, 10, 1681–1692. [Google Scholar] [CrossRef]
- Liang, H.; Yu, Y.; Wang, S.; Lian, Y.; Wang, T.; Wei, Y.; Gong, P.; Liu, X.; Fang, X.; Zhang, M. QTL Mapping of Isoflavone, Oil and Protein Contents in Soybean (Glycine max L. Merr.). Agric. Sci. China 2010, 9, 1108–1116. [Google Scholar] [CrossRef]
- Reinprecht, Y.; Poysa, V.W.; Yu, K.; Rajcan, I.; Ablett, G.R.; Pauls, K.P. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 2006, 49, 1510–1527. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, G.; Green, M.; Scott, R.A.; Song, Q.; Hyten, D.L.; Cregan, P.B. Identification and validation of quantitative trait loci for seed yield, oil and protein contents in two recombinant inbred line populations of soybean. Mol. Genet. Genom. 2014, 289, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Bailey, M.A.; Mian, M.A.; Shipe, E.R.; Ashley, D.A.; Parrott, W.A.; Hussey, R.S.; Boerma, H.R. RFLP loci associated with soybean seed protein and oil content across populations and locations. Theor. Appl. Genet. 1996, 93, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Bolon, Y.-T.; Joseph, B.; Cannon, S.B.; Graham, M.A.; Diers, B.W.; Farmer, A.D.; May, G.D.; Muehlbauer, G.J.; Specht, J.E.; Tu, Z.J.; et al. Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean. BMC Plant Biol. 2010, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.; Roux, F. Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat. Rev. Genet. 2010, 11, 867. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Kim, S.-R.; An, G. Rice aldehyde dehydrogenase7 is needed for seed maturation and viability. Plant Physiol. 2009, 149, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Ramasamy, R.K.; Ramasamy, S.; Bindroo, B.B.; Naik, V.G. STRUCTURE PLOT: A program for drawing elegant STRUCTURE bar plots in user friendly interface. SpringerPlus 2014, 3, 431. [Google Scholar] [CrossRef] [PubMed]
- Ligges, U.; Maechler, M. Scatterplot3d—An R package for visualizing multivariate data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- McCouch, S.; Cho, Y.; Yano, M.; Paul, E.; Blinstrub, M.; Morishima, H.; Kinoshita, T. Report on QTL nomenclature. Rice Genet. Newsl. 1997, 14, 11–13. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Year | Mean ± SD (%) | Range (%) | Heritability (%) | F-Values from ANOVA | ||
|---|---|---|---|---|---|---|---|
| Accession | Environment | Accession × Environment | |||||
| Protein content | 2015 | 42.6 ± 2.1 | 37.2–47.6 | 80.4 | 5.10 *** | 0.08 ns | 1.50 *** |
| 2016 | 42.6 ± 2.5 | 36.1–49.3 | |||||
| Oil content | 2015 | 18.5 ± 1.2 | 15.2–21.4 | 79.0 | 4.77 *** | 1.91 ns | 1.39 ** |
| 2016 | 18.2 ± 1.3 | 14.8–21.1 | |||||
| QTL | SNPLDB a | Allele Number | Log10 P | R2 (%) | QTL in SoyBase b | QTL in Previous GWAS c |
|---|---|---|---|---|---|---|
| qProt-1-1 | Gm01_50257226 | 2 | 11.7 | 2.8 | Seed protein 13-1,31-3, 36-9,36-10 | |
| qProt-2-1 | Gm02_BLOCK_34241156_34302885 | 3 | 3.8 | 1.0 | Seed protein 27-1 | |
| qProt-3-1 | Gm03_46437265 | 2 | 2.9 | 0.6 | Seed protein 27-4 | |
| qProt-4-1 | Gm04_BLOCK_2157397_2345500 | 5 | 8.2 | 2.4 | Seed protein 4-4,9-2, 36-5,36-6 | |
| qProt-4-2 | Gm04_8725710 | 2 | 2.1 | 0.4 | Seed protein 7-2,19-1 | |
| qProt-4-3 | Gm04_BLOCK_39987192_40167695 | 7 | 2.8 | 1.2 | Seed protein 36-4 | Zhang et al. [17] |
| qProt-6-1 | Gm06_44035193 | 2 | 5.2 | 1.1 | Seed protein 36-7,36-8, cqSeed protein-012 | |
| qProt-6-2 | Gm06_BLOCK_46724398_46724570 | 3 | 5.3 | 1.4 | Seed protein 13-2,24-1 | Bandillo et al. [16] |
| qProt-7-1 | Gm07_BLOCK_8657174_8845556 | 7 | 15.2 | 4.7 | Seed protein 24-4,33-5, cqSeed protein-009 | Hwang et al. [4] |
| qProt-8-1 | Gm08_BLOCK_16237107_16278243 | 11 | 24.1 | 8.0 | Novel | |
| qProt-8-2 | Gm08_BLOCK_42026048_42200505 | 6 | 3.2 | 1.2 | Seed protein 3-1,21-1 | |
| qProt-9-1 | Gm09_BLOCK_31004337_31004679 | 2 | 3.4 | 0.7 | Seed protein 36-28,36-29, 36-30,37-10,47-2 | |
| qProt-10-1 | Gm10_BLOCK_856393_1044958 | 5 | 3.0 | 1.0 | Seed protein 21-5 | Hwang et al. [4] |
| qProt-10-2 | Gm10_BLOCK_37907908_38079904 | 4 | 13.2 | 3.6 | Seed protein 27-5,36-39, 36-40,40-1 | |
| qProt-10-3 | Gm10_BLOCK_38679572_38679818 | 4 | 5.1 | 1.5 | Seed protein 12-5,36-38 | |
| qProt-13-1 | Gm13_BLOCK_2883529_3082036 | 4 | 5.3 | 1.5 | Novel | |
| qProt-15-1 | Gm15_3633885 | 2 | 3.0 | 0.6 | Seed protein 30-3 | Hwang et al. [4]; Vaughn et al. [29]; Zhang et al. [30] |
| qProt-15-2 | Gm15_31011761 | 2 | 5.9 | 1.3 | Seed protein 27-2 | |
| qProt-17-1 | Gm17_BLOCK_6574500_6577175 | 3 | 7.8 | 2.0 | Novel | |
| qProt-18-1 | Gm18_BLOCK_35961794_36027902 | 3 | 3.0 | 0.8 | Novel | |
| qProt-19-1 | Gm19_37500961 | 2 | 3.0 | 0.6 | Novel | |
| qProt-19-2 | Gm19_BLOCK_38886258_38944167 | 4 | 3.1 | 0.9 | Novel | |
| qProt-20-1 | Gm20_5531497 | 2 | 2.5 | 0.5 | Seed protein 1-3,1-4, 3-12,10-1,11-1,30-1, 36-26,37-8,47-8 | |
| qProt-20-2 | Gm20_BLOCK_27111387_27111623 | 3 | 3.6 | 0.9 | Seed protein 1-1,1-2, 15-1,26-5,31-1,34-11, 39-4,cqSeed protein-003 | |
| qProt-20-3 | Gm20_BLOCK_30995685_31177423 | 4 | 56.1 | 16.0 | Seed protein 1-1,1-2, 15-1, 31-1,34-11,39-4, cqSeed protein-003 | Hwang et al. [4]; Vaughn et al. [29]; Bandillo et al. [16]; Sonah et al. [31]; Zhang et al. [17] |
| qProt-20-4 | Gm20_BLOCK_43288485_43465351 | 4 | 5.7 | 1.6 | Novel | |
| Total | 26 | 98 | 58.3 | 19 (54) | 6 (12) |
| QTL | SNPLDB a | Allele No. | −Log10 P | R2 (%) | QTL in SoyBase b | QTL in Previous GWAS c |
|---|---|---|---|---|---|---|
| qOil-1-1 | Gm01_BLOCK_41522087_41713586 | 6 | 5.9 | 1.9 | Seed oil 24-19,39-5, mqSeed Oil-009 | |
| qOil-1-2 | Gm01_50257226 | 2 | 9.5 | 2.1 | Seed oil 42-21 | |
| qOil-3-1 | Gm03_BLOCK_4553710_4553720 | 3 | 5.0 | 1.2 | Seed oil 24-5,39-14 | |
| qOil-3-2 | Gm03_BLOCK_11906061_11923443 | 3 | 7.1 | 1.7 | Seed oil 39-15, cqSeed oil-005 | |
| qOil-4-1 | Gm04_21748147 | 2 | 5.3 | 1.1 | Novel | |
| qOil-4-2 | Gm04_41026444 | 2 | 3.2 | 0.6 | Zhang et al. [18] | |
| qOil-4-3 | Gm04_BLOCK_47957394_47957714 | 2 | 2.3 | 0.4 | mqSeed Oil-007 | |
| qOil-6-1 | Gm06_44035193 | 2 | 3.3 | 0.6 | Seed oil 23-1,31-2, 33-1,38-2 | |
| qOil-7-1 | Gm07_38954920 | 2 | 3.0 | 0.6 | Seed oil 34-7 | |
| qOil-8-1 | Gm08_BLOCK_14242705_14306849 | 6 | 10.7 | 3.2 | Seed oil 30-3,34-1, mqSeed Oil-004 | Han et al. [32] |
| qOil-8-2 | Gm08_BLOCK_16237107_16278243 | 11 | 15.4 | 5.2 | Novel | |
| qOil-8-3 | Gm08_BLOCK_18015046_18031943 | 4 | 5.7 | 1.6 | Zhang et al. [33] | |
| qOil-10-1 | Gm10_BLOCK_5509737_5559675 | 3 | 4.1 | 1.0 | Seed oil 34-6,43-33, 43-34 | |
| qOil-10-2 | Gm10_22446436 | 2 | 2.9 | 0.6 | Seed oil 19-3 | |
| qOil-10-3 | Gm10_BLOCK_38679572_38679818 | 4 | 9.9 | 2.6 | Novel | |
| qOil-10-4 | Gm10_BLOCK_46662161_46730774 | 3 | 18.4 | 4.6 | Seed oil 29-3 | |
| qOil-13-1 | Gm13_BLOCK_30751302_30790418 | 6 | 3.8 | 1.3 | Seed oil 37-8,38-4 | |
| qOil-15-1 | Gm15_BLOCK_10984687_11112792 | 3 | 2.6 | 0.6 | Seed oil 27-2,39-8 | Zhou et al. [34] |
| qOil-16-1 | Gm16_31945745 | 2 | 11.7 | 2.7 | Seed oil 39-12 | Zhang et al. [18] |
| qOil-17-1 | Gm17_BLOCK_5346273_5356960 | 2 | 8.0 | 1.8 | Seed oil 23-3 | Hwang et al. [4] |
| qOil-17-2 | Gm17_9078832 | 2 | 5.1 | 1.1 | Seed oil 43-12 | |
| qOil-18-1 | Gm18_BLOCK_11985000_12125810 | 4 | 4.9 | 1.4 | Seed oil 27-10,43-16 | |
| qOil-20-1 | Gm20_BLOCK_30995685_31177423 | 4 | 55.0 | 15.1 | Seed oil 2-1,2-2,15-1, 24-30,mqSeed Oil-020, cqSeed oil-004 | Hwang et al. [4]; Vaughn et al. [29]; Bandillo et al. [16]; Sonah et al. [31]; Cao et al. [35]; Zhang et al. [18] |
| Total | 23 | 80 | 53.1 | 18 (39) | 7 (12) |
| QTL | SNPLDB | Allele | Frequency | Protein Content (%) a | Oil Content (%) a | Protein vs. Oil Relationship b |
|---|---|---|---|---|---|---|
| qProt-1-1/qOil-1-2 | Gm01_50257226 | A | 197 | 43.0 * | 18.2 * | Negative |
| C | 82 | 41.8 | 18.8 | |||
| qProt-6-1/qOil-6-1 | Gm06_44035193 | C | 154 | 43.2 * | 18.1 * | Negative |
| T | 125 | 41.9 | 18.7 | |||
| qProt-8-1/qOil-8-2 | Gm08_BLOCK_16237107_16278243 | CGCCATT | 3 | 45.6 a | 16.8 f | Negative |
| CGCTGCA | 6 | 45.3 a | 17.1 ef | |||
| TGTCGTT | 4 | 44.6 ab | 17.2 ef | |||
| CGCTATT | 4 | 43.8 bc | 18.0 cde | |||
| CGCTACA | 82 | 43.7 bc | 17.8 de | |||
| CGTCGTT | 19 | 43.1 bcd | 18.2 cd | |||
| CGCCGCA | 5 | 42.4 cde | 18.3 bcd | |||
| TGCCGTT | 76 | 42.0 de | 18.6 abcd | |||
| CATCGTT | 57 | 41.8 de | 18.8 abc | |||
| CGCTGTT | 5 | 41.0 e | 19.1 ab | |||
| CGCCGTT | 18 | 40.9 e | 19.3 a | |||
| qProt-10-3/qOil-10-3 | Gm10_BLOCK_38679572_38679818 | GCCC | 5 | 46.4 a | 16.1 b | Negative |
| ACTC | 212 | 42.8 b | 18.3 a | |||
| GCCT | 44 | 42.2 bc | 18.6 a | |||
| GTCC | 18 | 41.2 c | 18.9 a | |||
| qProt-20-3/qOil-20-1 | Gm20_BLOCK_30995685_31177423 | CACCCAGGAATCACGGACGCGC | 20 | 45.0 a | 17.1 c | Negative |
| TTATTCAATGCTGTATCTATAT | 70 | 44.0 b | 17.7 b | |||
| CTATTCAATGCTGTATCTATAT | 109 | 42.1 c | 18.6 a | |||
| CTATTCAATGCTGTATCTATGT | 80 | 41.6 c | 18.9 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Xu, H.; Yang, J.; Zhao, T. Dissecting the Genetic Architecture of Seed Protein and Oil Content in Soybean from the Yangtze and Huaihe River Valleys Using Multi-Locus Genome-Wide Association Studies. Int. J. Mol. Sci. 2019, 20, 3041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123041
Li S, Xu H, Yang J, Zhao T. Dissecting the Genetic Architecture of Seed Protein and Oil Content in Soybean from the Yangtze and Huaihe River Valleys Using Multi-Locus Genome-Wide Association Studies. International Journal of Molecular Sciences. 2019; 20(12):3041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123041
Chicago/Turabian StyleLi, Shuguang, Haifeng Xu, Jiayin Yang, and Tuanjie Zhao. 2019. "Dissecting the Genetic Architecture of Seed Protein and Oil Content in Soybean from the Yangtze and Huaihe River Valleys Using Multi-Locus Genome-Wide Association Studies" International Journal of Molecular Sciences 20, no. 12: 3041. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123041