Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.)

 , ,
, , 
 and
and
Abstract
:1. Introduction
2. Results
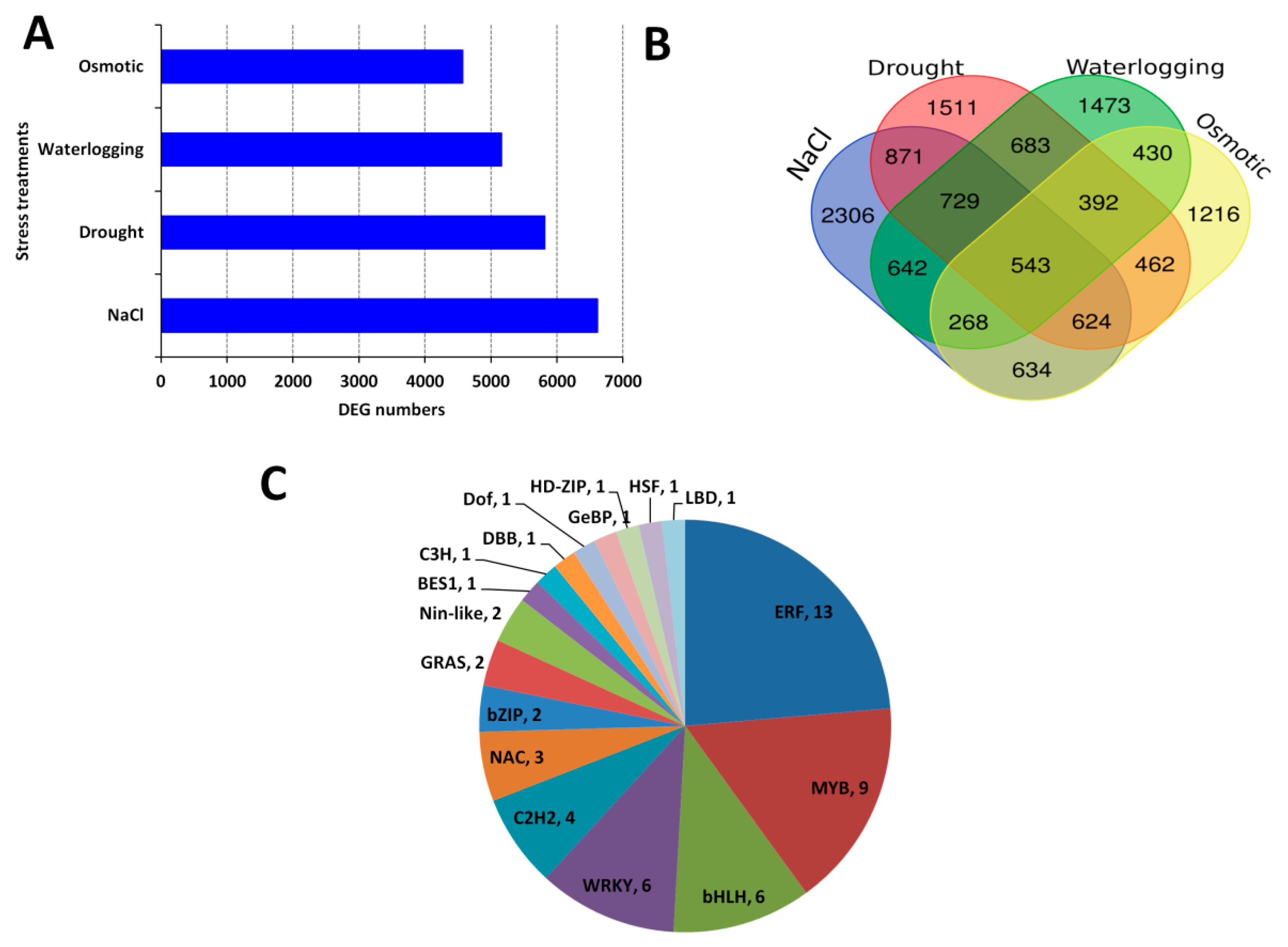
2.1. Overview of the Transcriptome Data and Gene Expression Profile under Various Stress Treatments
2.2. Identification of DEGs, Core Conserved DEGs in Response to Abiotic Stress
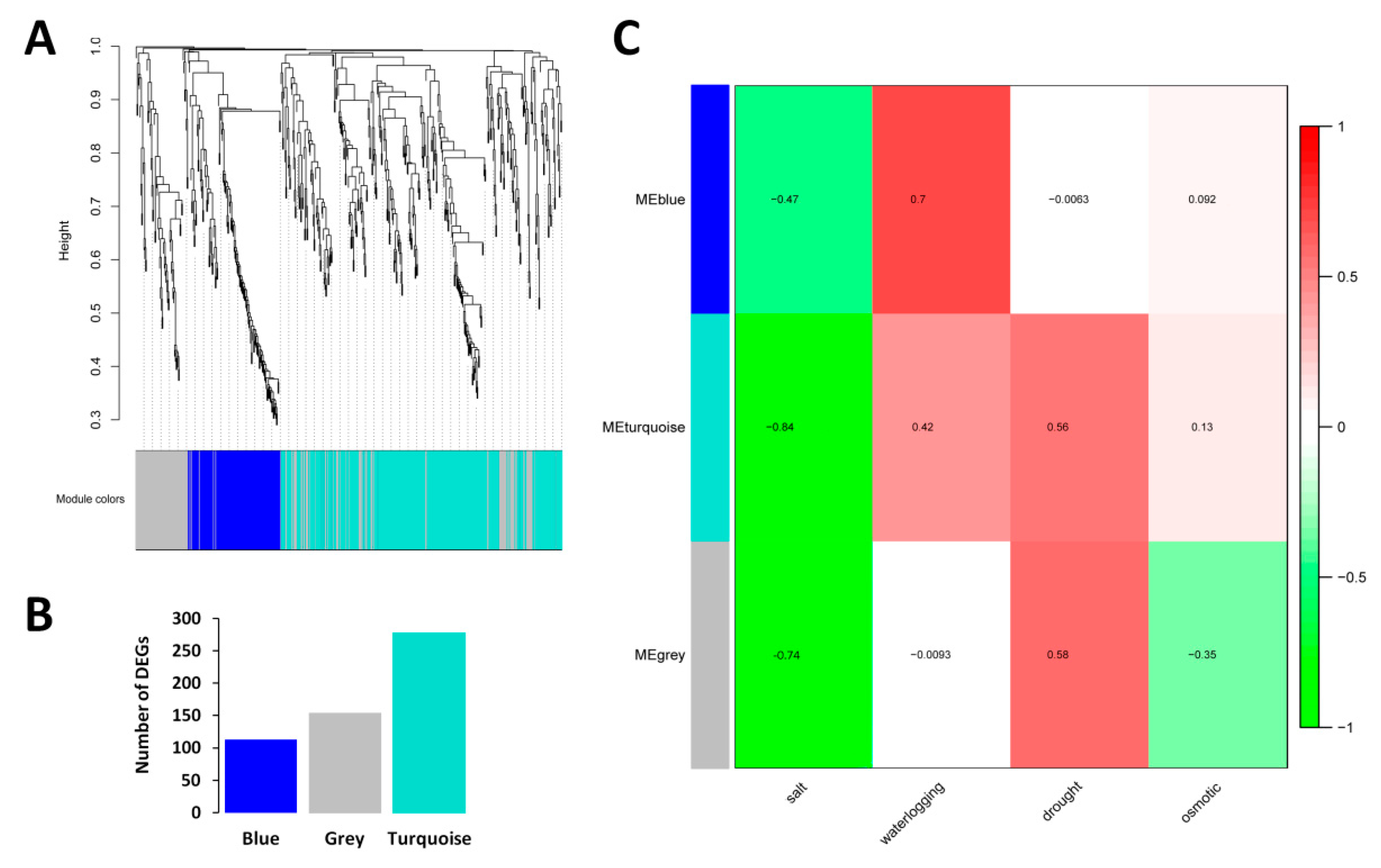
2.3. WGCNA and Detection of Functional Modules
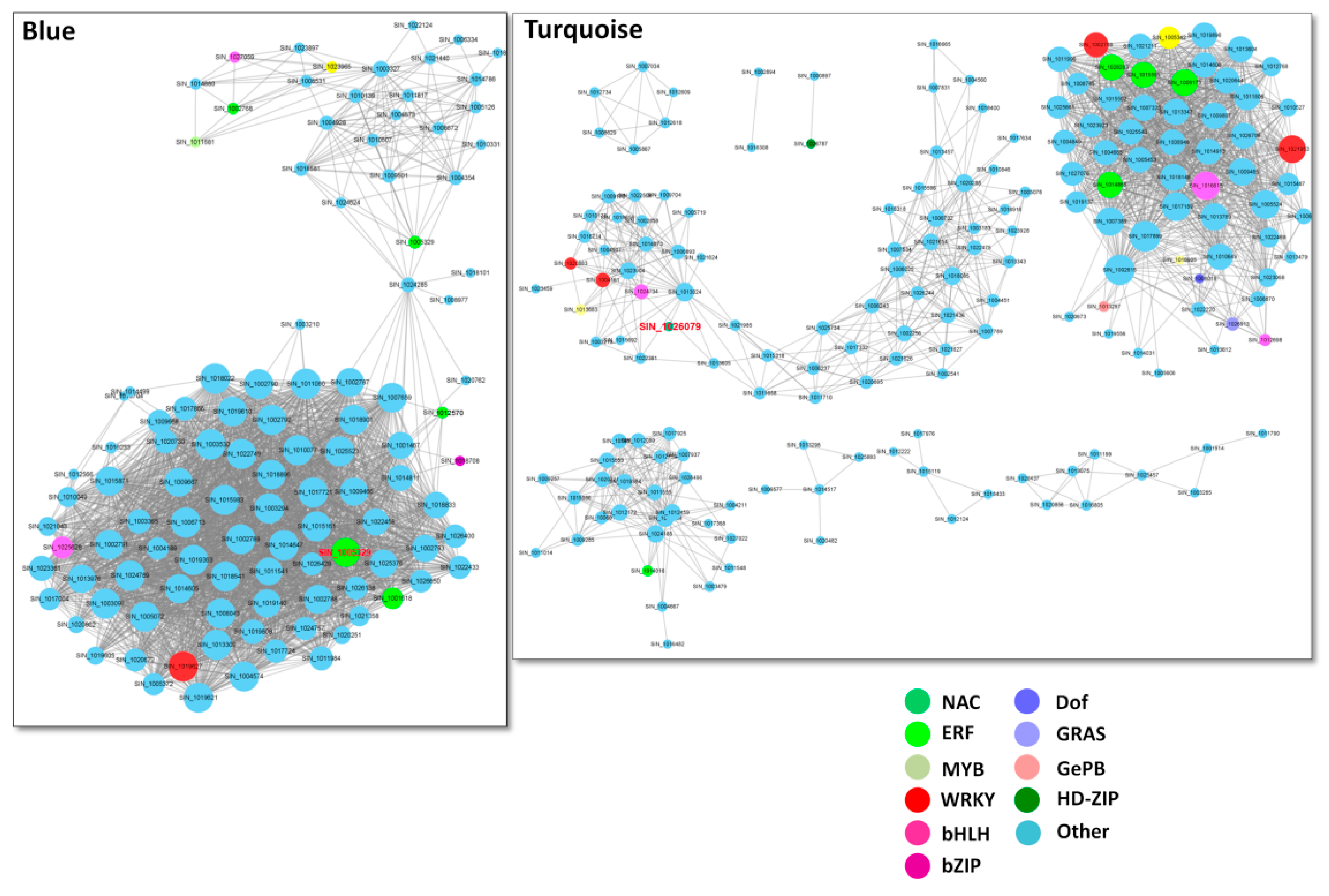
2.4. Networks Displaying Relationships among Genes within Co-Expressed Modules
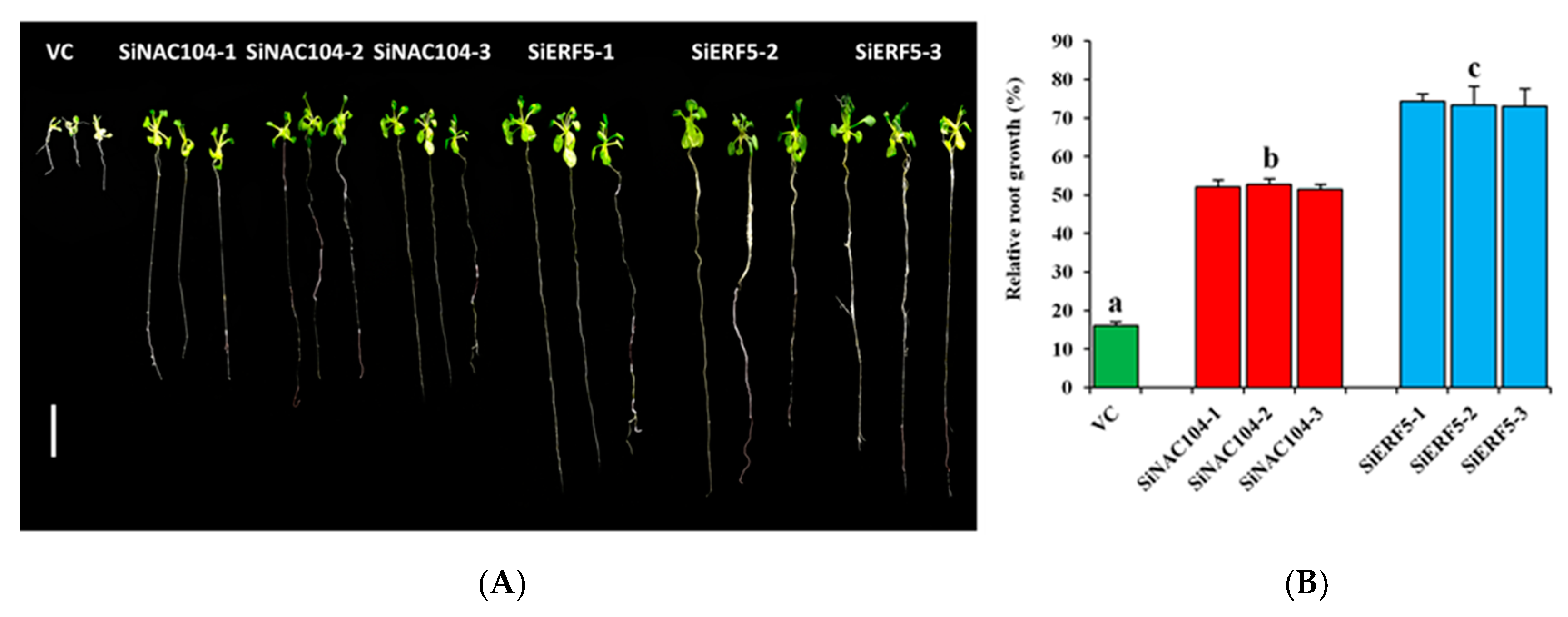
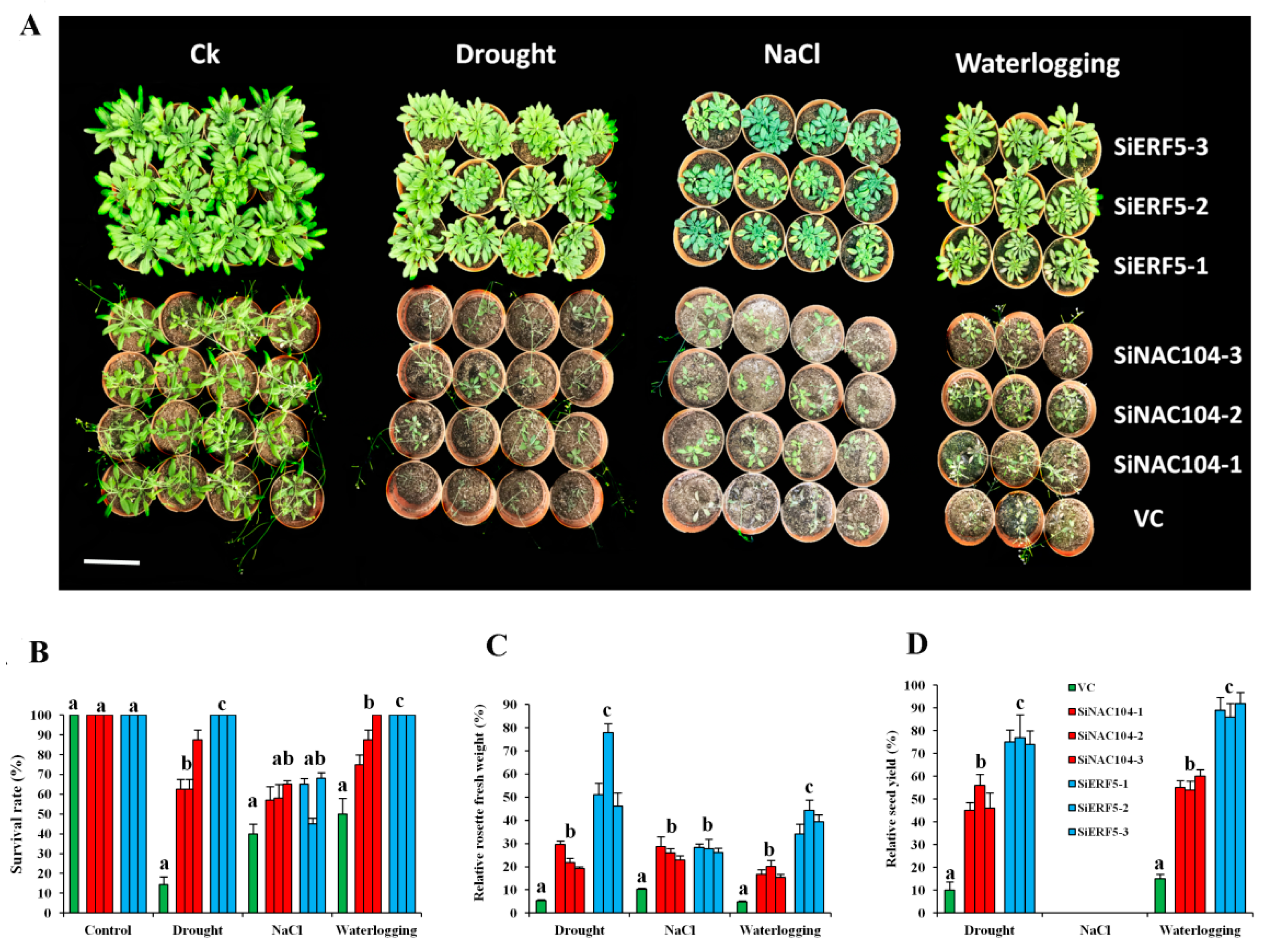
2.5. Validation of Hub and Non-Hub TFs from the Co-Expressed Modules of the Sesame CARGs in Transgenic Arabidopsis
3. Discussion
4. Materials and Methods
4.1. RNA-Sequencing Datasets of Abiotic Stressed Sesame Samples
4.2. Total RNA Isolation and Sequencing from the PEG6000-Treated Seedlings
4.3. RNA-Seq Data Analysis
4.4. Analysis of Co-Expression Modules Based on WGCNA
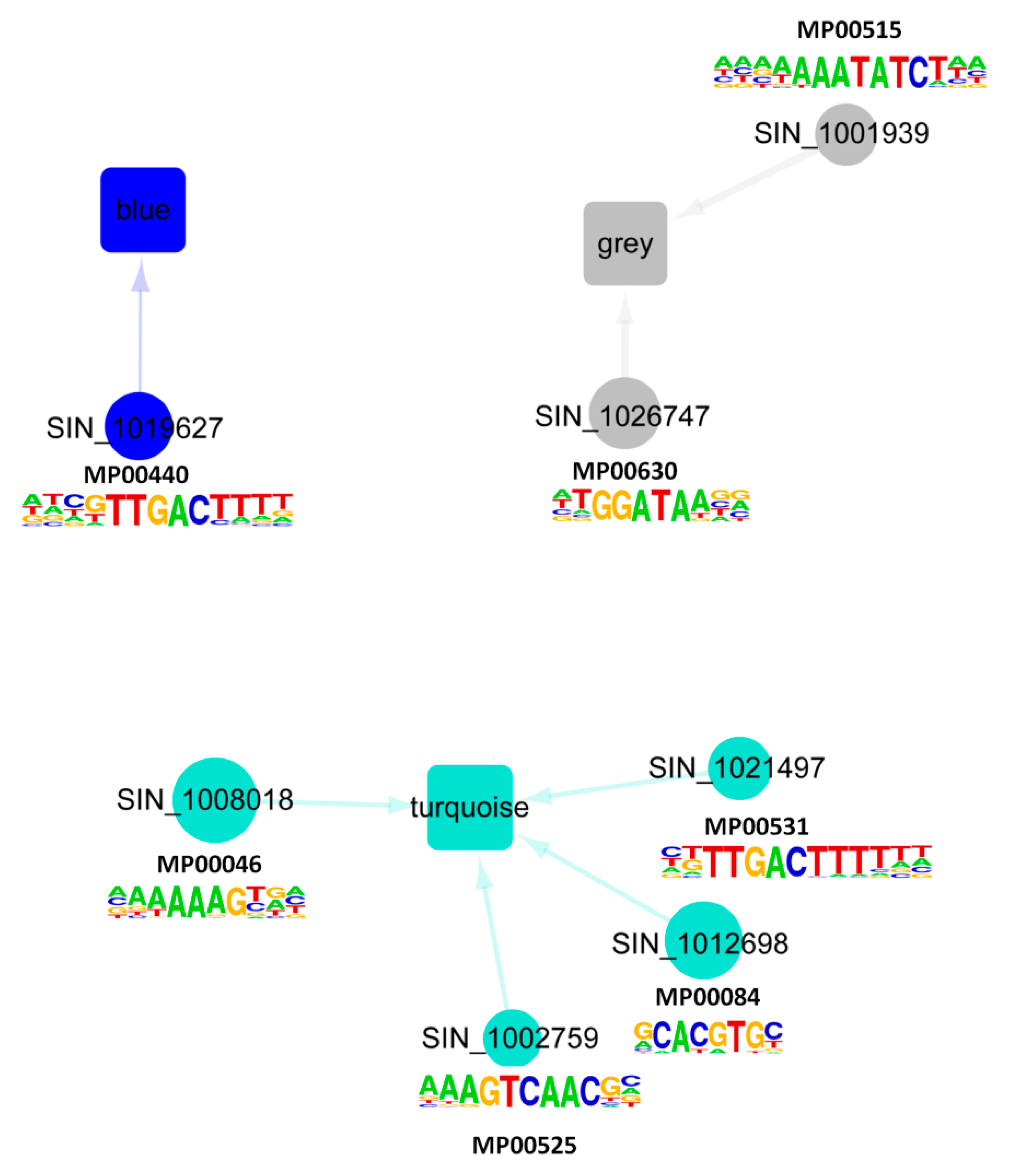
4.5. Enrichment Analysis of Cis-Regulatory Motifs
4.6. Vector Construction and Arabidopsis Genetic Transformation
4.7. Evaluation of Transgenic Lines Exposed to Osmotic, Salt, Waterlogging and Drought Stresses
4.8. Sesame Materials and Stress Treatments
4.9. Gene Expression Analysis in Arabidopsis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Foley, J.A.; Ramankutty, N.; Brauman, K.A.; Cassidy, E.S.; Gerber, J.S.; Johnston, M.; Mueller, N.D.; O’Connell, C.; Ray, D.K.; West, P.C.; et al. Solutions for a cultivated planet. Nature 2011, 478, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K. Regulons involved in osmotic stress-responsive and cold stress-responsive gene expression in plants. Physiol. Plant 2006, 126, 62–71. [Google Scholar] [CrossRef]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought stress responses in crops. Funct. Integr. Genom. 2014, 14, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, D.; Zhang, Y.; Gao, Y.; Yu, J.; Wei, X.; Zhang, X. Tolerant and susceptible sesame genotypes reveal waterlogging stress response patterns. PLoS ONE 2016, 11, e0149912. [Google Scholar] [CrossRef] [PubMed]
- Dossa, K.; Li, D.; Wang, L.; Zheng, X.; Liu, A.; Yu, J.; Wei, X.; Zhou, R.; Fonceka, D.; Diouf, D.; et al. Transcriptomic, biochemical and physio-anatomical investigations shed more light on responses to drought stress in two contrasting sesame genotypes. Sci. Rep. 2017, 7, 8755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, D.; Zhou, R.; Wang, X.; Dossa, K.; Wang, L.; Zhang, Y.; Yu, J.; Gong, H.; Zhang, X.; et al. Transcriptome and metabolome analyses of two contrasting sesame genotypes reveal the crucial biological pathways involved in rapid adaptive response to salt stress. BMC Plant Biol. 2019, 1, 66. [Google Scholar] [CrossRef]
- Mittler, R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006, 11, 15–19. [Google Scholar] [CrossRef]
- Prasad, P.V.V.; Pisipati, S.R.; Momcilovic, I.; Ristic, Z. Independent and combined effects of high temperature and drought stress during grain filling on plant yield and chloroplast EF-Tu expression in spring wheat. J. Agron. Crop Sci. 2011, 197, 430–441. [Google Scholar] [CrossRef]
- Takeda, S.; Matsuoka, M. Genetic approaches to crop improvement: Responding to environmental and population changes. Nat. Rev. Genet. 2008, 9, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Johnson, S.N.; Gregory, P.J. Implications of climate change for diseases, crop yields and food security. Euphytica 2011, 179, 3–18. [Google Scholar] [CrossRef]
- Hu, W.; Ding, Z.; Tie, W.; Yan, Y.; Liu, Y.; Wu, C.; Liu, J.; Wang, J.; Peng, M.; Xu, B.; et al. Comparative physiological and transcriptomic analyses provide integrated insight into osmotic, cold, and salt stress tolerance mechanisms in banana. Sci. Rep. 2017, 7, 43007. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed]
- Causton, H.C.; Ren, B.; Koh, S.S.; Harbison, C.T.; Kanin, E.; Jennings, E.G.; Lee, T.I.; True, H.L.; Lander, E.S.; Young, R.A. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 2001, 12, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Toone, W.M.; Mata, J.; Lyne, R.; Burns, G.; Kivinen, K.; Brazma, A.; Jones, N.; Bahler, J. Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell 2003, 14, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P. Comparative genomics of the environmental stress response in ascomycete fungi. Yeast 2007, 24, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.C.; Kissinger, M.T.; Burke, P.V.; Kwast, K.E. Comparison of the transcriptomic “stress response” evoked by antimycin A and oxygen deprivation in Saccharomyces cerevisiae. BMC Genom. 2008, 9, 627. [Google Scholar] [CrossRef]
- Kim, D.; Kim, M.S.; Cho, K.H. The core regulation module of stress-responsive regulatory networks in yeast. Nucleic Acids Res. 2012, 40, 8793–8802. [Google Scholar] [CrossRef] [Green Version]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. Cell Mol. Biol. 2007, 50, 347–363. [Google Scholar] [CrossRef]
- Wanke, D.; Berendzen, K.W.; Kilian, J.; Harter, K. Insights into the Arabidopsis abiotic stress response from the AtGenExpress expression profile dataset. In Plant Stress Biology; Hirst, H., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar] [CrossRef]
- Hahn, A.; Kilian, J.; Mohrholz, A.; Ladwig, F.; Peschke, F.; Dautel, R.; Harter, K.; Berendzen, K.W.; Wanke, D. Plant core environmental stress response genes are systemically coordinated during abiotic stresses. Int. J. Mol. Sci. 2013, 14, 7617–7641. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Danyluk, J.; Charron, J.B.; Sarhan, F. Expression profiling and bioinformatic analyses of a novel stress-regulated multispanning transmembrane protein family from cereals and Arabidopsis. Plant Physiol. 2003, 132, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Mangelsen, E.; Kilian, J.; Harter, K.; Jansson, C.; Wanke, D.; Sundberg, E. Transcriptome analysis of high-temperature stress in developing barley caryopses: Early stress responses and effects on storage compound biosynthesis. Mol. Plant 2011, 4, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Tanaka, M.; Seki, M.; Ham le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Differential gene expression in soybean leaf tissues at late developmental stages under drought stress revealed by genome-wide transcriptome analysis. PLoS ONE 2012, 7, e49522. [Google Scholar] [CrossRef] [PubMed]
- Priest, H.D.; Fox, S.E.; Rowley, E.R.; Murray, J.R.; Michael, T.P.; Mockler, T.C. Analysis of global gene expression in Brachypodium distachyon reveals extensive network plasticity in response to abiotic stress. PLoS ONE 2014, 9, e87499. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Leach, J.E. Abiotic and biotic stresses induce a core transcriptome response in rice. Sci. Rep. 2019, 9, 6273. [Google Scholar] [CrossRef]
- Noguchi, T.; Ikeda, K.; Sasaki, Y.; Yamamoto, J.; Yamori, Y. Effects of vitamin E and sesamin on hypertension and cerebral thrombogenesis in stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2001, 24, 735–742. [Google Scholar] [CrossRef]
- Sankar, D.; Sambandam, G.; Ramakrishna, R.M.; Pugalendi, K.V. Modulation of blood pressure, lipid profiles and redox status in hypertensive patients taking different edible oils. Clin. Chem. Acta 2005, 355, 97–104. [Google Scholar] [CrossRef]
- Cheng, F.C.; Jinn, T.R.; Hou, R.C.W.; Tzen, J.T.C. Neuroprotective effects of sesamin and sesamolin on gerbil brain in cerebral ischemia. Int. J. Biomed. Sci. 2006, 2, 284–288. [Google Scholar]
- Hibasami, H.; Fujikawa, T.; Takeda, H.; Nishibe, S.; Satoh, T.; Fujisawa, T.; Nakashima, K. Induction of apoptosis by Acanthopanax senticosus HARMS and its component, sesamin in human stomach cancer KATO III cells. Oncol. Rep. 2000, 7, 1213–1216. [Google Scholar] [CrossRef]
- Miyahara, Y.; Hibasami, H.; Katsuzaki, H.; Imai, K.; Komiya, T. Sesamolin from sesame seed inhibits proliferation by inducing apoptosis in human lymphoid leukemia Molt 4B cells. Int. J. Mol. Med. 2001, 7, 369–371. [Google Scholar] [CrossRef]
- Dossa, K.; Diouf, D.; Wang, L.; Wei, X.; Zhang, Y.; Niang, M.; Fonceka, D.; Yu, J.; Mmadi, M.A.; Yehouessi, L.W.; et al. The emerging oilseed crop Sesamum indicum enters the “Omics” era. Front. Plant Sci. 2017, 8, 1154. [Google Scholar]
- Sarkar, P.K.; Khatun, A.; Singha, A. Effect of duration of water-logging on crop stand and yield of sesame. Int. J. Innov. Appl. Stud. 2016, 14, 1–6. [Google Scholar]
- Witcombe, J.R.; Hollington, P.A.; Howarth, C.J.; Reader, S.M.; Steele, K. Breeding for abiotic stresses for sustainable agriculture. Philos. Trans. B 2008, 363, 703–716. [Google Scholar] [CrossRef]
- Zhang, H.; Miao, H.; Ju, M. Potential for adaptation to climate change through genomic breeding in sesame. In Genomic Designing of Climate-Smart Oilseed Crops; Kole, C., Ed.; Springer Nature: Switzerland, Basel, 2019; pp. 371–444. [Google Scholar] [CrossRef]
- Li, D.; Dossa, K.; Zhang, Y.; Wei, X.; Wang, L.; Zhang, Y.; Liu, A.; Zhou, R.; Zhang, X. GWAS uncovers differential genetic bases for drought and salt tolerances in sesame at the germination stage. Genes 2018, 9, 87. [Google Scholar] [CrossRef]
- Dossa, K.; Li, D.; Yu, J.; Wang, L.; Zhang, Y.; You, J.; Zhou, R.; Mmadi, M.A.; Li, A.; Fonceka, D.; et al. The genetic basis of drought tolerance in the high oil crop Sesamum indicum. Plant Biotechnol. J. 2019, 1–16. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shahan, R.; Zawora, C.; Wight, H.; Sittmann, J.; Wang, W.; Mount, S.M.; Liu, Z. Consensus coexpression network analysis identifies key regulators of flower and fruit development in wild strawberry. Plant Physiol. 2018, 178, 202–216. [Google Scholar] [CrossRef]
- Childs, K.L.; Davidson, R.M.; Buell, C.R. Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS ONE 2011, 6, e22196. [Google Scholar] [CrossRef]
- Weston, D.J.; Karve, A.A.; Gunter, L.E.; Jawdy, S.S.; Yang, X.; Allen, S.M.; Wullschleger, S.D. Comparative physiology and transcriptional networks underlying the heat shock response in Populus trichocarpa, Arabidopsis thaliana and Glycine max. Plant Cell Environ. 2011, 34, 1488–1506. [Google Scholar] [CrossRef]
- Downs, G.S.; Bi, Y.M.; Colasanti, J.; Wu, W.; Chen, X.; Zhu, T.; Rothstein, S.J.; Lukens, L.N. A developmental transcriptional network for Zea mays defines coexpression modules. Plant Physiol. 2013, 161, 1830–1843. [Google Scholar] [CrossRef]
- Ma, S.; Ding, Z.; Li, P. Maize network analysis revealed gene modules involved in development, nutrients utilization, metabolism, and stress response. BMC Plant Biol. 2017, 17, 131. [Google Scholar] [CrossRef]
- Shen, P.; Hour, A.; Liu, L.D. Microarray meta-analysis to explore abiotic stress-specific gene expression patterns in Arabidopsis. Bot. Stud. 2017, 58, 22. [Google Scholar] [CrossRef] [Green Version]
- He, H.; He, L. Heme oxygenase 1 and abiotic stresses in plants. Acta Physiol. Plant. 2013, 36, 581–588. [Google Scholar] [CrossRef]
- Tripathi, D.K.; Singh, S.; Gaur, S.; Singh, S.; Yadav, V.; Liu, S.; Singh, V.P.; Sharma, S.; Srivastava, P.; Prasad, S.M.; et al. Acquisition and homeostasis of iron in higher plants and their probable role in abiotic stress tolerance. Front. Environ. Sci. 2018, 5, 86. [Google Scholar] [CrossRef]
- Zhu, Y.-N.; Shi, D.-Q.; Ruan, M.-B.; Zhang, L.-L.; Meng, Z.-H.; Liu, J.; Yang, W.-C. Transcriptome analysis reveals crosstalk of responsive genes to multiple abiotic stresses in cotton (Gossypium hirsutum L.). PLoS ONE 2013, 8, e80218. [Google Scholar] [CrossRef]
- Tseng, G.C.; Ghosh, D.; Feingold, E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012, 40, 3785–3799. [Google Scholar] [CrossRef] [Green Version]
- Shaar-Moshe, L.; Hübner, S.; Peleg, Z. Identification of conserved drought-adaptive genes using a cross-species meta-analysis approach. BMC Plant Biol. 2015, 15, 111. [Google Scholar] [CrossRef]
- Ashrafi-Dehkordi, E.; Alemzadeh, A.; Tanaka, N.; Razi, H. Meta-analysis of transcriptomic responses to biotic and abiotic stress in tomato. PeerJ 2018, 6, e4631. [Google Scholar] [CrossRef]
- Tahmasebi, A.E.; Ashrafi-Dehkordi, A.G.; Shahriari, S.M.; Mazloomi, E. Ebrahimie Integrative meta-analysis of transcriptomic responses to abiotic stress in cotton. Prog. Biophys. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Olveracarrillo, Y.; Campos, F.; Reyes, J.L.; Garciarrubio, A.; Covarubias, A.A. Functional analysis of the group 4 Late Embryogenesis Abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 2010, 154, 373–390. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, H.; Cheng, W.; Liu, Y.; Cheng, X.; Sun, J.; Gill, S.S.; Tuteja, N. Polyamines and abiotic stress tolerance in plants. Plant Signal. Behav. 2010, 5, 26–33. [Google Scholar] [Green Version]
- Huang, Z.; Zhong, X.J.; He, J.; Jin, S.H.; Guo, H.D.; Yu, X.F.; Zhou, Y.J.; Li, X.; Ma, M.D.; Chen, Q.B.; et al. Genome-wide identification, characterization, and stress-responsive expression profiling of genes encoding lea (Late Embryogenesis Abundant) proteins in moso bamboo (Phyllostachys edulis). PLoS ONE 2016, 11, e0165953. [Google Scholar] [CrossRef]
- Muvunyi, B.P.; Yan, Q.; Wu, F.; Min, X.; Yan, Z.Z.; Kanzana, G.; Wang, Y.; Zhang, J. Mining Late Embryogenesis Abundant (LEA) family genes in Cleistogenes songorica, a xerophyte perennial desert plant. Int. J. Mol. Sci. 2018, 19, 3430. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Zhang, B.; Yi, J.; Yang, Y.; Chunyan, K.; Lei, C.; Gong, M. The role of the Late Embryogenesis Abundant (LEA) protein family in development and the abiotic stress response: A comprehensive expression analysis of potato (Solanum tuberosum). Genes 2019, 10, 148. [Google Scholar] [CrossRef]
- Chang, C.; Meyerowitz, E.M. Molecular cloning and DNA sequence of the Arabidopsis thaliana alcohol dehydrogenase gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1408–1412. [Google Scholar] [CrossRef]
- Chung, H.J.; Ferl, R.J. Arabidopsis alcohol dehydrogenase expression in both shoots and roots is conditioned by root growth environment. Plant Physiol. 1999, 121, 429–436. [Google Scholar] [CrossRef]
- Matton, D.P.; Constabel, P.; Brisson, N. Alcohol dehydrogenase gene expression in potato following elicitor and stress treatment. Plant Mol. Biol. 1990, 14, 775–783. [Google Scholar] [CrossRef]
- Christie, P.J.; Hahn, M.; Walbot, V. Low-temperature accumulation of alcohol dehydrogenase-1 mRNA and protein activity in maize and rice seedlings. Plant Physiol. 1991, 95, 699–706. [Google Scholar] [CrossRef]
- Tesniere, C.; Torregrosa, L.; Pradal, M.; Souquet, J.-M.; Gilles, C.; Santos, K.D.; Chatelet, P.; Gunata, Z. Effects of genetic manipulation of alcohol dehydrogenase levels on the response to stress and the synthesis of secondary metabolites in grapevine leaves. J. Exp. Bot. 2006, 57, 91–99. [Google Scholar] [CrossRef]
- Yang, C.Y. Hydrogen peroxide controls transcriptional responses of ERF73/HRE1 and ADH1 via modulation of ethylene signaling during hypoxic stress. Planta 2014, 239, 877–885. [Google Scholar] [CrossRef]
- Shi, H.; Liu, W.; Yao, Y.; Wei, Y.; Chan, Z. Alcohol dehydrogenase 1 (ADH1) confers both abiotic and biotic stress resistance in Arabidopsis. Plant Sci. 2017, 262, 24–31. [Google Scholar] [CrossRef]
- Sauter, M.; Rzewuski, G.; Marwedel, T.; Lorbiecke, R.A. The novel ethylene-regulated gene OsUsp1 from rice encodes a member of plant protein family related to prokaryotic universal stress proteins. J. Exp. Bot. 2002, 53, 2325–2331. [Google Scholar] [CrossRef]
- Sun, W.; Van Montagu, M.; Verbruggen, N. Small heat shock proteins and stress tolerance in plants. Biochim. Biophys. Acta 2002, 157, 1–9. [Google Scholar] [CrossRef]
- Dubouzet, J.G.; Sakuma, Y.; Ito, Y.; Kasuga, M.; Dubouzet, E.G.; Miura, S.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. OsDREB genes in rice, Oryza sativa L.; encode transcription activators that function in drought, high salt and cold responsive gene expression. Plant J. 2003, 33, 751–763. [Google Scholar] [CrossRef]
- Kawano, T. Role of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell Rep. 2003, 21, 829–837. [Google Scholar]
- Mittova, V.; Guy, M.; Tal, M.; Volokita, M. Salinity up-regulates the antioxidative system in root mitochondria and peroxisomes of the wild salt-tolerant tomato species Lycopersicon pennellii. J. Exp. Bot. 2004, 55, 1105–1113. [Google Scholar] [CrossRef]
- Valerio, L.; Meyer, M.D.; Penel, C.; Dunand, C. Expression analysis of the Arabidopsis peroxidase multigene family. Phytochemistry 2004, 65, 1331–1342. [Google Scholar] [CrossRef]
- Passardi, F.; Cosio, C.; Penel, C.; Dunand, C. Peroxidases have more functions than a Swiss army knife. Plant Cell Rep. 2005, 24, 255–265. [Google Scholar] [CrossRef]
- Zahur, M.; Maqbool, A.; Irfan, M.; Barozai, M.Y.K.; Rashid, B.; Riazuddin, S.; Husnain, T. Isolation and functional analysis of cotton universal stress protein promoter in response to phytohormones and abiotic stresses. Mol. Biol. 2009, 43, 578–585. [Google Scholar] [CrossRef]
- Chen, J.H.; Jiang, H.W.; Hsieh, E.J.; Chen, H.Y.; Chien, C.T.; Hsieh, H.L.; Lin, T.P. Drought and salt stress tolerance of an Arabidopsis glutathione S-transferase U17 knockout mutant are attributed to the combined effect of glutathione and abscisic acid. Plant Physiol. 2012, 158, 340–351. [Google Scholar] [CrossRef]
- Yoon, G.M.; Kieber, J.J. 1-Aminocyclopropane-1-carboxylic acid as a signaling molecule in plants. AoB Plants 2013, 5, plt017. [Google Scholar] [CrossRef]
- Chen, D.; Ma, X.; Li, C.; Zhang, W.; Xia, G.; Wang, M. A wheat aminocyclopropane-1-carboxylate oxidase gene, TaACO1, negatively regulates salinity stress in Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 1815–1827. [Google Scholar] [CrossRef]
- Gonzali, S.; Loreti, E.; Cardarelli, F.; Novi, G.; Parlanti, S.; Pucciariello, C.; Bassolino, L.; Banti, V.; Licausi, F.; Perata, P. Universal stress protein HRU1 mediates ROS homeostasis under anoxia. Nat. Plants 2015, 1, 15151. [Google Scholar] [CrossRef]
- Udawat, P.; Jha, R.K.; Sinha, D.; Mishra, A.; Jha, B. Overexpression of a cytosolic abiotic stress responsive universal stress protein (SbUSP) mitigates salt and osmotic stress in transgenic tobacco plants. Front. Plant Sci. 2016, 7, 518. [Google Scholar] [CrossRef]
- Mishra, D.; Shekhar, S.; Singh, D.; Chakraborty, S.; Chakraborty, N. Heat shock protein and abiotic stress tolerance in plants. In Regulation of Heat Shock Protein Responses; Asea, A., Kaur, P., Eds.; Springer: Cham, Switzerland, 2018; Volume 13, pp. 41–69. [Google Scholar]
- Kumar, S.; Trivedi, P.K. Gluthatione S-transferase: Role in combating abiotic stresses including arsenic detoxification in plants. Front. Plant Sci. 2018, 9, 751. [Google Scholar] [CrossRef]
- Anirban, B. Bananas tackling drought and heat- with DREBs and more. Physiol. Plant. 2019, 165, 128–130. [Google Scholar]
- Jangale, B.L.; Chaudhari, R.S.; Azeez, A.; Sane, P.V.; Sane, A.P.; Krishna, B. Independent and combined abiotic stresses affect the physiology and expression patterns of DREB genes differently in stress-susceptible and resistant genotypes of banana. Physiol. Plant 2019, 165, 303–318. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, Z.; Xie, B.; Chen, Q.; Tian, X.; Zhang, X.; Zhang, H.; Lu, X.; Huang, D.; Huang, R. The ethylene-, jasmonate-, abscisic acid-and NaCl-responsive tomato transcription factor JERF1 modulates expression of GCC box-containing genes and salt tolerance in tobacco. Planta 2004, 220, 262–270. [Google Scholar] [CrossRef]
- Mishra, M.; Das, R.; Pandey, G.K. Role of ethylene responsive factors (ERFs) in abiotic stress mediated signaling in plants. J. Biol. Sci. 2009, 1, 133–146. [Google Scholar]
- Zhang, Z.; Li, F.; Li, D.; Zhang, H.; Huang, R. Expression of ethylene response factor JERF1 in rice improves tolerance to drought. Planta 2010, 232, 765–774. [Google Scholar] [CrossRef]
- Müller, M.; Munné-Bosh, S. Ethylene response factors: A key regulatory hub in hormone and stress signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef]
- Klay, I.; Gouia, S.; Liu, M.; Mila, I.; Khoudi, H.; Bernadac, A.; Bouzayen, M.; Pirrello, J. Ethylene response factors (ERF) are differentially regulated by different abiotic stress types in tomato plants. Plant Sci. 2018, 274, 137–145. [Google Scholar] [CrossRef]
- Li, C.; Ng, C.K.Y.; Fan, L.M. MYB transcription factors, active players in abiotic stress signaling. Environ. Exp. Bot. 2015, 114, 80–91. [Google Scholar] [CrossRef]
- Roy, S. Function of MYB domain transcription factors in abiotic stress and epigenetic control of stress response in plant genome. Plant Signal Behav. 2016, 11, e1117723. [Google Scholar] [CrossRef] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Sui, N. Transcription regulation of bHLH during plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 397–401. [Google Scholar] [CrossRef]
- Beer, M.A.; Tavazoie, S. Predicting gene expression from sequence. Cell 2004, 117, 185–198. [Google Scholar] [CrossRef]
- Danko, C.G.; Pertsov, A.M. Identification of gene co-regulatory modules and associated cis-elements involved in degenerative heart disease. BMC Med. Genom. 2009, 2, 31. [Google Scholar] [CrossRef]
- Helsen, J.; Frickel, J.; Jeller, R.; Verstrepen, K.J. Network hubs affect evolvability. PLoS Biol. 2019, 17, e3000111. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, S.; Tong, C.; Zhao, Y.; Liu, Y.; Song, C.; Zhang, Y.; Zhang, X.; Wang, Y.; Hua, W.; et al. Genome sequencing of the high oil crop sesame. Genome Biol. 2014, 15, R39. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Tarazona, S.; Garcia-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.; Tang, J.; Han, Y.; Wang, D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp. Eye Res. 2018, 166, 13–20. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Wei, B.; Peng, C.; Wang, L.; Hu, G.; Kong, D.; Du, C. Candidate biomarkers and molecular mechanism investigation for Glioblastoma multiforme utilizing WGCNA. BioMed Res. Int. 2018, 2018, 4246703. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 1–24. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef]
- Dossa, K.; Mmadi, M.A.; Zhou, R.; Zhou, Q.; Yang, M.; Cisse, N.; Diouf, D.; Wang, L.; Zhang, X. The contrasting response to drought and waterlogging is underpinned by divergent DNA methylation programs associated with transcript accumulation in sesame. Plant Sci. 2018, 277, 207–217. [Google Scholar]
- Raineri, J.; Ribichich, K.F.; Chan, R.L. The sunflower transcription factor HaWRKY76 confers drought and flood tolerance to Arabidopsis thaliana plants without yield penalty. Plant Cell Rep. 2015, 34, 2065–2080. [Google Scholar] [CrossRef]
- Lalitha, S. Primer premier 5. Biotech Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Salt | Drought | Waterlogging | Osmotic | Total |
|---|---|---|---|---|---|
| Number of samples | 30 | 24 | 6 | 12 | 72 |
| Clean reads (Gb) | 230 | 160 | 13.5 | 83.1 | 486.6 |
| Total expressed genes | 23,415 | 24,113 | 21,064 | 22,418 | 25,319 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dossa, K.; Mmadi, M.A.; Zhou, R.; Zhang, T.; Su, R.; Zhang, Y.; Wang, L.; You, J.; Zhang, X. Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2019, 20, 3930. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163930
Dossa K, Mmadi MA, Zhou R, Zhang T, Su R, Zhang Y, Wang L, You J, Zhang X. Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.). International Journal of Molecular Sciences. 2019; 20(16):3930. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163930
Chicago/Turabian StyleDossa, Komivi, Marie A. Mmadi, Rong Zhou, Tianyuan Zhang, Ruqi Su, Yujuan Zhang, Linhai Wang, Jun You, and Xiurong Zhang. 2019. "Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.)" International Journal of Molecular Sciences 20, no. 16: 3930. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163930