New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
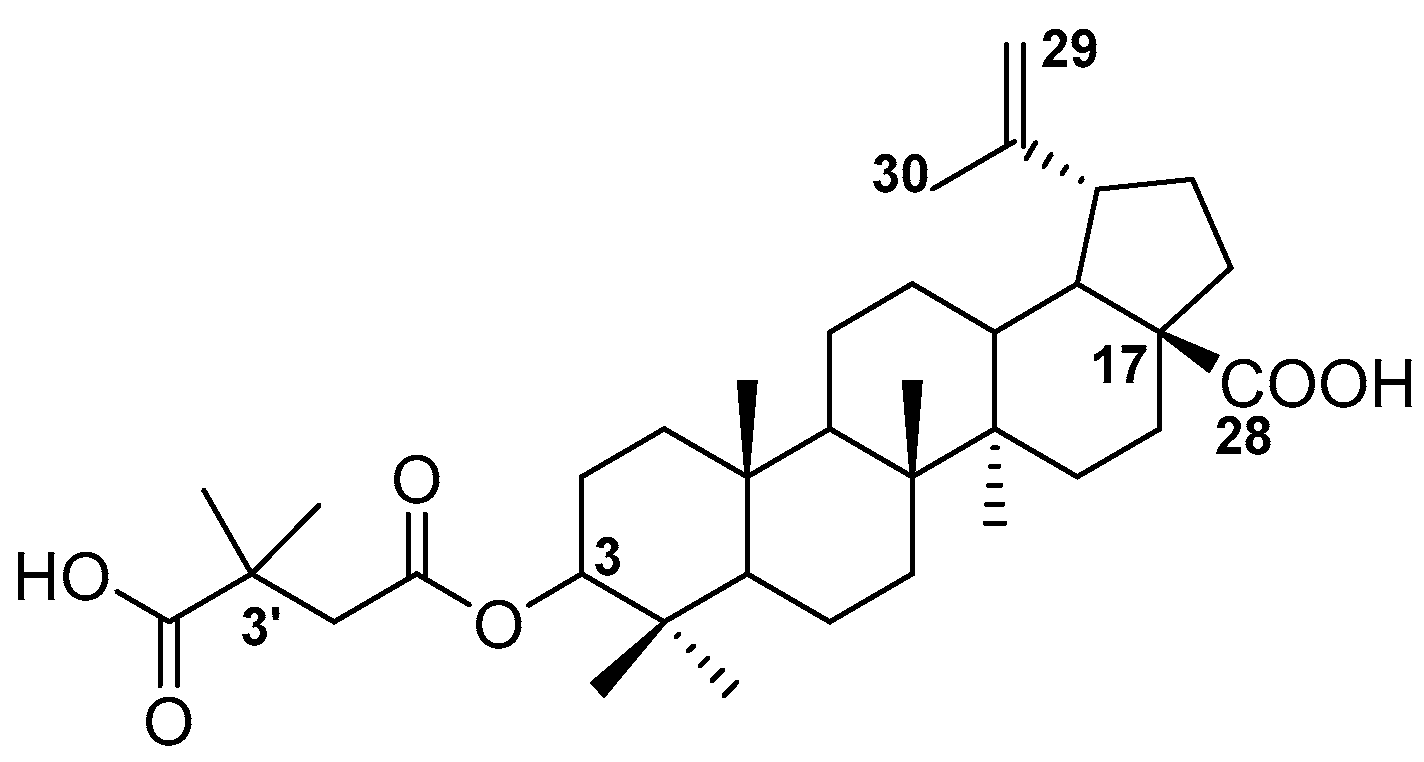
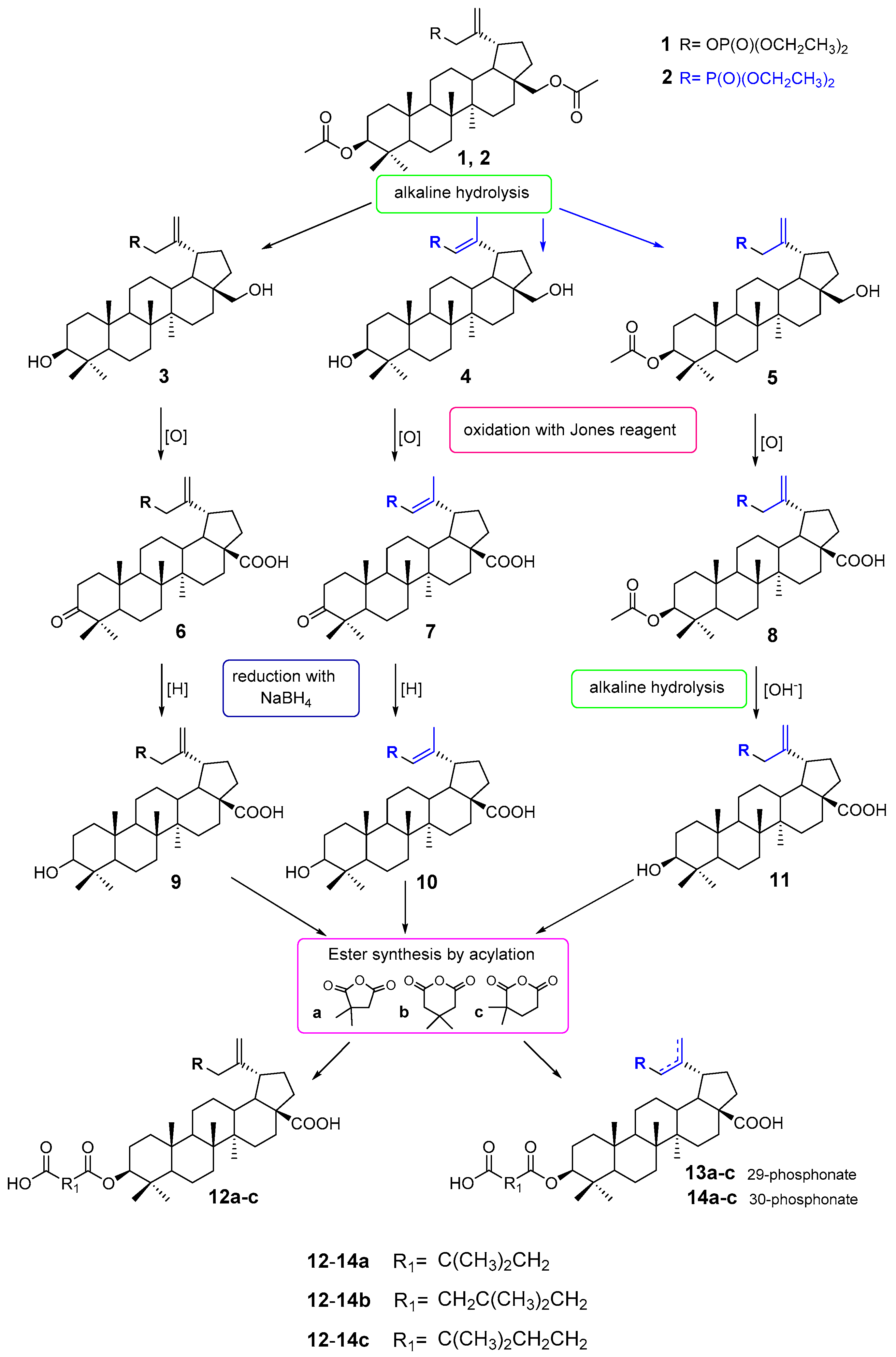
2.1. Chemistry
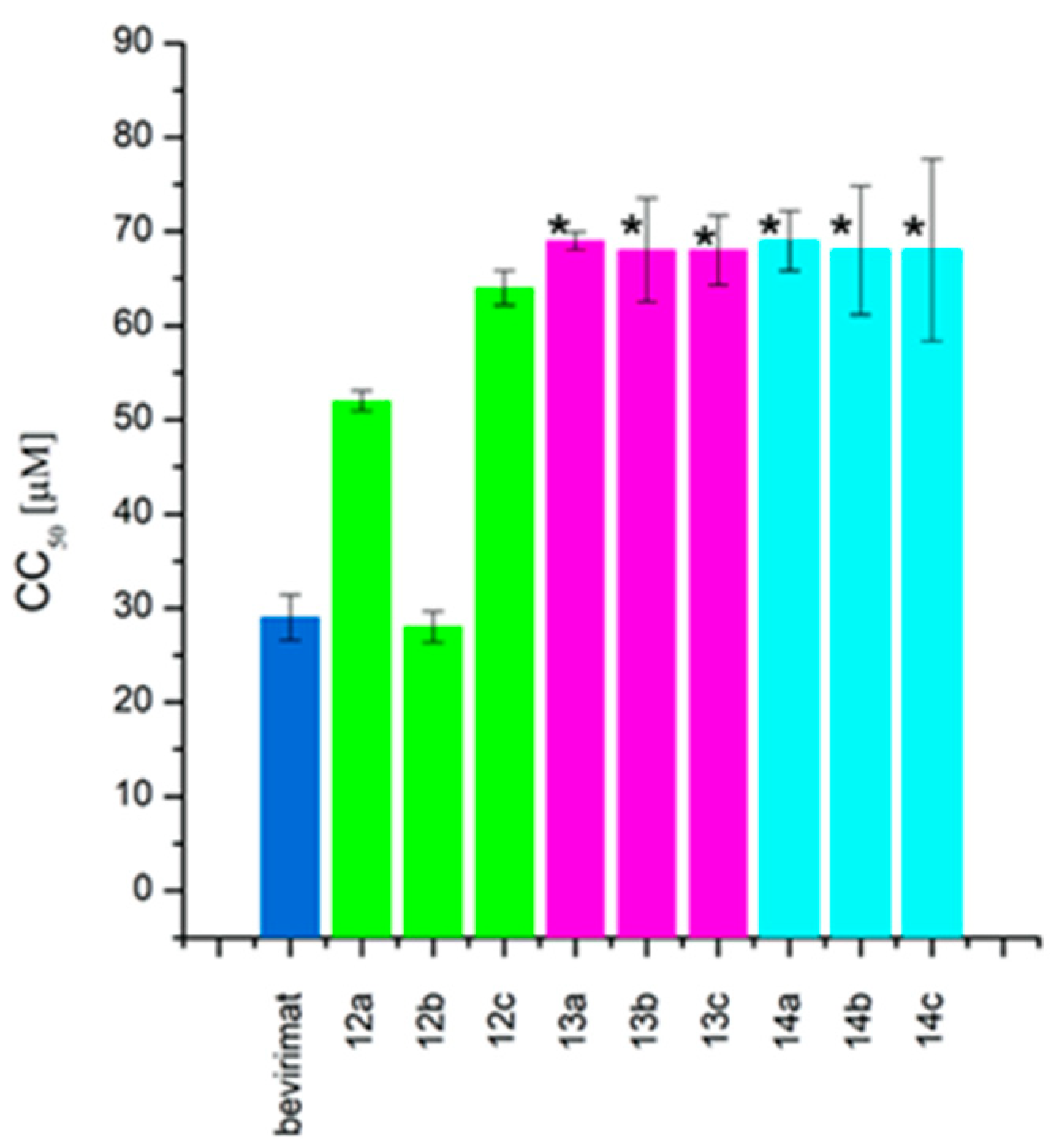
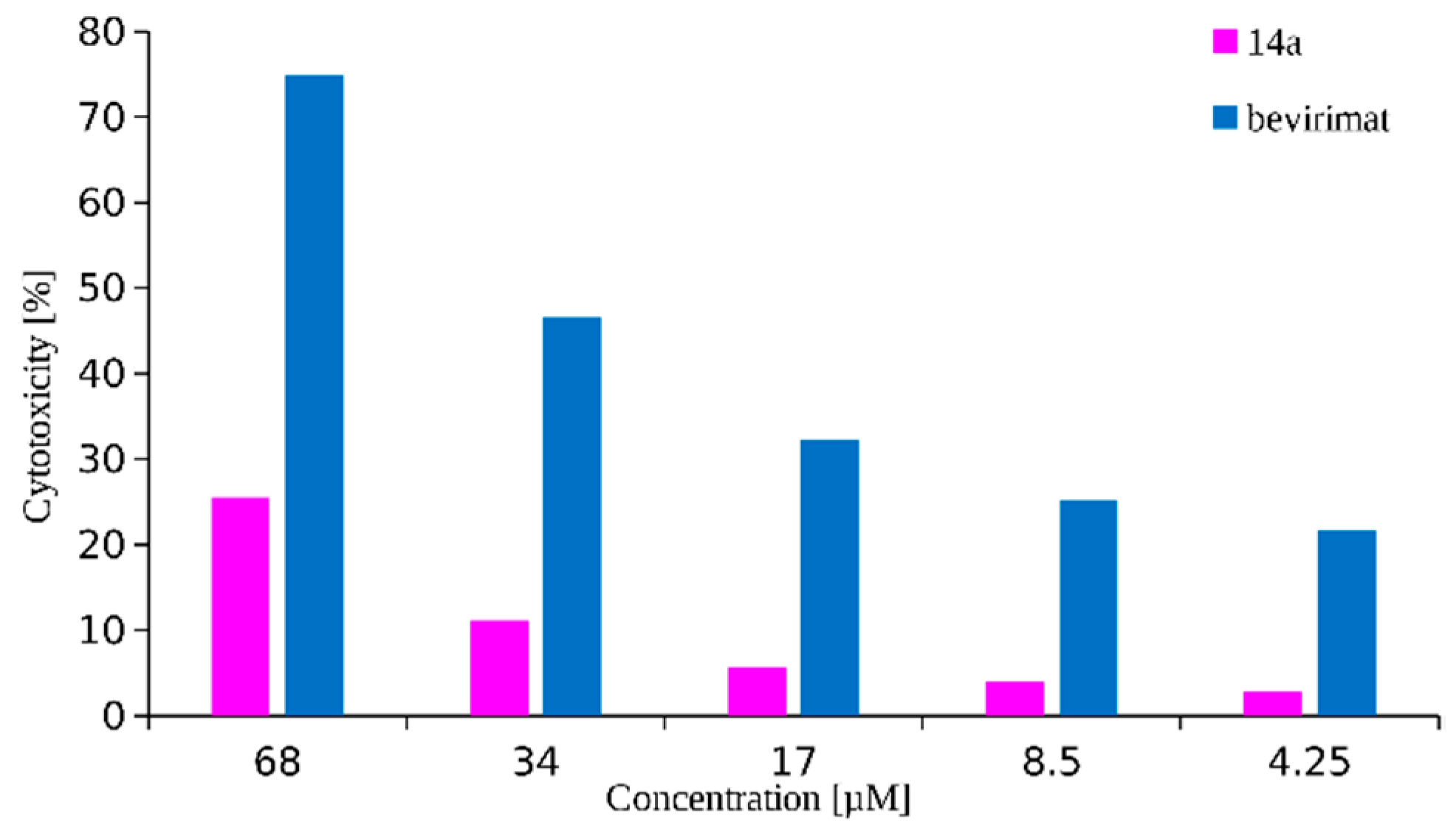
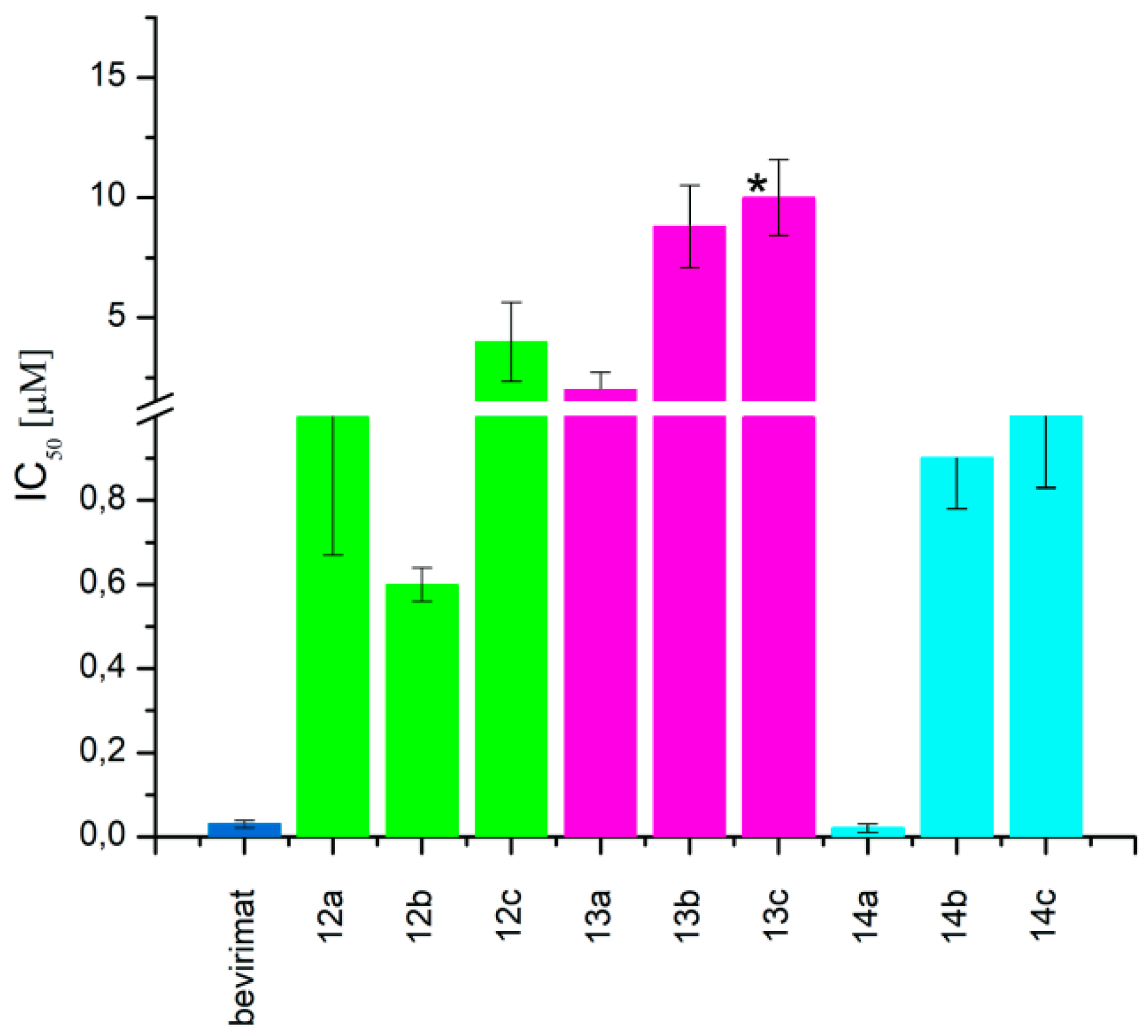
2.2. Cytotoxicity and Antiretroviral Activity
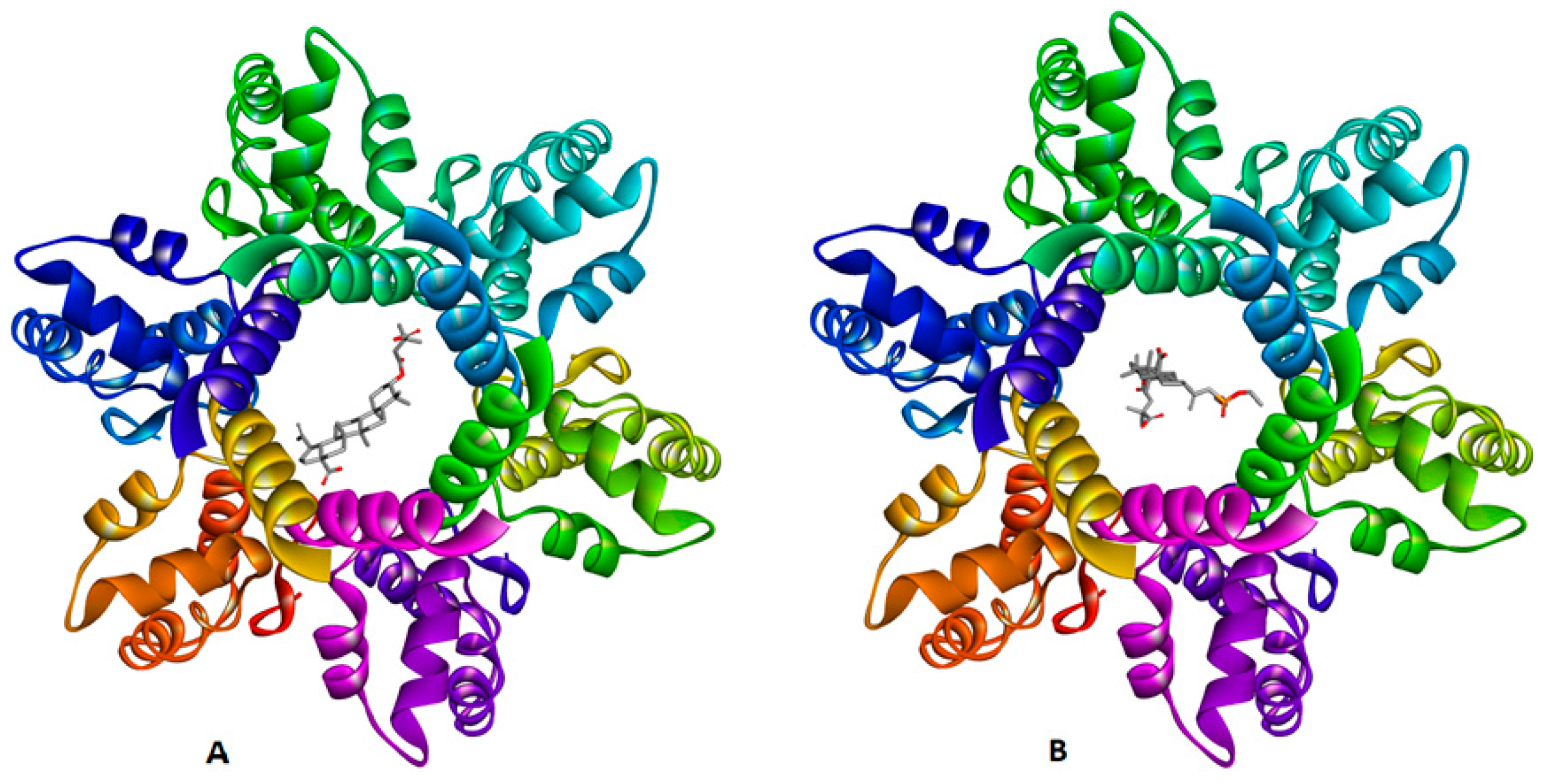
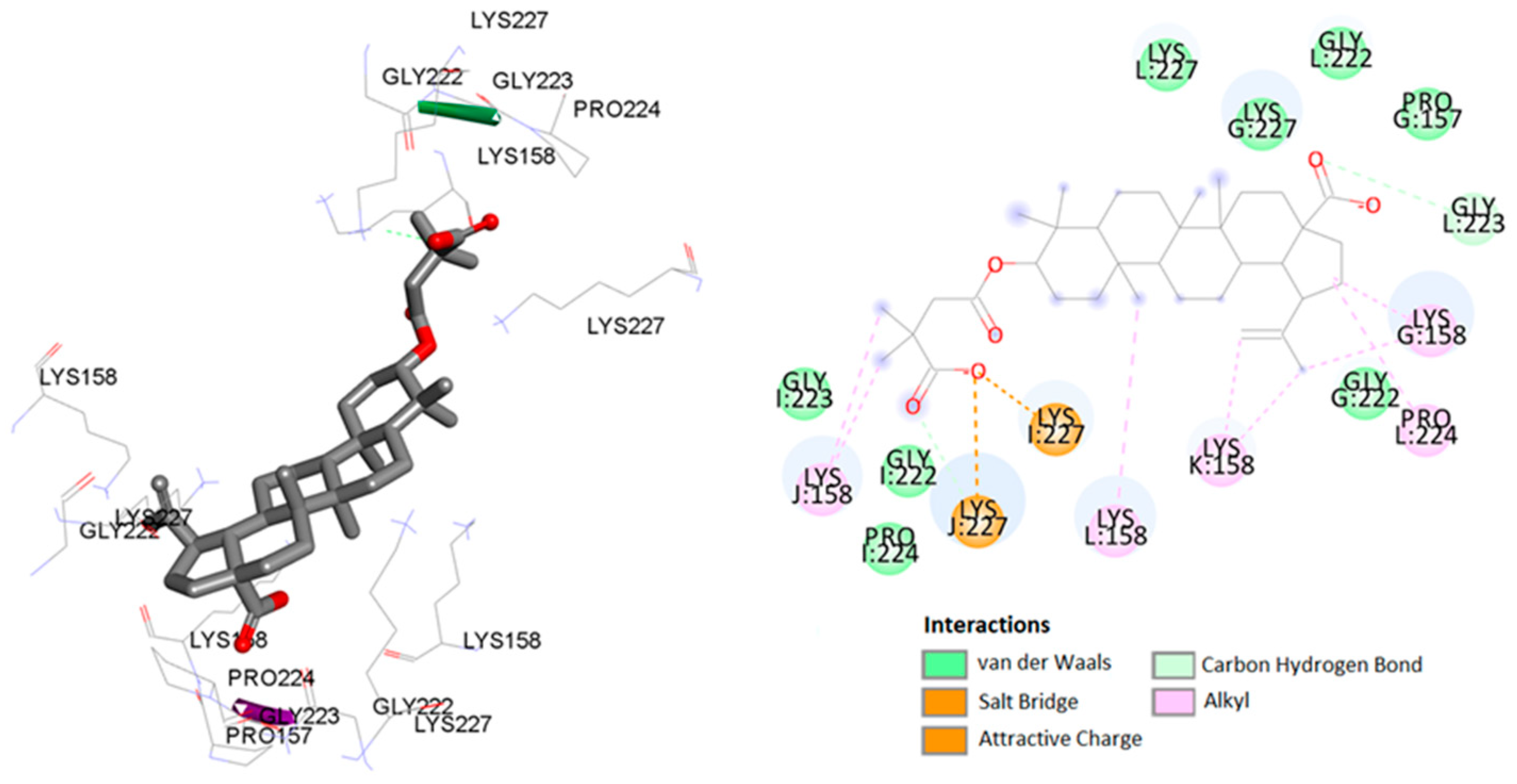
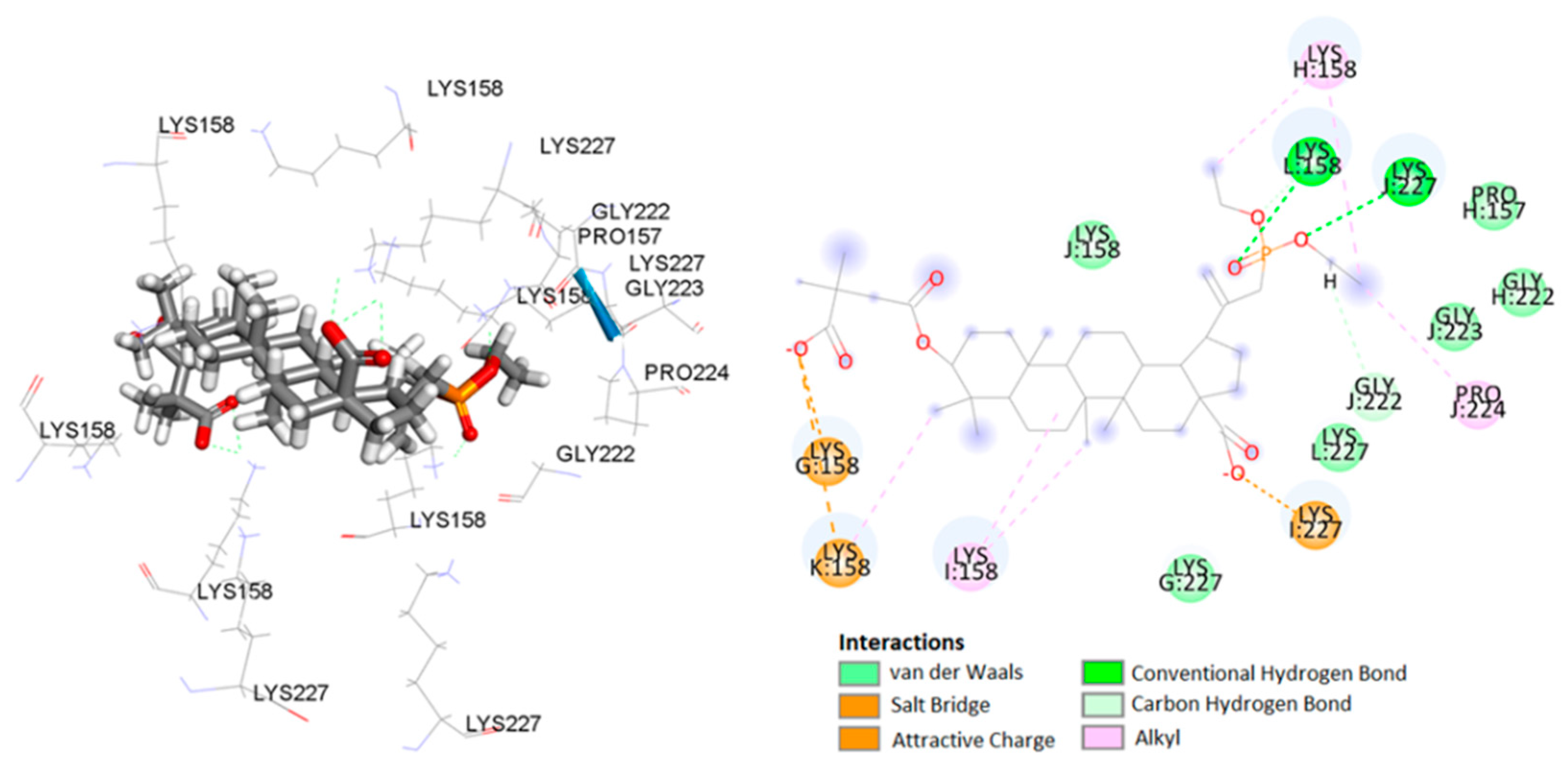
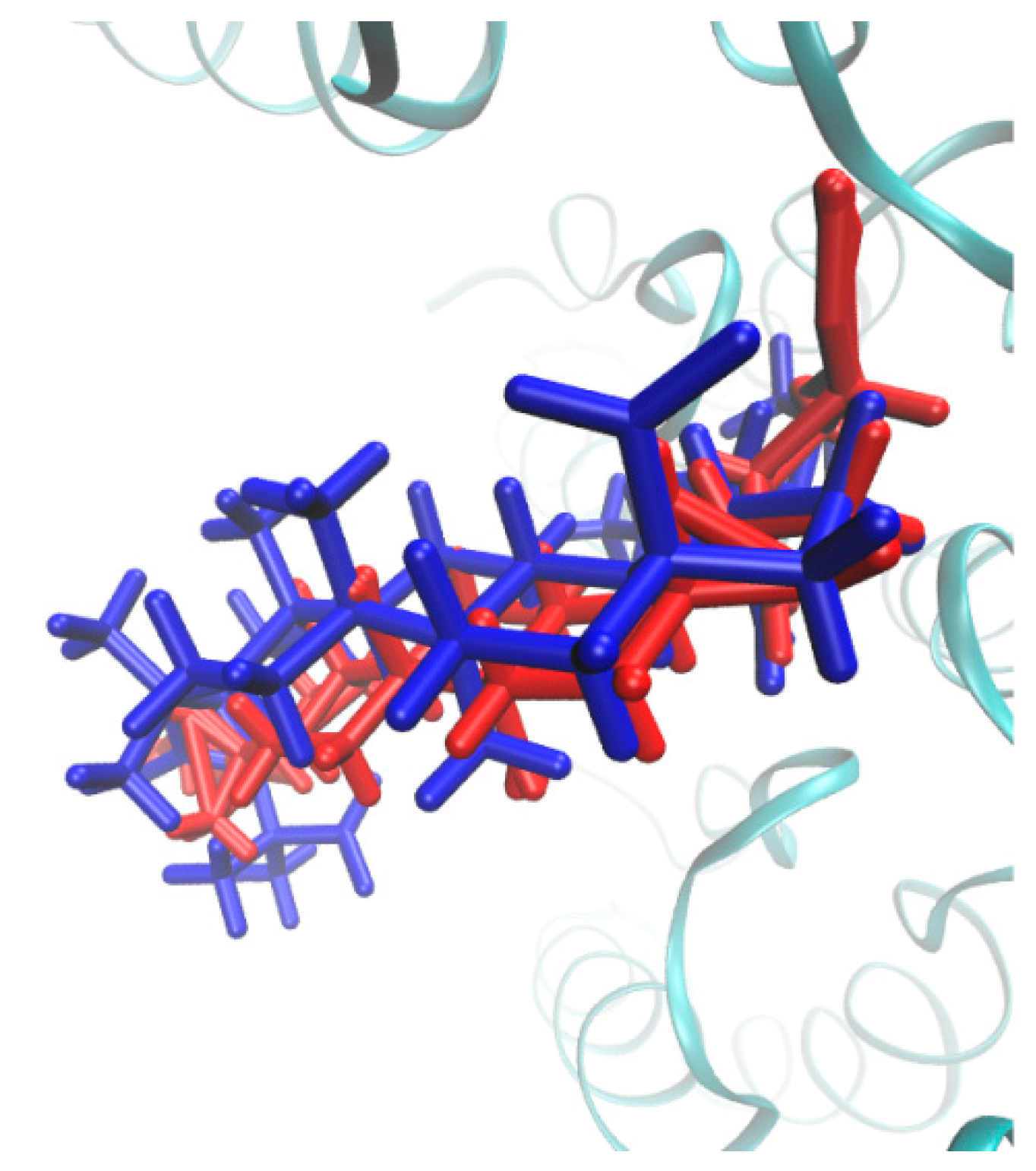
2.3. Molecular Docking Study
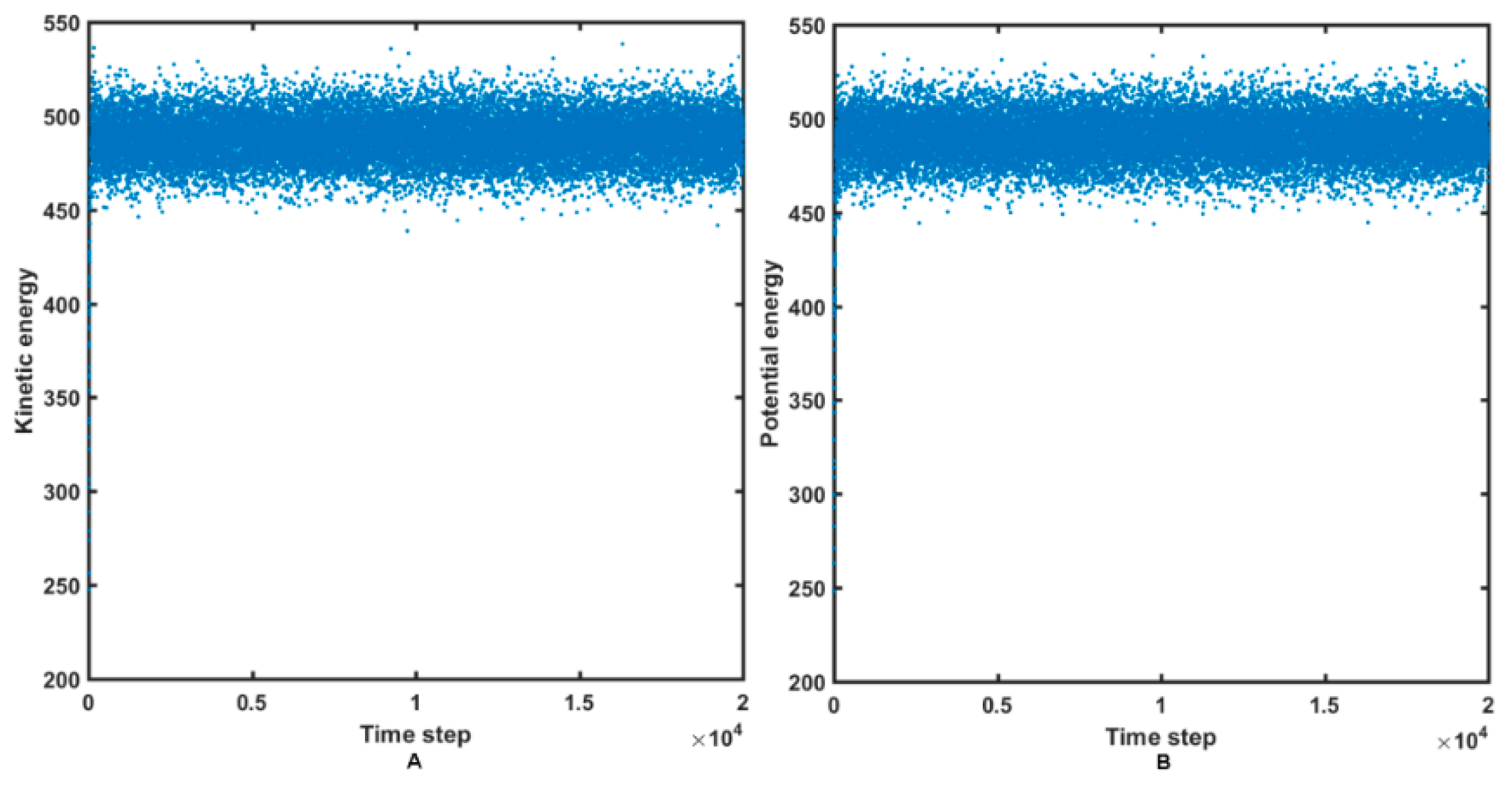
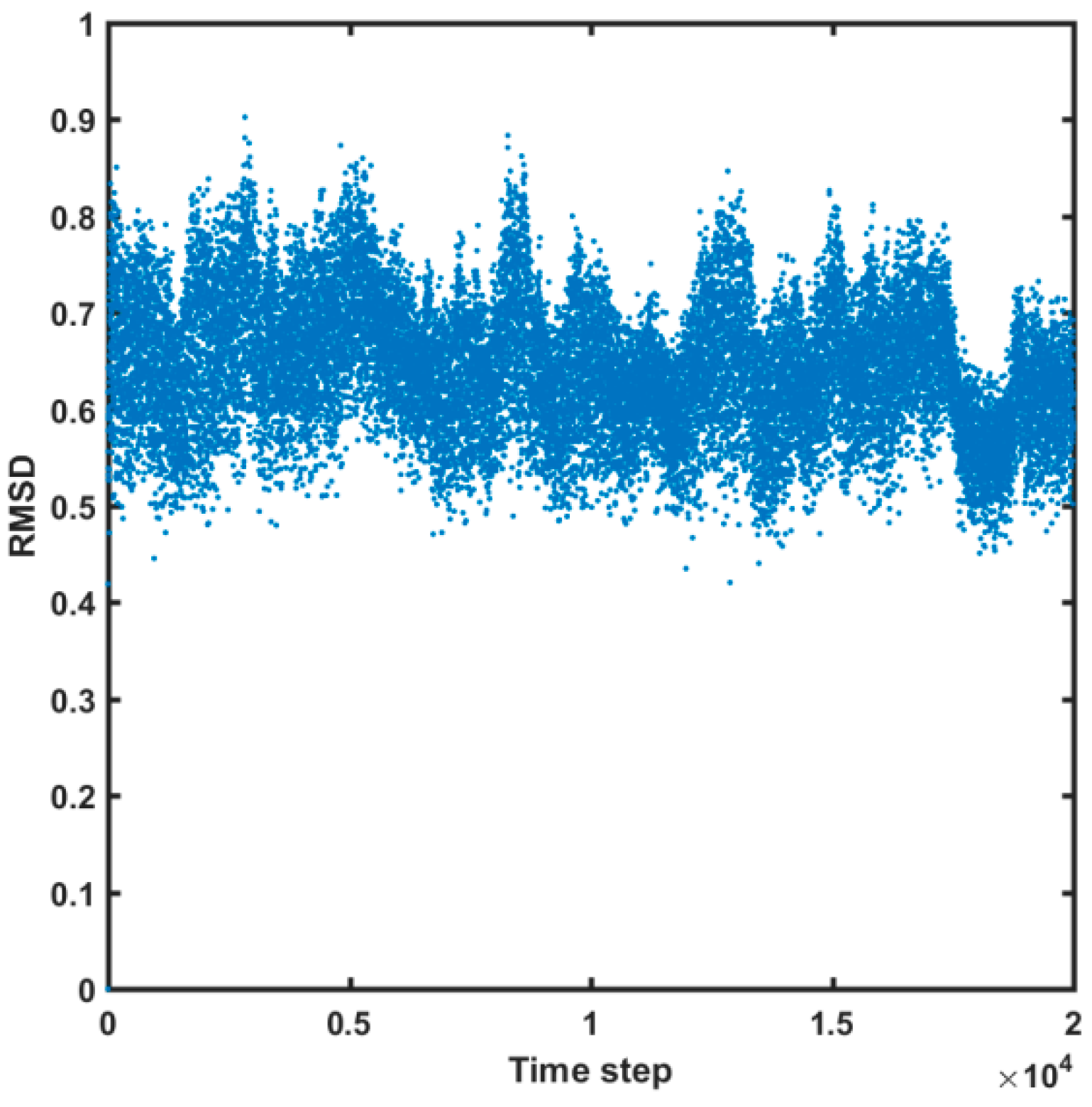
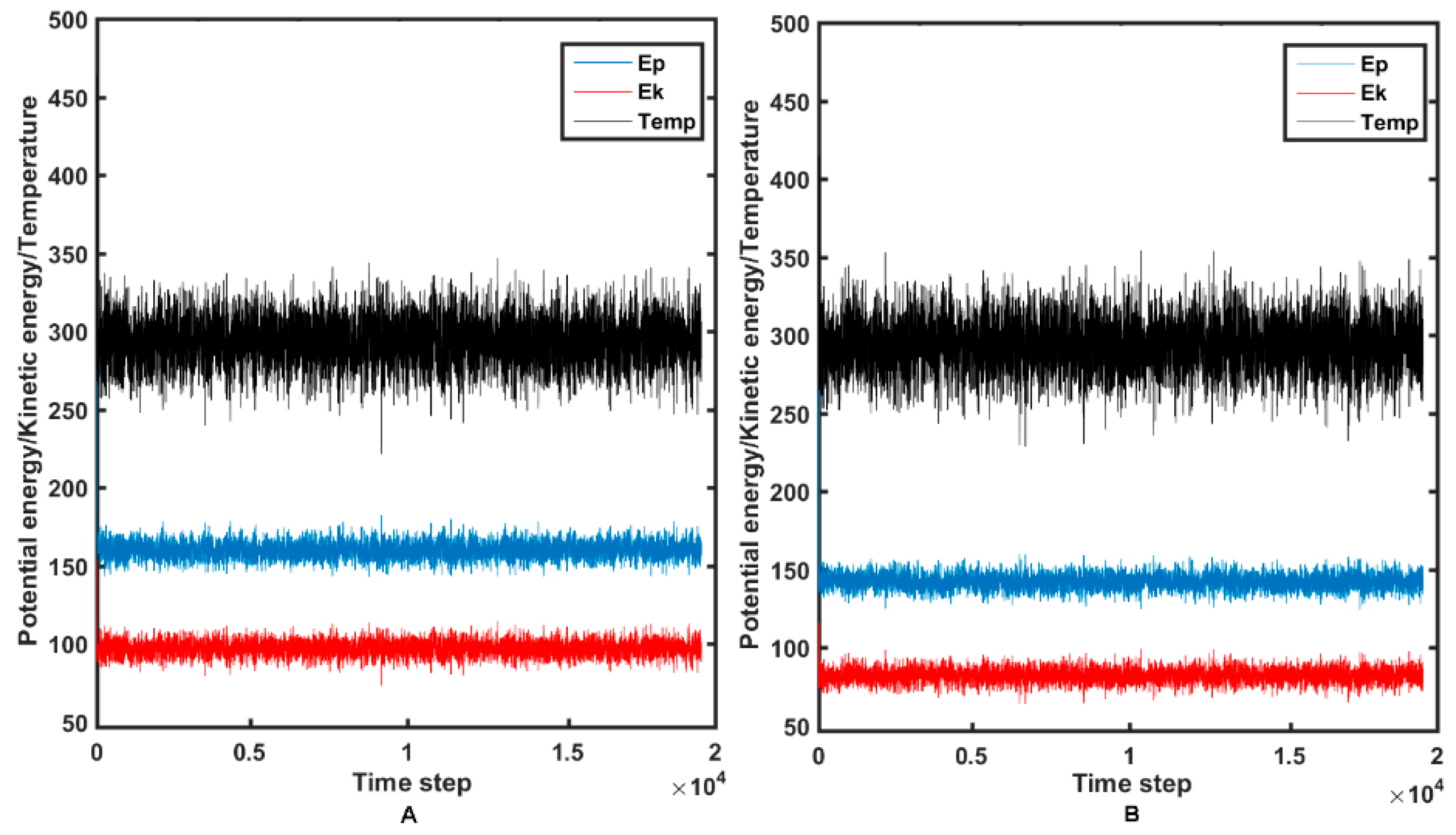
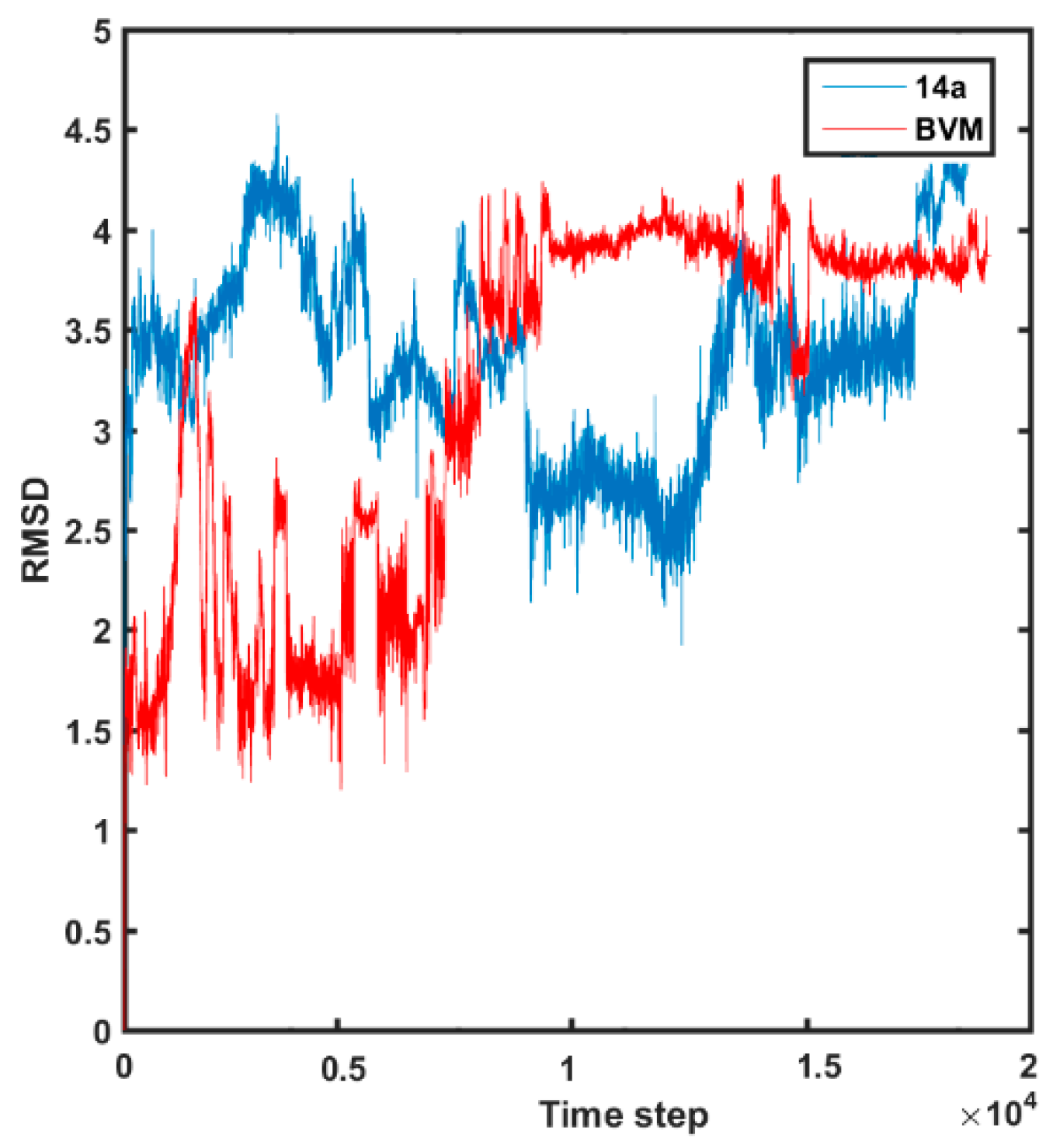
2.4. Molecular Dynamics Simulations
3. Materials and Method
3.1. Biological Activity
3.1.1. Cytotoxicity
3.1.2. Anti-HIV Activity
3.2. Synthesis
3.2.1. Synthesis of 3-Acetyl-30-Diethoxyphosphorylbetulin 5
3.2.2. General Procedures for Oxidation with the Jones Reagent
3.2.3. General Procedures for the Sodium Borohydride Reduction (Synthesis of Compounds 9 and 10)
3.2.4. Synthesis of 30-Diethoxyphosphorylbetulinic Acid 11
3.2.5. General Method of Synthesis 3-Carboxyacyl Derivatives 12a–c, 13a–c and 14a–c
3.3. Molecular Docking Study
3.4. Molecular Dynamic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BVM | Bevirimat |
| EC50 | Half maximal effective concentration—the concentration of a compound at which 50% of the Population exhibit a response |
| TI | Therapeutic index |
| CC50 | Cytotoxicity concentration of 50%—concentration required for the reduction of cell viability by 50% |
| IC50 | Half maximal inhibitory concentration—the concentration of a compound required to inhibit 50% of a specific biological or biochemical function |
| HSV | Herpes simplex virus |
| HBV | Hepatitis B virus |
| ELISA | Enzyme-linked immunosorbent assay |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2 5-diphenyltetrazolium bromide |
References
- UNAIDS DATA 2018. Available online: https://www.unaids.org/sites/default/files/media_asset/unaids-data-2018_en.pdf (accessed on 28 June 2019).
- Chrobak, E.; Kadela-Tomanek, M.; Bębenek, E.; Marciniec, K.; Wietrzyk, J.; Trynda, J.; Pawełczak, B.; Kusz, J.; Kasperczyk, J.; Chodurek, E.; et al. New phosphate derivatives of betulin as anticancer agents: Synthesis, crystal structure, and molecular docking study. Bioorg. Chem. 2019, 87, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khademe, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, T.; Kashiwada, Y. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-J.; Tan, G.T.; Hoang, V.D.; Hung, N.V.; Cuong, N.M.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H.S. Natural anti-HIV agents. part IV anti-HIV constituents from Vatica cinerea. J. Nat. Prod. 2003, 66, 263–268. [Google Scholar] [CrossRef]
- Mayaux, J.F.; Bousseau, A.; Pauwels, R.; Huet, T.; Hénin, Y.; Dereu, N.; Evers, M.; Soler, F.; Poujade, C.; De Clercq, E.; et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3564–3568. [Google Scholar] [CrossRef]
- Dang, Z.; Qian, K.; Ho, P.; Zhu, L.; Lee, K.-H.; Huang, L.; Chen, C.-H. Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1 and bevirimat-resistance HIV-1 variants. Bioorg. Med. Chem. Lett. 2012, 22, 5190–5194. [Google Scholar] [CrossRef]
- Soler, F.; Poujade, C.; Evers, M.; Carry, J.-C.; Hénin, Y.; Bousseau, A.; Huet, T.; Pauwels, R.; De Clercq, E.; Mayaux, J.-F.; et al. Betulinic acid derivatives: A new class of specific inhibitors of human immunodeficiency virus type 1 entry. J. Med. Chem. 1996, 39, 1069–1083. [Google Scholar] [CrossRef]
- Hashimoto, F.; Kashiwada, Y.; Cosentino, L.M.; Chen, C.-H.; Garrett, P.E.; Lee, K.-H. Anti-AIDS agents-XXVII. Synthesis and anti-HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorg. Med. Chem. 1997, 5, 2133–2143. [Google Scholar] [CrossRef]
- Kanamoto, T.; Kashiwada, Y.; Kanbara, K.; Gotoh, K.; Yoshimori, M.; Goto, T.; Sano, K.; Nakashima, H. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob. Agents Chemother. 2001, 45, 1225–1230. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Brik, A.; Wong, Ch.-H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- McCallister, S.; Lalezari, J.; Richmond, G.; Thompson, M.; Harrigan, R.; Martin, D.; Salzwedel, K.; Allaway, G. HIV-1 Gag polymorphisms determine treatment response to bevirimat (PA-457). Antivir. Ther. 2008, 13, A10. [Google Scholar]
- Wang, D.; Lu, W.; Li, F. Pharmacological intervention of HIV-1 maturation. Acta Pharm. Sin. B. 2015, 5, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quian, K.; Yu, D.; Chen, C.-H.; Huang, L.; Morris-Natschke, S.L.; Nitz, T.J.; Salzwedel, K.; Reddick, M.; Allaway, G.P.; Lee, K.-H. Anti-AIDS agents. 78. Design, synthesis, metabolic stability assessment, and antiviral evaluation of novel betulinic acid derivatives as potent anti-human immunodeficiency virus (HIV) agents. J. Med. Chem. 2009, 52, 3248–3258. [Google Scholar] [CrossRef] [PubMed]
- Coric, P.; Turcaud, S.; Souquet, F.; Briant, L.; Gay, B.; Royer, J.; Chazal, N.; Bouaziz, S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur. J. Med. Chem. 2013, 62, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Stacey, A.; Jones, S.A.; Jeffery, J.L.; Miranda, S.R.; Galardi, C.M.; Irlbeck, D.M.; Brown, K.W.; McDanal, C.B.; Han, N.; et al. Synthesis and biological evaluation of macrocyclized betulin derivatives as a novel class of anti-HIV-1 maturation inhibitors. Open Med. Chem. J. 2014, 8, 23–27. [Google Scholar] [CrossRef]
- Tang, J.; Jones, S.A.; Jeffrey, J.L.; Miranda, S.R.; Galardi, C.M.; Irlbeck, D.M.; Brown, K.W.; McDanal, C.B.; Johns, B.A. Discovery of a novel and potent class of anti-HIV-1 maturation inhibitors with improved virology profile against gag polymorphisms. Bioorg. Med. Chem. Lett. 2017, 27, 2689–2694. [Google Scholar] [CrossRef]
- Roussos, N.; Karageorgopoulos, D.E.; Samonis, G.; Falagas, M.E. Clinical significance of the pharmacokinetic and pharmacodynamic characteristics of fosfomycin for the treatment of patients with systemic infections. Int. J. Antimicrob. Agents 2009, 34, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Lell, B.; Ruangweerayut, R.; Wiesner, J.; Missinou, M.A.; Schindler, A.; Baranek, T.; Hintz, M.; Hutchinson, D.; Jomaa, H.; Kremsner, P.G. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 2003, 47, 735–738. [Google Scholar] [CrossRef]
- Borisova, S.A.; Circello, B.T.; Zhang, J.K.; van der Donk, W.A.; Metcalf, W.W. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem. Biol. 2010, 17, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Laber, B.; Lindell, S.D.; Pohlenz, H.-D. Inactivation of Escherichia coli threonine synthase by DL-Z-2-amino-5-phosphono-3-pentenoic acid. Arch. Microbiol. 1994, 161, 400–403. [Google Scholar] [PubMed]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Dracinsky, M.; Krečmerová, M.; Holý, A. Study of chemical stability of antivirally active 5-azacytosine acyclic nucleoside phosphonates using NMR spectroscopy. Bioorg. Med. Chem. 2008, 16, 6778–6782. [Google Scholar] [CrossRef] [PubMed]
- Broganelli, P.; Chiaretta, A.; Fragnelli, B.; Bernengo, M.G. Intralesional cidofovir for the treatment of multiple and recalcitrant cutaneous viral warts. Dermatol. Ther. 2012, 25, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-H.; Fang, J.-M.; Cheng, Y.-S.; Shie, J.-J. Zanamivir Phosphonate Congeners with Anti-Influenza Activity and Determining Oseltamivir Susceptibility of Influenza Viruses. U.S. Patent 2013/0225532 A1, 29 August 2013. [Google Scholar]
- Boojamra, C.G.; Cannizzaro, C.E.; Chen, J.M.; Chen, X.; Cho, A.; Chong, L.S.; Fardis, M.; Jin, H.; Hirshmann, R.; Huang, A.X.; et al. Gilead Sciences Inc., Antiviral Phosphonate Analogs. U.S. Patent 2015/0025039 A1, 22 January 2015. [Google Scholar]
- Feng, Q.; Lu, F.; Jiangxi Qingfeng Pharmaceutical Inc. Lupane Triterpenoid Derivatives and Pharmaceutical Use Thereof. U.S. Patent 2016/9428542 B2, 30 August 2016. [Google Scholar]
- Nitz, T.J.; Montalbetti, C.; Mears, R.; Gai, X.; Gleen, E. Extended Triterpene Derivatives. U.S. Patent 2012/0046291 A1, 23 February 2012. [Google Scholar]
- Rodriguez, J.B.; Gallo-Rodriguez, C. The role of the phosphorus atom in drug design. Chem. Med. Chem. 2019, 14, 190–216. [Google Scholar] [CrossRef]
- Yuan, H.; Li, N.; Lai, Y. Evaluation of in vitro models for screening alkaline phosphatase-mediated bioconversion of phosphate ester prodrugs. Drug Metab. Dispos. 2009, 37, 1443–1447. [Google Scholar] [CrossRef]
- Pezzuto, J.M.; Kosmeder, J.W.; Xu, Z.-Q.; Zhou, N.E.; Goldsmith, M.E. Method of Preparing and Use of Prodrugs of Betulinic Acid Dervatives. U.S. Patent 2003/6569842B2, 27 May 2003. [Google Scholar]
- Boryczka, S.; Chrobak, E.; Szymura, A.; Latocha, M.; Kadela, M.; Bębenek, E. Acetylene Derivatives of Betulin 30-phosphate with Anti-Tumor Activity, Method of Their Preparation and Application. RP Patent PL 230002B1, 13 April 2018. [Google Scholar]
- Chrobak, E.; Bębenek, E.; Kadela-Tomanek, M.; Latocha, M.; Jelsch, C.; Wenger, E.; Boryczka, S. Betulin phosphonates; synthesis, structure, and cytotoxic activity. Molecules 2016, 21, 1123. [Google Scholar] [CrossRef]
- Boryczka, S.; Chrobak, E.; Bębenek, E.; Kadela-Tomanek, M.; Dąbrowska, A.; Chilmonczyk, Z.; Wiktorska, K.; Milczarek, M.; Jastrzębska, M. Phosphonate Derivatives of 3-carboxyacylbetulinic Acid with Antiviral Activity, Method for Their Preparation and Their Application. RP Patent Application No. P.425755, 29 May 2018. [Google Scholar]
- Sun, I.-C.; Shen, J.-K.; Wang, H.-K.; Cosentino, L.M.; Lee, K.-H. Anti-AIDS agents. 32. Synthesis and anti-HIV activity of betulin derivatives. Bioorg. Med. Chem. Lett. 1998, 8, 1267–1272. [Google Scholar] [CrossRef]
- Urano, E.; Ablan, S.D.; Mandt, R.; Pauly, G.T.; Sigano, D.M.; Schneider, J.P.; Martin, D.E.; Nitz, T.J.; Wild, C.T.; Freed, E.O. Alkyl amine bevirimat derivatives are potent and broadly active HIV-1 maturation inhibitors. Antimicrob. Agents Chemother. 2016, 60, 190–197. [Google Scholar] [CrossRef]
- Yu, D.; Lee, K.-H. Recent progress and prospects on plant-derived anti-HIV agents and analogs. In Medicinal Chemistry of Bioactive Natural Products; Lian, X.-T., Fang, W.-S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 379–391. [Google Scholar]
- Aiken, C.; Chen, C.H. Betulinic acid derivatives as HIV-1 antivirals. Trends Mol. Med. 2005, 11, 31–36. [Google Scholar] [CrossRef] [PubMed]
- ACD labs 2015 Release (Build 2726. Nov 2014); Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2015.
- Wang, M.; Quinn, C.M.; Perilla, J.R.; Zhang, H.; Shirra, R., Jr.; Hou, G.; Byeon, I.-J.; Suiter, C.L.; Ablan, S.; Urano, E.; et al. Quenching protein dynamics interferes with HIV capsid maturation. Nat. Commun. 2017, 24, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Zadrozny, K.K.; Chrustowicz, J.; Purdy, M.D.; Yeager, M.; Ganser-Pornillos, B.K.; Pornillos, O. Crystal structure of an HIV assembly and maturation switch. eLife 2016, 5, e17063. [Google Scholar] [CrossRef] [PubMed]
- Voltz, K.; Trylska, J.; Tozzini, V.; Kurkal-Siebert, V.; Langovski, J.; Smith, J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comp. Chem. 2008, 29, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V.; Trylska, J.; Chang, C.E.; McCammon, J.A. Flap opening dynamics in HIV-1 protease explored with a coarse-grained model. J. Struct. Biol. 2007, 157, 606–615. [Google Scholar] [CrossRef]
- Leonarski, F.; Trylska, J. RedMDStream: Parameterization and simulation toolbox for coarse-grained molecular dynamics models. Biophys. J. 2015, 108, 1843–1847. [Google Scholar] [CrossRef]
- Pauwels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyter, J.; De Clercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 1988, 20, 309–321. [Google Scholar] [CrossRef]
- Viscidi, R.; Farzadegan, H.; Leister, F.; Francisco, M.L.; Yolken, R. Enzyme immunoassay for detection of human immunodeficiency virus antigens in cells cultures. J. Clin. Microbiol. 1988, 26, 453–458, PMC266312. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, A. 03. 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Dessault Systemes BIOVIA. Discovery Studio Modeling Environment; Release 2017; Dessault Systemes: San Diego, CA, USA, 2016; Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery- studio/ (accessed on 28 June 2019).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solvent | Temperature (Temp.) [°C] | Time [h] | Yield [%] |
|---|---|---|---|---|
| 4 | KOH in CH3CH2OH | Reflux | 2 | 61 |
| 5 | NaOH in CH3OH/H2O/THF | Room temp. | 0.5 | 61 |
| 11 | NaOH in CH3OH/H2O/THF | Room temp. | 24 | 66 |
| Compound | CC50 (μM) | IC50 (μM) | TI | |
|---|---|---|---|---|
| bevirimat betulinic acid betulin | 29 ± 2.43 | 0.03 ± 0.009 | 967 | |
| 5 ± 2.44 | NO | - | ||
| 46±3.01 | NO | - | ||
| 9 |  | 18 ± 1.79 | NO | - |
| 10 |  | 80 ± 1.3 | >10 | <8 |
| 11 |  | >42 | >10 | >4 |
| 12a |  | 52 ± 1.07 | 1 ± 0.33 | 52 |
| 12b |  | 28 ± 1.64 | 0.6 ± 0.04 | 47 |
| 12c |  | 64 ± 1.84 | 4 ± 1.65 | 16 |
| 13a |  | >68 | 2 ± 0.72 | >35 |
| 13b |  | >68 | 8.8 ± 1.70 | >8 |
| 13c |  | >68 | >10 | >7 |
| 14a |  | >68 | 0.02 ± 0.01 | 3450 |
| 14b |  | >68 | 0.9 ± 0.12 | >75 |
| 14c |  | >68 | 1 ± 0.17 | >68 |
| Compound | 12a | 12b | 12c | 13a | 13b | 13c | 14a | 14b | 14c | BVM |
|---|---|---|---|---|---|---|---|---|---|---|
| Binding energy ΔG [kcal/mol] | −6.13 | −5.61 | −7.14 | −6.73 | −7.55 | −8.20 | −7.30 | −6.89 | −6.74 | −8.12 |
| Protein | Ligand | Interaction | |||
|---|---|---|---|---|---|
| Name | Chain: Residue | Name | Residue | Type | Distance [Å] |
| CTD-SP1 (5I4T) | I:Lys227 | BVM | carboxylate | salt bridge | 1.90 |
| J:Lys227 | carboxylate | attractive charge | 3.67 | ||
| I:Pro224 | C-21 of betulin | carbon–hydrogen bond | 2.90 | ||
| J:Lys227 | carboxylate | alkyl–alkyl | 2.11 | ||
| L:Gly223 | carboxylate | alkyl–alkyl | 2.30 | ||
| G:Lys158 | C-21 of betulin | alkyl–alkyl | 4.69 | ||
| L:Pro224 | C-21 of betulin | alkyl–alkyl | 5.41 | ||
| L:Lys158 | C-25 of betulin | alkyl–alkyl | 5.28 | ||
| K:Lys158 | C-29 of betulin | alkyl–alkyl | 3.67 | ||
| K:Lys158 | C-30 of betulin | alkyl–alkyl | 3.89 | ||
| G:Lys158 | C-21 of betulin | alkyl–alkyl | 4.55 | ||
| J:Lys158 | C-5′ of BVM | alkyl–alkyl | 3.71 | ||
| J:Lys158 | C-6′ of BVM | alkyl–alkyl | 4.76 | ||
| I:Lys227 | 14a | carboxylate | salt bridge | 1.99 | |
| G:Lys158 | carboxylate | carbon–hydrogen bond | 2.98 | ||
| K:Lys158 | carboxylate | carbon–hydrogen bond | 5.18 | ||
| J:Lys227 | phosphonate | conventional hydrogen bond | 2.23 | ||
| L:Lys158 | phosphonate | conventional hydrogen bond | 1.97 | ||
| J:Lys227 | ethyl | carbon–hydrogen bond | 2.40 | ||
| L:Lys158 | phosphonate | carbon–hydrogen bond | 2.19 | ||
| J:Gly222 | ethyl | carbon–hydrogen bond | 3.59 | ||
| I:Lys158 | C-25 of betulin | alkyl–alkyl | 5.23 | ||
| K:Lys158 | C-24 of betulin | alkyl–alkyl | 4.94 | ||
| I:Lys158 | C-7 of betulin | alkyl–alkyl | 4.38 | ||
| H:Lys158 | ethyl | alkyl–alkyl | 5.23 | ||
| J:Pro224 | ethyl | alkyl–alkyl | 4.08 | ||
| H:Lys158 | ethyl | alkyl–alkyl | 3.69 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrobak, E.; Marciniec, K.; Dąbrowska, A.; Pęcak, P.; Bębenek, E.; Kadela-Tomanek, M.; Bak, A.; Jastrzębska, M.; Boryczka, S. New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study. Int. J. Mol. Sci. 2019, 20, 5209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205209
Chrobak E, Marciniec K, Dąbrowska A, Pęcak P, Bębenek E, Kadela-Tomanek M, Bak A, Jastrzębska M, Boryczka S. New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study. International Journal of Molecular Sciences. 2019; 20(20):5209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205209
Chicago/Turabian StyleChrobak, Elwira, Krzysztof Marciniec, Aleksandra Dąbrowska, Paweł Pęcak, Ewa Bębenek, Monika Kadela-Tomanek, Andrzej Bak, Maria Jastrzębska, and Stanisław Boryczka. 2019. "New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study" International Journal of Molecular Sciences 20, no. 20: 5209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205209